SIRT2 Contributes to the Resistance of Melanoma Cells to the Multikinase Inhibitor Dasatinib

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Identification of SIRT2-Dependent Genetic Information in Melanoma Cells
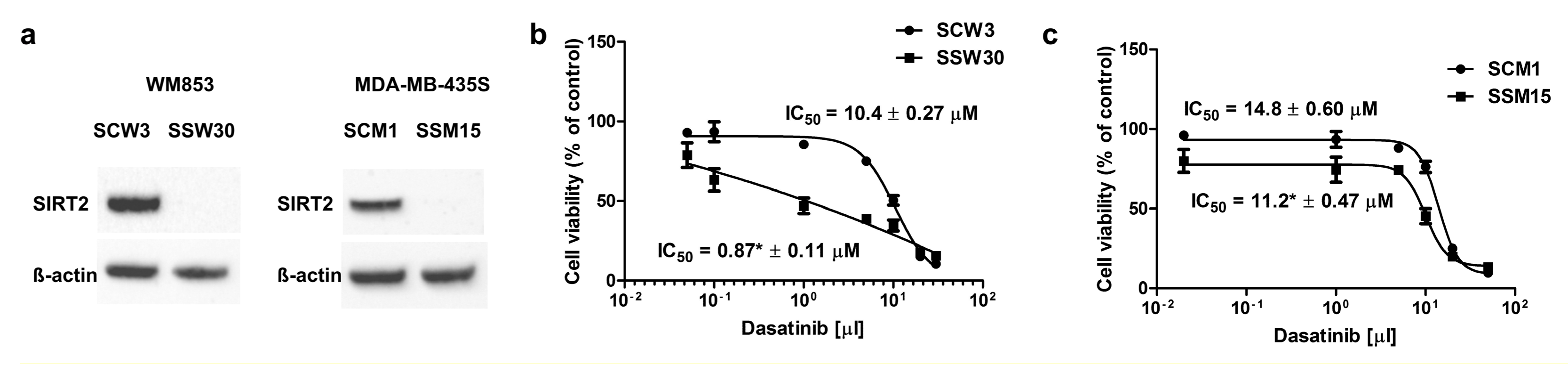
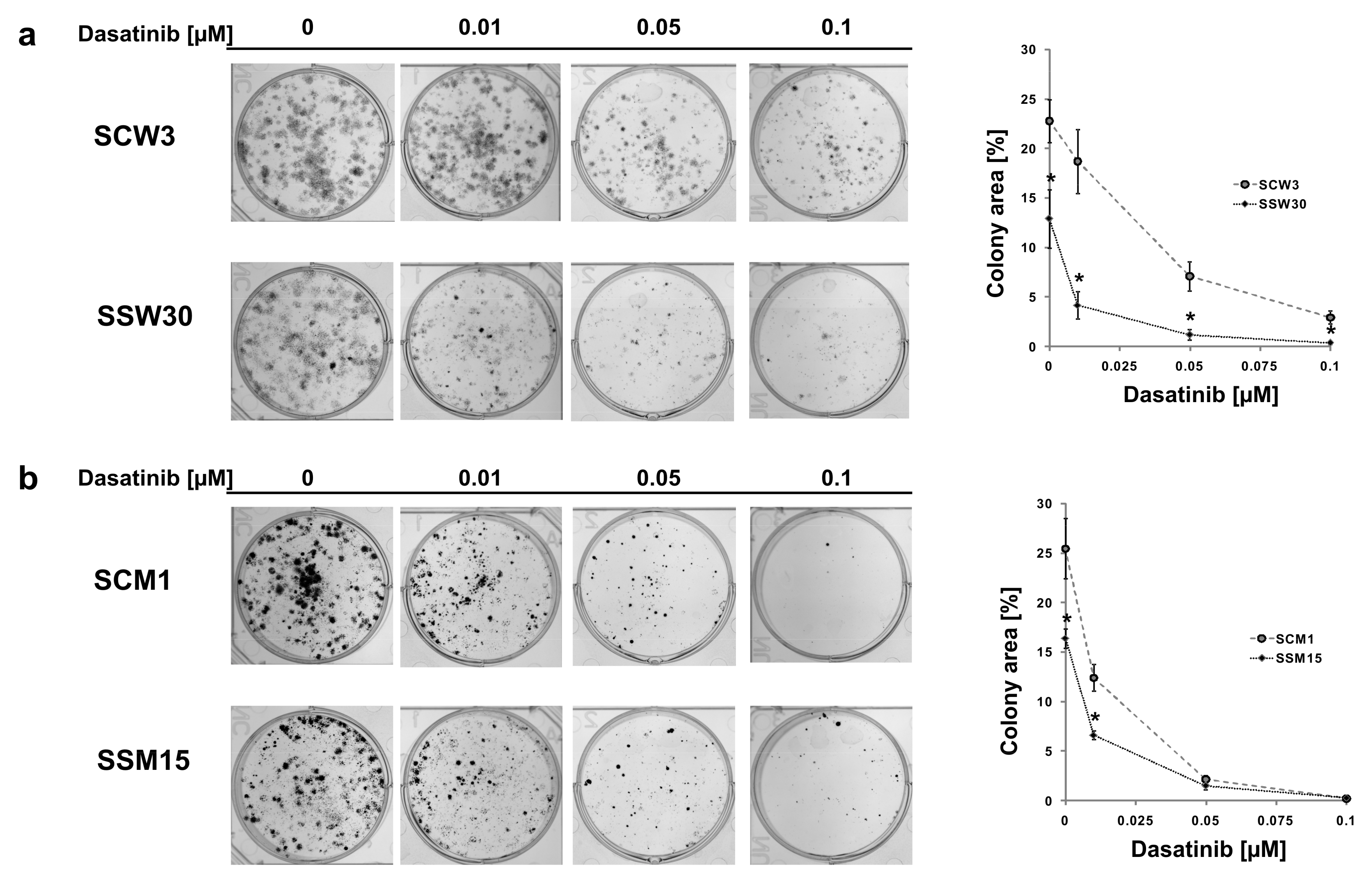
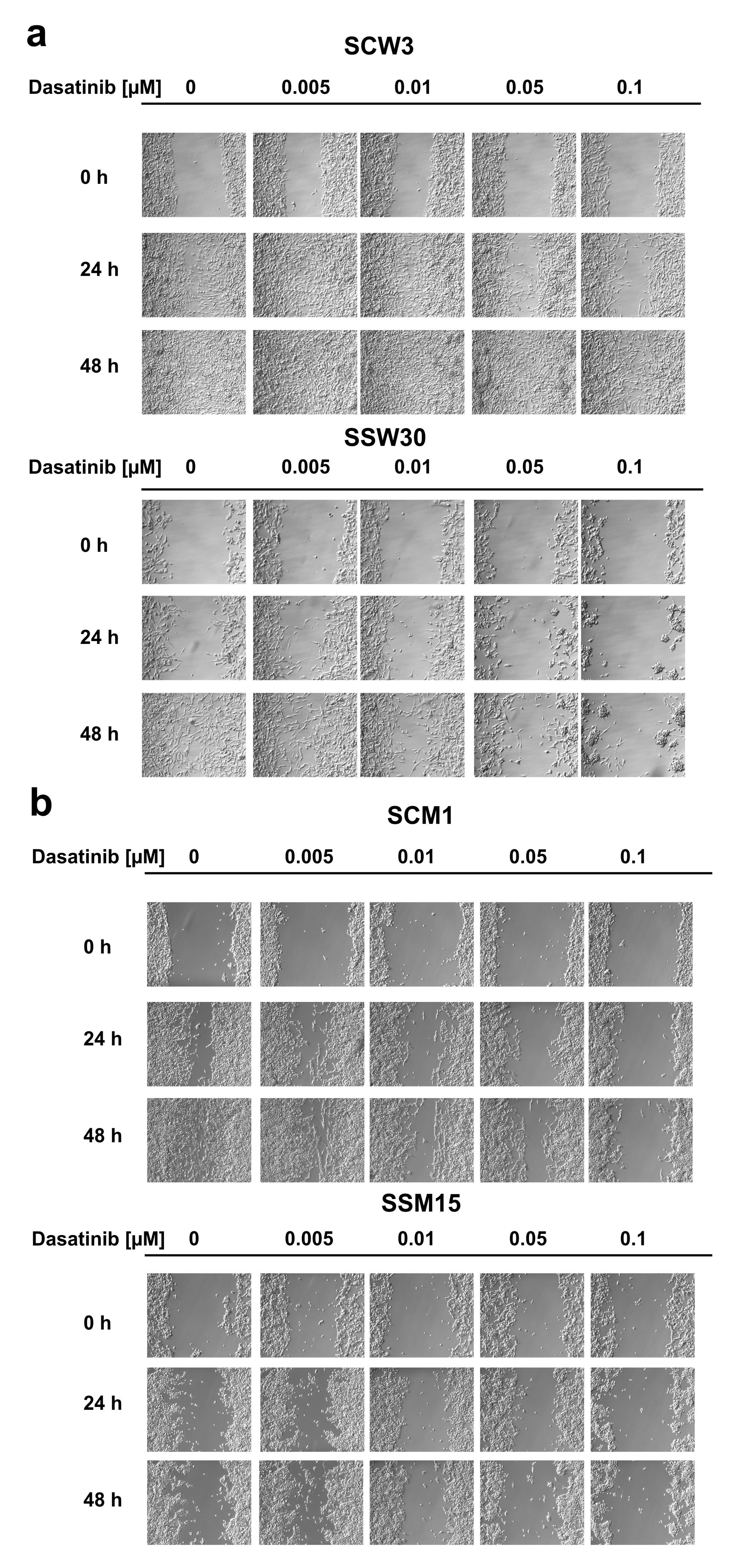
2.2. SIRT2-Inhibition Sensitizes Melanoma Cells to Dasatinib
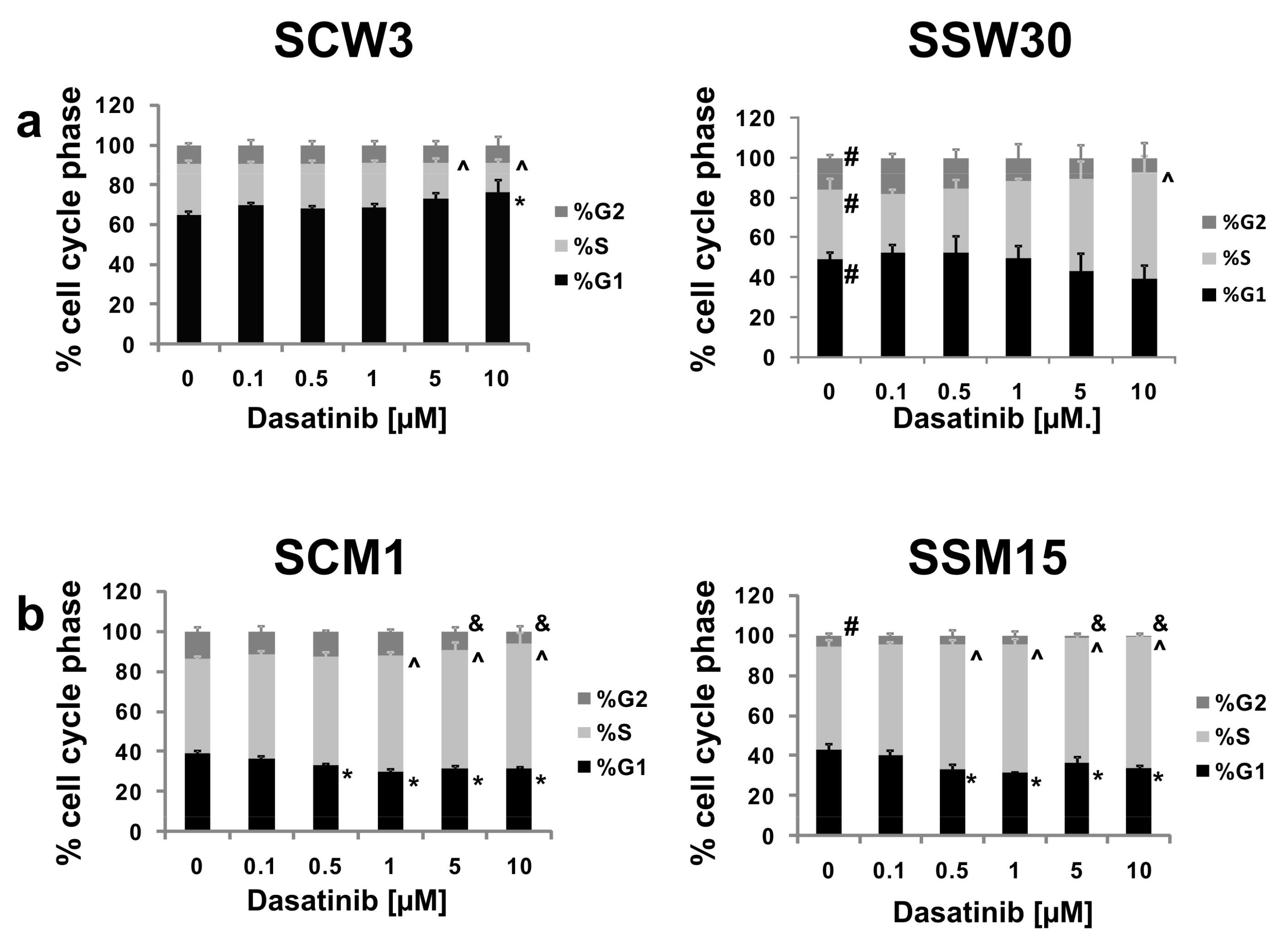
2.3. Effects of SIRT2 Inhibition and Dasatinib on the Cell Cycle Distribution
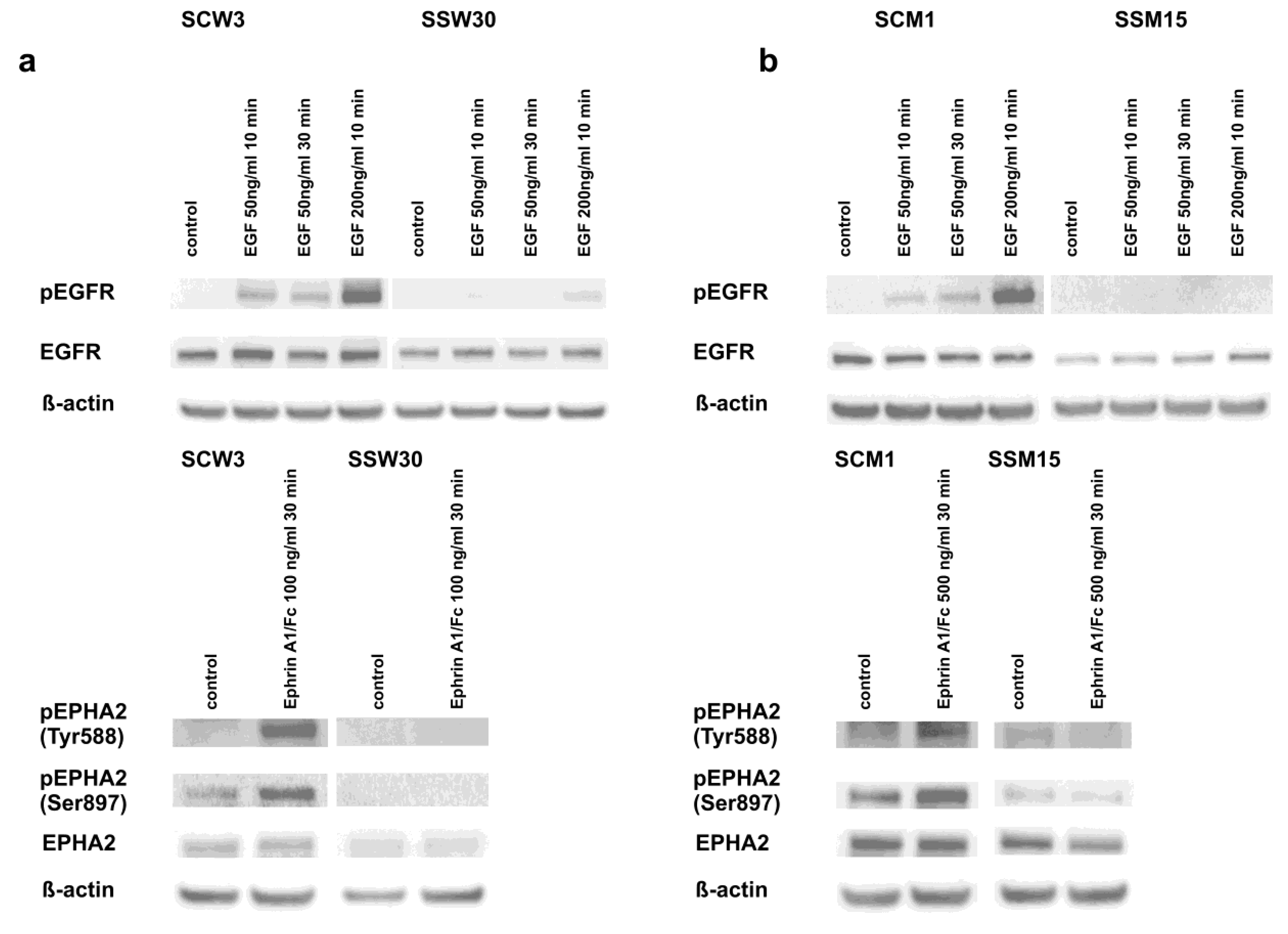
2.4. Modulation of Signaling Pathways by SIRT2 and Dasatinib
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Cell Culture and Reagents
4.2. Stable SIRT2 Knockdown Melanoma Cell Line Generation
4.3. RNA Extraction, Reverse Transcription and Quantitative PCR
4.4. Western Blotting
4.5. Preparation of Libraries, Sequencing and RNA-seq Data Analysis
4.6. Gene Ontology Analysis
4.7. Cell Cycle Analysis
4.8. In Vitro Scratch Assay
4.9. Detection of Early Cellular Membrane Damage
4.10. Cell Viability Measurements
4.11. Caspase 3/7 Activity Measurements
4.12. Colony Formation Assay
4.13. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef]
- Serrone, L.; Zeuli, M.; Sega, F.M.; Cognetti, F. Dacarbazine-based chemotherapy for metastatic melanoma: Thirty-year experience overview. J. Exp. Clin. Cancer Res. 2000, 19, 21–34. [Google Scholar] [PubMed]
- Jilaveanu, L.B.; Aziz, S.A.; Kluger, H.M. Chemotherapy and biologic therapies for melanoma: Do they work? Clin. Dermatol. 2009, 27, 614–625. [Google Scholar] [CrossRef]
- Atkins, M.B.; Lotze, M.T.; Dutcher, J.P.; Fisher, R.I.; Weiss, G.; Margolin, K.; Abrams, J.; Sznol, M.; Parkinson, D.; Hawkins, M.; et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: Analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 1999, 17, 2105–2116. [Google Scholar] [CrossRef]
