IL-8 Released from Human Pancreatic Cancer and Tumor-Associated Stromal Cells Signals through a CXCR2-ERK1/2 Axis to Induce Muscle Atrophy

 , , and
, , and
Abstract
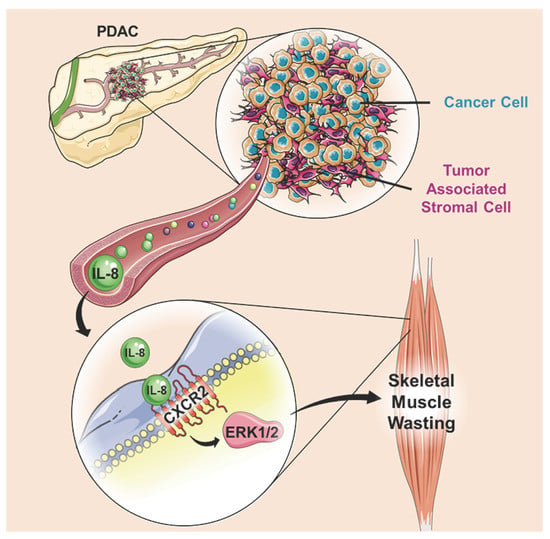
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Conditioned Medium from Human Pancreatic Cancer and Stromal Cells Induces Myotube Atrophy
2.2. Identification of Cytokines and Chemokines Released from Human Panceratic Cancer Cells and Human Tumor Associated Stromal Cells
2.3. Interleukin-8 is Sufficient to Induce Skeletal Muscle Atrophy
2.4. Biological Processes in Skeletal Muscle Regulated by IL-8
2.5. Interleukin-8 Activates STAT, Smad and ERK Signaling in Myotubes
2.6. ERK Inhibition Attenuates Interleukin-8 Induced Skeletal Muscle Wasting
2.7. Myotube Atrophy Induced by Human Pancreatic Cancer Cell CM Requires Active Interleukin-8/CXCR2
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Conditioned Media
4.3. Cytokine and Chemokine Analysis
4.4. Myotube Treatments and Diameter Measurements
4.5. Plasmids
4.6. Luciferase Reporter Assays
4.7. Western Blots
4.8. Animals
4.9. IL-8 Treatment of Mice
4.10. Muscle Immunohistochemistry and Measurements
4.11. Microarray and Gene Expression Analysis
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Evans, W.J.; Morley, J.E.; Argiles, J.; Bales, C.; Baracos, V.; Guttridge, D.; Jatoi, A.; Kalantar-Zadeh, K.; Lochs, H.; Mantovani, G.; et al. Cachexia: A new definition. Clin. Nutr. 2008, 27, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Fouladiun, M.; Korner, U.; Gunnebo, L.; Sixt-Ammilon, P.; Bosaeus, I.; Lundholm, K. Daily physical-rest activities in relation to nutritional state, metabolism, and quality of life in cancer patients with progressive cachexia. Clin. Cancer Res. 2007, 13, 6379–6385. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Hendifar, A.E.; Chang, J.I.; Huang, B.Z.; Tuli, R.; Wu, B.U. Cachexia, and not obesity, prior to pancreatic cancer diagnosis worsens survival and is negated by chemotherapy. J. Gastrointest Oncol. 2018, 9, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Kandarian, S.C.; Nosacka, R.L.; Delitto, A.E.; Judge, A.R.; Judge, S.M.; Ganey, J.D.; Moreira, J.D.; Jackman, R.W. Tumour-derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in c26 tumour-bearing mice. J. Cachexia Sarcopenia Muscle 2018, 9, 1109–1120. [Google Scholar] [CrossRef]
- Zhang, G.; Jin, B.; Li, Y.P. C/ebpbeta mediates tumour-induced ubiquitin ligase atrogin1/mafbx upregulation and muscle wasting. EMBO J. 2011, 30, 4323–4335. [Google Scholar] [CrossRef]
- Bohnert, K.R.; Goli, P.; Roy, A.; Sharma, A.K.; Xiong, G.; Gallot, Y.S.; Kumar, A. The toll-like receptor/myd88/xbp1 signaling axis mediates skeletal muscle wasting during cancer cachexia. Mol. Cell Biol. 2019, 39, MCB-00184. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, Z.; Ding, H.; Zhou, Y.; Doan, H.A.; Sin, K.W.T.; Zhu, Z.J.; Flores, R.; Wen, Y.; Gong, X.; et al. Tumor induces muscle wasting in mice through releasing extracellular hsp70 and hsp90. Nat. Commun. 2017, 8, 589. [Google Scholar] [CrossRef]
- Lopes, M.A.; Oliveira Franco, F.; Henriques, F.; Peres, S.B.; Batista, M.L., Jr. Llc tumor cells-derivated factors reduces adipogenesis in co-culture system. Heliyon 2018, 4, e00708. [Google Scholar] [CrossRef]