- Robinson, W.A.; Mughal, T.I.; Thomas, M.R.; Johnson, M.; Spiegel, R.J. Treatment of metastatic malignant melanoma with recombinant interferon alpha 2. Immunobiology 1986, 172, 275–282. [Google Scholar] [CrossRef]
- Banzi, M.; De Blasio, S.; Lallas, A.; Longo, C.; Moscarella, E.; Alfano, R.; Argenziano, G. Dabrafenib: A new opportunity for the treatment of BRAF v600-positive melanoma. Oncol. Targets Ther. 2016, 9, 2725–2733. [Google Scholar] [PubMed]
- Legha, S.S.; Ring, S.; Papadopoulos, N.; Plager, C.; Chawla, S.; Benjamin, R. A prospective evaluation of a triple-drug regimen containing cisplatin, vinblastine, and dacarbazine (CVD) for metastatic melanoma. Cancer 1989, 64, 2024–2029. [Google Scholar] [CrossRef]
- Del Prete, S.A.; Maurer, L.H.; O’Donnell, J.; Forcier, R.J.; LeMarbre, P. Combination chemotherapy with cisplatin, carmustine, dacarbazine, and tamoxifen in metastatic melanoma. Cancer Treat. Rep. 1984, 68, 1403–1405. [Google Scholar] [PubMed]
- Fierro, M.T.; Bertero, M.; Novelli, M.; Appino, A.; Doveil, G.C.; Colonna, S.; Bernengo, M.G. Therapy for metastatic melanoma: Effective combination of dacarbazine, carmustine, cisplatin and tamoxifen. Melanoma Res. 1993, 3, 127–131. [Google Scholar] [PubMed]
- Chen, K.G.; Leapman, R.D.; Zhang, G.; Lai, B.; Valencia, J.C.; Cardarelli, C.O.; Vieira, W.D.; Hearing, V.J.; Gottesman, M.M. Influence of melanosome dynamics on melanoma drug sensitivity. J. Natl. Cancer Inst. 2009, 101, 1259–1271. [Google Scholar] [CrossRef]
- Chen, K.G.; Valencia, J.C.; Gillet, J.P.; Hearing, V.J.; Gottesman, M.M. Involvement of ABC transporters in melanogenesis and the development of multidrug resistance of melanoma. Pigment. Cell Melanoma Res. 2009, 22, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Jansen, B.; Wacheck, V.; Heere-Ress, E.; Schlagbauer-Wadl, H.; Hoeller, C.; Lucas, T.; Hoermann, M.; Hollenstein, U.; Wolff, K.; Pehamberger, H. Chemosensitisation of malignant melanoma by BCL2 antisense therapy. Lancet 2000, 356, 1728–1733. [Google Scholar] [CrossRef]
- Wolter, K.G.; Verhaegen, M.; Fernandez, Y.; Nikolovska-Coleska, Z.; Riblett, M.; de la Vega, C.M.; Wang, S.; Soengas, M.S. Therapeutic window for melanoma treatment provided by selective effects of the proteasome on BCL-2 proteins. Cell Death Differ. 2007, 14, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Tron, V.A.; Reed, J.C.; Mah, K.J.; Krajewska, M.; Li, G.; Zhou, X.; Ho, V.C.; Trotter, M.J. Expression of apoptosis regulators in cutaneous malignant melanoma. Clin. Cancer Res. 1998, 4, 1865–1871. [Google Scholar]
- Nikolaou, V.A.; Stratigos, A.J.; Flaherty, K.T.; Tsao, H. Melanoma: New insights and new therapies. J. Investig. Dermatol. 2012, 132, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Chartrain, M.; Riond, J.; Stennevin, A.; Vandenberghe, I.; Gomes, B.; Lamant, L.; Meyer, N.; Gairin, J.E.; Guilbaud, N.; Annereau, J.P. Melanoma chemotherapy leads to the selection of ABCB5-expressing cells. PLoS ONE 2012, 7, e36762. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Khaled, M.; Fisher, D.E. Mitf: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef]