- Norden, D.M.; Devine, R.; McCarthy, D.O.; Wold, L.E. Storage conditions and passages alter il-6 secretion in c26 adenocarcinoma cell lines. MethodsX 2015, 2, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Strassmann, G.; Fong, M.; Kenney, J.S.; Jacob, C.O. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J. Clin. Invest. 1992, 89, 1681–1684. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.S.; Ley, K. Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 283, R7–R28. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Bussard, K.M.; Mutkus, L.; Stumpf, K.; Gomez-Manzano, C.; Marini, F.C. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016, 18, 84. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. Stellatum: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef]
- Hou, Y.C.; Wang, C.J.; Chao, Y.J.; Chen, H.Y.; Wang, H.C.; Tung, H.L.; Lin, J.T.; Shan, Y.S. Elevated serum interleukin-8 level correlates with cancer-related cachexia and sarcopenia: An indicator for pancreatic cancer outcomes. J. Clin. Med. 2018, 7, 502. [Google Scholar] [CrossRef]
- Pfitzenmaier, J.; Vessella, R.; Higano, C.S.; Noteboom, J.L.; Wallace, D., Jr.; Corey, E. Elevation of cytokine levels in cachectic patients with prostate carcinoma. Cancer 2003, 97, 1211–1216. [Google Scholar] [CrossRef]
- Krzystek-Korpacka, M.; Matusiewicz, M.; Diakowska, D.; Grabowski, K.; Blachut, K.; Kustrzeba-Wojcicka, I.; Banas, T. Impact of weight loss on circulating il-1, il-6, il-8, tnf-alpha, vegf-a, vegf-c and midkine in gastroesophageal cancer patients. Clin. Biochem. 2007, 40, 1353–1360. [Google Scholar] [CrossRef]
- Roberts, B.M.; Ahn, B.; Smuder, A.J.; Al-Rajhi, M.; Gill, L.C.; Beharry, A.W.; Powers, S.K.; Fuller, D.D.; Ferreira, L.F.; Judge, A.R. Diaphragm and ventilatory dysfunction during cancer cachexia. FASEB J. 2013, 27, 2600–2610. [Google Scholar] [CrossRef] [PubMed]
- Kays, J.K.; Shahda, S.; Stanley, M.; Bell, T.M.; O’Neill, B.H.; Kohli, M.D.; Couch, M.E.; Koniaris, L.G.; Zimmers, T.A. Three cachexia phenotypes and the impact of fat-only loss on survival in folfirinox therapy for pancreatic cancer. J. Cachexia Sarcopenia Muscle 2018, 9, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using david bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Kamei, Y.; Miura, S.; Suzuki, M.; Kai, Y.; Mizukami, J.; Taniguchi, T.; Mochida, K.; Hata, T.; Matsuda, J.; Aburatani, H.; et al. Skeletal muscle foxo1 (fkhr) transgenic mice have less skeletal muscle mass, down-regulated type i (slow twitch/red muscle) fiber genes, and impaired glycemic control. J. Biol. Chem. 2004, 279, 41114–41123. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Reed, S.A.; Sandesara, P.B.; Senf, S.M.; Judge, A.R. Inhibition of foxo transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J. 2012, 26, 987–1000. [Google Scholar] [CrossRef]
- Judge, S.M.; Wu, C.L.; Beharry, A.W.; Roberts, B.M.; Ferreira, L.F.; Kandarian, S.C.; Judge, A.R. Genome-wide identification of foxo-dependent gene networks in skeletal muscle during c26 cancer cachexia. BMC Cancer 2014, 14, 997. [Google Scholar] [CrossRef]
- Khal, J.; Wyke, S.M.; Russell, S.T.; Hine, A.V.; Tisdale, M.J. Expression of the ubiquitin-proteasome pathway and muscle loss in experimental cancer cachexia. Br. J. Cancer 2005, 93, 774–780. [Google Scholar] [CrossRef]
- Judge, S.M.; Nosacka, R.L.; Delitto, D.; Gerber, M.H.; Cameron, M.E.; Trevino, J.G.; Judge, A.R. Skeletal muscle fibrosis in pancreatic cancer patients with respect to survival. JNCI Cancer Spectr. 2018, 2, pky043. [Google Scholar] [CrossRef]
- Cai, D.; Frantz, J.D.; Tawa, N.E., Jr.; Melendez, P.A.; Oh, B.C.; Lidov, H.G.; Hasselgren, P.O.; Frontera, W.R.; Lee, J.; Glass, D.J.; et al. Ikkbeta/nf-kappab activation causes severe muscle wasting in mice. Cell 2004, 119, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. Jak/stat3 pathway inhibition blocks skeletal muscle wasting downstream of il-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.L.; Walton, K.L.; Hagg, A.; Colgan, T.D.; Johnson, K.; Qian, H.; Gregorevic, P.; Harrison, C.A. Specific targeting of tgf-beta family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc. Natl. Acad. Sci. USA 2017, 114, E5266–E5275. [Google Scholar] [PubMed] [Green Version]