- Kaur, A.; Webster, M.R.; Marchbank, K.; Behera, R.; Ndoye, A.; Kugel, C.H., 3rd; Dang, V.M.; Appleton, J.; O’Connell, M.P.; Cheng, P.; et al. sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature 2016, 532, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Winder, M.; Viros, A. Mechanisms of drug resistance in melanoma. Handb. Exp. Pharmacol. 2018, 249, 91–108. [Google Scholar]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the RAF-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF v600e mutation. N Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Hauschild, A.; Grob, J.J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H., Jr.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.J.; et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J. Clin. Oncol 2013, 31, 3205–3211. [Google Scholar] [CrossRef]
- Medina, T.M.; Lewis, K.D. The evolution of combined molecular targeted therapies to advance the therapeutic efficacy in melanoma: A highlight of vemurafenib and cobimetinib. Oncol. Targets Ther. 2016, 9, 3739–3752. [Google Scholar]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(v600e) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Lito, P.; Rosen, N.; Solit, D.B. Tumor adaptation and resistance to RAF inhibitors. Nat. Med. 2013, 19, 1401–1409. [Google Scholar] [CrossRef]
- Maertens, O.; Johnson, B.; Hollstein, P.; Frederick, D.T.; Cooper, Z.A.; Messiaen, L.; Bronson, R.T.; McMahon, M.; Granter, S.; Flaherty, K.; et al. Elucidating distinct roles for nf1 in melanomagenesis. Cancer Discov. 2013, 3, 338–349. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(v600e). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Wilking, M.J.; Singh, C.K.; Nihal, M.; Ndiaye, M.A.; Ahmad, N. Sirtuin deacetylases: A new target for melanoma management. Cell Cycle 2014, 13, 2821–2826. [Google Scholar] [CrossRef]
- Garcia-Peterson, L.M.; Wilking-Busch, M.J.; Ndiaye, M.A.; Philippe, C.G.A.; Setaluri, V.; Ahmad, N. Sirtuins in skin and skin cancers. Skin Pharmacol. Physiol. 2017, 30, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, C.B.; Sherman, J.M.; Devine, S.E.; Cameron, E.E.; Pillus, L.; Boeke, J.D. The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 1995, 9, 2888–2902. [Google Scholar] [CrossRef]
- Tanny, J.C.; Dowd, G.J.; Huang, J.; Hilz, H.; Moazed, D. An enzymatic activity in the yeast SIR2 protein that is essential for gene silencing. Cell 1999, 99, 735–745. [Google Scholar] [CrossRef]
- Frye, R.A. Phylogenetic classification of prokaryotic and eukaryotic SIR2-like proteins. Biochem. Biophys. Res. Commun. 2000, 273, 793–798. [Google Scholar] [CrossRef]
- Nakagawa, T.; Guarente, L. Sirtuins at a glance. J. Cell Sci. 2011, 124, 833–838. [Google Scholar] [CrossRef]
- Huang, J.Y.; Hirschey, M.D.; Shimazu, T.; Ho, L.; Verdin, E. Mitochondrial sirtuins. Biochim. Biophys. Acta 2010, 1804, 1645–1651. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian sirtuins: Biological insights and disease relevance. Annu. Rev. Pathol. 2010, 5, 253–295. [Google Scholar] [CrossRef] [PubMed]
- Mendes, K.L.; Lelis, D.F.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef]
- Li, W.; Zhang, B.; Tang, J.; Cao, Q.; Wu, Y.; Wu, C.; Guo, J.; Ling, E.A.; Liang, F. Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J. Neurosci. 2007, 27, 2606–2616. [Google Scholar] [CrossRef]
- Wang, C.; Wang, M.W.; Tashiro, S.; Onodera, S.; Ikejima, T. Roles of SIRT1 and phosphoinositide 3-Oh kinase/protein kinase C pathways in evodiamine-induced human melanoma A375-S2 cell death. J. Pharmacol. Sci. 2005, 97, 494–500. [Google Scholar] [CrossRef]