- Knall, C.; Young, S.; Nick, J.A.; Buhl, A.M.; Worthen, G.S.; Johnson, G.L. Interleukin-8 regulation of the ras/raf/mitogen-activated protein kinase pathway in human neutrophils. J. Biol. Chem. 1996, 271, 2832–2838. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.P.; Liu, C.; Chiang, F.Y.; Wang, L.F.; Lee, K.W.; Chen, W.T.; Kuo, P.L.; Liang, C.H. Il-8 promotes inflammatory mediators and stimulates activation of p38 mapk/erk-nf-kappab pathway and reduction of jnk in hnscc. Oncotarget 2017, 8, 56375–56388. [Google Scholar] [CrossRef] [Green Version]
- MacManus, C.F.; Pettigrew, J.; Seaton, A.; Wilson, C.; Maxwell, P.J.; Berlingeri, S.; Purcell, C.; McGurk, M.; Johnston, P.G.; Waugh, D.J. Interleukin-8 signaling promotes translational regulation of cyclin d in androgen-independent prostate cancer cells. Mol. Cancer Res. 2007, 5, 737–748. [Google Scholar] [CrossRef] [Green Version]
- Luppi, F.; Longo, A.M.; de Boer, W.I.; Rabe, K.F.; Hiemstra, P.S. Interleukin-8 stimulates cell proliferation in non-small cell lung cancer through epidermal growth factor receptor transactivation. Lung Cancer 2007, 56, 25–33. [Google Scholar] [CrossRef]
- Venkatakrishnan, G.; Salgia, R.; Groopman, J.E. Chemokine receptors cxcr-1/2 activate mitogen-activated protein kinase via the epidermal growth factor receptor in ovarian cancer cells. J. Biol. Chem. 2000, 275, 6868–6875. [Google Scholar] [CrossRef] [Green Version]
- Penna, F.; Costamagna, D.; Fanzani, A.; Bonelli, G.; Baccino, F.M.; Costelli, P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by erk inhibition. PLoS ONE 2010, 5, e13604. [Google Scholar] [CrossRef]
- Park, J.I.; Strock, C.J.; Ball, D.W.; Nelkin, B.D. The ras/raf/mek/extracellular signal-regulated kinase pathway induces autocrine-paracrine growth inhibition via the leukemia inhibitory factor/jak/stat pathway. Mol. Cell Biol. 2003, 23, 543–554. [Google Scholar] [CrossRef] [Green Version]
- Jinnin, M.; Ihn, H.; Tamaki, K. Characterization of sis3, a novel specific inhibitor of smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol. Pharmacol. 2006, 69, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Kennedy, C.L.; Lee, M.; Gao, Y.; Xia, H.; Olguin, F.; Fraga, D.A.; Ayers, K.; Choi, S.; Kim, M.; et al. Smad3 initiates oxidative stress and proteolysis that underlies diaphragm dysfunction during mechanical ventilation. Sci. Rep. 2017, 7, 14530. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Patera, A.C.; Pong-Kennedy, A.; Deno, G.; Gonsiorek, W.; Manfra, D.J.; Vassileva, G.; Zeng, M.; Jackson, C.; Sullivan, L.; et al. Murine cxcr1 is a functional receptor for gcp-2/cxcl6 and interleukin-8/cxcl8. J. Biol Chem 2007, 282, 11658–11666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delitto, D.; Delitto, A.E.; DiVita, B.B.; Pham, K.; Han, S.; Hartlage, E.R.; Newby, B.N.; Gerber, M.H.; Behrns, K.E.; Moldawer, L.L.; et al. Human pancreatic cancer cells induce a myd88-dependent stromal response to promote a tumor-tolerant immune microenvironment. Cancer Res. 2017, 77, 672–683. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.L.; Walton, K.L.; Qian, H.; Colgan, T.D.; Hagg, A.; Watt, M.J.; Harrison, C.A.; Gregorevic, P. Differential effects of il6 and activin a in the development of cancer-associated cachexia. Cancer Res. 2016, 76, 5372–5382. [Google Scholar] [CrossRef] [Green Version]