- Kunimoto, R.; Jimbow, K.; Tanimura, A.; Sato, M.; Horimoto, K.; Hayashi, T.; Hisahara, S.; Sugino, T.; Hirobe, T.; Yamashita, T.; et al. SIRT1 regulates lamellipodium extension and migration of melanoma cells. J. Investig. Dermatol. 2017, 134, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Ohanna, M.; Bonet, C.; Bille, K.; Allegra, M.; Davidson, I.; Bahadoran, P.; Lacour, J.P.; Ballotti, R.; Bertolotto, C. SIRT1 promotes proliferation and inhibits the senescence-like phenotype in human melanoma cells. Oncotarget 2014, 5, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Karwaciak, I.; Gorzkiewicz, M.; Ryba, K.; Dastych, J.; Pulaski, L.; Ratajewski, M. AC-93253 triggers the downregulation of melanoma progression markers and the inhibition of melanoma cell proliferation. Chem. Biol. Interact. 2015, 236, 9–18. [Google Scholar] [CrossRef]
- Bajpe, P.K.; Prahallad, A.; Horlings, H.; Nagtegaal, I.; Beijersbergen, R.; Bernards, R. A chromatin modifier genetic screen identifies SIRT2 as a modulator of response to targeted therapies through the regulation of MEK kinase activity. Oncogene 2015, 34, 531–536. [Google Scholar] [CrossRef]
- Lai, Y.H.; Lin, S.Y.; Wu, Y.S.; Chen, H.W.; Chen, J.J.W. Ac-93253 iodide, a novel SRC inhibitor, suppresses NSCLC progression by modulating multiple SRC-related signaling pathways. J. Hematol. Oncol. 2017, 10, 172. [Google Scholar] [CrossRef]
- McGlynn, L.M.; Zino, S.; MacDonald, A.I.; Curle, J.; Reilly, J.E.; Mohammed, Z.M.; McMillan, D.C.; Mallon, E.; Payne, A.P.; Edwards, J.; et al. SIRT2: Tumour suppressor or tumour promoter in operable breast cancer? Eur. J. Cancer 2014, 50, 290–301. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DEseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Rofstad, E.K. Integrins as therapeutic targets in the organ-specific metastasis of human malignant melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 92. [Google Scholar] [CrossRef] [PubMed]