- Molotkov, A.; Satoh, M.; Tohyama, C. Tumor growth and food intake in interleukin-6 gene knock-out mice. Cancer Lett. 1998, 132, 187–192. [Google Scholar] [CrossRef]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef]
- Matsushima, K.; Oppenheim, J.J. Interleukin 8 and mcaf: Novel inflammatory cytokines inducible by il 1 and tnf. Cytokine 1989, 1, 2–13. [Google Scholar] [CrossRef]
- Bazzoni, F.; Cassatella, M.A.; Rossi, F.; Ceska, M.; Dewald, B.; Baggiolini, M. Phagocytosing neutrophils produce and release high amounts of the neutrophil-activating peptide 1/interleukin 8. J. Exp. Med. 1991, 173, 771–774. [Google Scholar] [CrossRef]
- Gregory, H.; Young, J.; Schroder, J.M.; Mrowietz, U.; Christophers, E. Structure determination of a human lymphocyte derived neutrophil activating peptide (lynap). Biochem. Biophys. Res. Commun. 1988, 151, 883–890. [Google Scholar] [CrossRef]
- Schroder, J.M.; Sticherling, M.; Henneicke, H.H.; Preissner, W.C.; Christophers, E. Il-1 alpha or tumor necrosis factor-alpha stimulate release of three nap-1/il-8-related neutrophil chemotactic proteins in human dermal fibroblasts. J. Immunol. 1990, 144, 2223–2232. [Google Scholar] [PubMed]
- Gimbrone, M.A., Jr.; Obin, M.S.; Brock, A.F.; Luis, E.A.; Hass, P.E.; Hebert, C.A.; Yip, Y.K.; Leung, D.W.; Lowe, D.G.; Kohr, W.J.; et al. Endothelial interleukin-8: A novel inhibitor of leukocyte-endothelial interactions. Science 1989, 246, 1601–1603. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Chauveau, C.; Brouillet, J.P.; Lucas, A.; Lacroix, M.; Licznar, A.; Vignon, F.; Lazennec, G. Il-8 expression and its possible relationship with estrogen-receptor-negative status of breast cancer cells. Oncogene 2003, 22, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Slaton, J.W.; Kim, S.J.; Perrotte, P.; Eve, B.Y.; Bar-Eli, M.; Radinsky, R.; Dinney, C.P. Interleukin 8 expression regulates tumorigenicity and metastasis in human bladder cancer. Cancer Res. 2000, 60, 2290–2299. [Google Scholar] [CrossRef] [PubMed]
- Kitadai, Y.; Haruma, K.; Sumii, K.; Yamamoto, S.; Ue, T.; Yokozaki, H.; Yasui, W.; Ohmoto, Y.; Kajiyama, G.; Fidler, I.J.; et al. Expression of interleukin-8 correlates with vascularity in human gastric carcinomas. Am. J. Pathol. 1998, 152, 93–100. [Google Scholar] [PubMed]
- Alfaro, C.; Sanmamed, M.F.; Rodriguez-Ruiz, M.E.; Teijeira, A.; Onate, C.; Gonzalez, A.; Ponz, M.; Schalper, K.A.; Perez-Gracia, J.L.; Melero, I. Interleukin-8 in cancer pathogenesis, treatment and follow-up. Cancer Treat. Rev. 2017, 60, 24–31. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Carranza-Rua, O.; Alfaro, C.; Onate, C.; Martin-Algarra, S.; Perez, G.; Landazuri, S.F.; Gonzalez, A.; Gross, S.; Rodriguez, I.; et al. Serum interleukin-8 reflects tumor burden and treatment response across malignancies of multiple tissue origins. Clin. Cancer Res. 2014, 20, 5697–5707. [Google Scholar] [CrossRef] [Green Version]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The il-8/il-8r axis: A double agent in tumor immune resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Krzystek-Korpacka, M.; Matusiewicz, M.; Diakowska, D.; Grabowski, K.; Blachut, K.; Konieczny, D.; Kustrzeba-Wojcicka, I.; Terlecki, G.; Banas, T. Elevation of circulating interleukin-8 is related to lymph node and distant metastases in esophageal squamous cell carcinomas--implication for clinical evaluation of cancer patient. Cytokine 2008, 41, 232–239. [Google Scholar] [CrossRef]