- Ciolczyk-Wierzbicka, D.; Laidler, P. The inhibition of invasion of human melanoma cells through N-cadherin knock-down. Med. Oncol. 2018, 35, 42. [Google Scholar] [CrossRef]
- Yamada, M.; Yanaba, K.; Hasegawa, M.; Matsushita, Y.; Horikawa, M.; Komura, K.; Matsushita, T.; Kawasuji, A.; Fujita, T.; Takehara, K.; et al. Regulation of local and metastatic host-mediated anti-tumour mechanisms by l-selectin and intercellular adhesion molecule-1. Clin. Exp. Immunol. 2006, 143, 216–227. [Google Scholar] [CrossRef]
- Wang, C.; Tseng, T.; Jhang, Y.; Tseng, J.; Hsieh, C.; Wu, W.G.; Lee, S. Loss of cell invasiveness through PKC-mediated syndecan-1 downregulation in melanoma cells under anchorage independency. Exp. Dermatol. 2014, 23, 843–849. [Google Scholar] [CrossRef]
- Sheppard, K.E.; McArthur, G.A. The cell-cycle regulator CDK4: An emerging therapeutic target in melanoma. Clin. Cancer Res. 2013, 19, 5320–5328. [Google Scholar] [CrossRef]
- Iida, M.; Omata, Y.; Nakano, C.; Yajima, I.; Tsuzuki, T.; Ishikawa, K.; Hori, M.; Kato, M. Decreased expression levels of cell cycle regulators and matrix metalloproteinases in melanoma from RET-transgenic mice by single irradiation of non-equilibrium atmospheric pressure plasmas. Int. J. Clin. Exp. Pathol. 2015, 8, 9326–9331. [Google Scholar] [PubMed]
- Abreu Velez, A.M.; Howard, M.S. Tumor-suppressor Genes, Cell Cycle Regulatory Checkpoints, and the Skin. N. Am. J. Med. Sci. 2015, 7, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Hu, J.; He, B.; Negron Abril, Y.L.; Stupinski, J.; Weiser, K.; Carbonaro, M.; Chiang, Y.L.; Southard, T.; Giannakakou, P.; et al. A SIRT2-selective inhibitor promotes c-MYC oncoprotein degradation and exhibits broad anticancer activity. Cancer Cell 2016, 29, 297–310. [Google Scholar] [CrossRef]