- Delitto, D.; Judge, S.M.; George, T.J., Jr.; Sarosi, G.A.; Thomas, R.M.; Behrns, K.E.; Hughes, S.J.; Judge, A.R.; Trevino, J.G. A clinically applicable muscular index predicts long-term survival in resectable pancreatic cancer. Surgery 2017, 161, 930–938. [Google Scholar] [CrossRef]
- Bozic, C.R.; Gerard, N.P.; von Uexkull-Guldenband, C.; Kolakowski, L.F.; Conklyn, M.J.; Breslow, R.; Showell, H.J.; Gerard, C. The murine interleukin 8 type b receptor homologue and its ligands. Expression and biological characterization. J. Biol. Chem. 1994, 269, 29355–29358. [Google Scholar] [PubMed]
- Rovai, L.E.; Herschman, H.R.; Smith, J.B. The murine neutrophil-chemoattractant chemokines lix, kc, and mip-2 have distinct induction kinetics, tissue distributions, and tissue-specific sensitivities to glucocorticoid regulation in endotoxemia. J. Leukoc. Biol. 1998, 64, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef] [PubMed]
- Cury, S.S.; de Moraes, D.; Freire, P.P.; de Oliveira, G.; Marques, D.V.P.; Fernandez, G.J.; Dal-Pai-Silva, M.; Hasimoto, E.N.; Dos Reis, P.P.; Rogatto, S.R.; et al. Tumor transcriptome reveals high expression of il-8 in non-small cell lung cancer patients with low pectoralis muscle area and reduced survival. Cancers 2019, 11, 1251. [Google Scholar] [CrossRef] [Green Version]
- Knall, C.; Worthen, G.S.; Johnson, G.L. Interleukin 8-stimulated phosphatidylinositol-3-kinase activity regulates the migration of human neutrophils independent of extracellular signal-regulated kinase and p38 mitogen-activated protein kinases. Proc. Natl. Acad. Sci. USA 1997, 94, 3052–3057. [Google Scholar] [CrossRef] [Green Version]
- Britschgi, A.; Andraos, R.; Brinkhaus, H.; Klebba, I.; Romanet, V.; Muller, U.; Murakami, M.; Radimerski, T.; Bentires-Alj, M. Jak2/stat5 inhibition circumvents resistance to pi3k/mtor blockade: A rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell 2012, 22, 796–811. [Google Scholar] [CrossRef] [Green Version]
- Pruijt, J.F.; Verzaal, P.; van Os, R.; de Kruijf, E.J.; van Schie, M.L.; Mantovani, A.; Vecchi, A.; Lindley, I.J.; Willemze, R.; Starckx, S.; et al. Neutrophils are indispensable for hematopoietic stem cell mobilization induced by interleukin-8 in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 6228–6233. [Google Scholar] [CrossRef] [Green Version]
- Simonet, W.S.; Hughes, T.M.; Nguyen, H.Q.; Trebasky, L.D.; Danilenko, D.M.; Medlock, E.S. Long-term impaired neutrophil migration in mice overexpressing human interleukin-8. J. Clin. Invest. 1994, 94, 1310–1319. [Google Scholar] [CrossRef] [Green Version]
- Kucharzik, T.; Williams, I.R. Neutrophil migration across the intestinal epithelial barrier--summary of in vitro data and description of a new transgenic mouse model with doxycycline-inducible interleukin-8 expression in intestinal epithelial cells. Pathobiology 2002, 70, 143–149. [Google Scholar] [CrossRef]
- Asfaha, S.; Dubeykovskiy, A.N.; Tomita, H.; Yang, X.; Stokes, S.; Shibata, W.; Friedman, R.A.; Ariyama, H.; Dubeykovskaya, Z.A.; Muthupalani, S.; et al. Mice that express human interleukin-8 have increased mobilization of immature myeloid cells, which exacerbates inflammation and accelerates colon carcinogenesis. Gastroenterology 2013, 144, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Manna, S.K.; Ramesh, G.T. Interleukin-8 induces nuclear transcription factor-kappab through a traf6-dependent pathway. J. Biol. Chem. 2005, 280, 7010–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, D.N.; Kandarian, S.C.; Jackman, R.W. A key role for leukemia inhibitory factor in c26 cancer cachexia. J. Biol. Chem. 2015, 290, 19976–19986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pham, K.; Delitto, D.; Knowlton, A.E.; Hartlage, E.R.; Madhavan, R.; Gonzalo, D.H.; Thomas, R.M.; Behrns, K.E.; George, T.J., Jr.; Hughes, S.J.; et al. Isolation of pancreatic cancer cells from a patient-derived xenograft model allows for practical expansion and preserved heterogeneity in culture. Am. J. Pathol. 2016, 186, 1537–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, C.J.; Harbison, M.T.; Kuniyasu, H.; Eue, I.; Fidler, I.J. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia 1999, 1, 50–62. [Google Scholar] [CrossRef] [Green Version]
- Trevino, J.G.; Summy, J.M.; Gray, M.J.; Nilsson, M.B.; Lesslie, D.P.; Baker, C.H.; Gallick, G.E. Expression and activity of src regulate interleukin-8 expression in pancreatic adenocarcinoma cells: Implications for angiogenesis. Cancer Res. 2005, 65, 7214–7222. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Delitto, D.; Zhang, D.; Sorenson, H.L.; Sarosi, G.A.; Thomas, R.M.; Behrns, K.E.; Wallet, S.M.; Trevino, J.G.; Hughes, S.J. Primary outgrowth cultures are a reliable source of human pancreatic stellate cells. Lab. Invest. 2015, 95, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Hunter, R.B.; Stevenson, E.; Koncarevic, A.; Mitchell-Felton, H.; Essig, D.A.; Kandarian, S.C. Activation of an alternative nf-kappab pathway in skeletal muscle during disuse atrophy. FASEB J. 2002, 16, 529–538. [Google Scholar] [CrossRef]
- Storz, P.; Doppler, H.; Toker, A. Protein kinase d mediates mitochondrion-to-nucleus signaling and detoxification from mitochondrial reactive oxygen species. Mol. Cell Biol. 2005, 25, 8520–8530. [Google Scholar] [CrossRef] [Green Version]
- Piek, E.; Westermark, U.; Kastemar, M.; Heldin, C.H.; van Zoelen, E.J.; Nister, M.; Ten Dijke, P. Expression of transforming-growth-factor (tgf)-beta receptors and smad proteins in glioblastoma cell lines with distinct responses to tgf-beta1. Int. J. Cancer 1999, 80, 756–763. [Google Scholar] [CrossRef]
- Besser, D.; Bromberg, J.F.; Darnell, J.E., Jr.; Hanafusa, H. A single amino acid substitution in the v-eyk intracellular domain results in activation of stat3 and enhances cellular transformation. Mol. Cell Biol. 1999, 19, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Senf, S.M.; Dodd, S.L.; McClung, J.M.; Judge, A.R. Hsp70 overexpression inhibits nf-kappab and foxo3a transcriptional activities and prevents skeletal muscle atrophy. FASEB J. 2008, 22, 3836–3845. [Google Scholar] [CrossRef] [PubMed]
- Krämer, A.; Green, J.; Pollard, J.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Callaway, C.S.; Delitto, A.E.; D’Lugos, A.C.; Patel, R.; Nosacka, R.L.; Delitto, D.; Deyhle, M.R.; Trevino, J.G.; Judge, S.M.; Judge, A.R. IL-8 Released from Human Pancreatic Cancer and Tumor-Associated Stromal Cells Signals through a CXCR2-ERK1/2 Axis to Induce Muscle Atrophy. Cancers 2019, 11, 1863. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121863
Callaway CS, Delitto AE, D’Lugos AC, Patel R, Nosacka RL, Delitto D, Deyhle MR, Trevino JG, Judge SM, Judge AR. IL-8 Released from Human Pancreatic Cancer and Tumor-Associated Stromal Cells Signals through a CXCR2-ERK1/2 Axis to Induce Muscle Atrophy. Cancers. 2019; 11(12):1863. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121863
Chicago/Turabian StyleCallaway, Chandler S., Andrea E. Delitto, Andrew C. D’Lugos, Rohan Patel, Rachel L. Nosacka, Daniel Delitto, Michael R. Deyhle, Jose G. Trevino, Sarah M. Judge, and Andrew R. Judge. 2019. "IL-8 Released from Human Pancreatic Cancer and Tumor-Associated Stromal Cells Signals through a CXCR2-ERK1/2 Axis to Induce Muscle Atrophy" Cancers 11, no. 12: 1863. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11121863