- Spiegelman, N.A.; Price, I.R.; Jing, H.; Wang, M.; Yang, M.; Cao, J.; Hong, J.Y.; Zhang, X.; Aramsangtienchai, P.; Sadhukhan, S.; et al. Direct comparison of SIRT2 inhibitors: Potency, specificity, activity-dependent inhibition, and on-target anticancer activities. ChemMedChem 2018, 13, 1890–1894. [Google Scholar] [CrossRef]
- Rix, U.; Hantschel, O.; Durnberger, G.; Remsing Rix, L.L.; Planyavsky, M.; Fernbach, N.V.; Kaupe, I.; Bennett, K.L.; Valent, P.; Colinge, J.; et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007, 110, 4055–4063. [Google Scholar] [CrossRef]
- Li, J.; Rix, U.; Fang, B.; Bai, Y.; Edwards, A.; Colinge, J.; Bennett, K.L.; Gao, J.; Song, L.; Eschrich, S.; et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat. Chem. Biol. 2010, 6, 291–299. [Google Scholar] [CrossRef]
- Ivanov, V.N.; Hei, T.K. Combined treatment with EGFR inhibitors and arsenite upregulated apoptosis in human EGFR-positive melanomas: A role of suppression of the PI3K-AKT pathway. Oncogene 2005, 24, 616–626. [Google Scholar] [CrossRef]
- Miao, B.; Ji, Z.; Tan, L.; Taylor, M.; Zhang, J.; Choi, H.G.; Frederick, D.T.; Kumar, R.; Wargo, J.A.; Flaherty, K.T.; et al. Eα2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 2015, 5, 274–287. [Google Scholar] [CrossRef]
- Fu, Y.M.; Yu, Z.X.; Pelayo, B.A.; Ferrans, V.J.; Meadows, G.G. Focal adhesion kinase-dependent apoptosis of melanoma induced by tyrosine and phenylalanine deficiency. Cancer Res. 1999, 59, 758–765. [Google Scholar]
- Liu, J.D.; Chen, S.H.; Lin, C.L.; Tsai, S.H.; Liang, Y.C. Inhibition of melanoma growth and metastasis by combination with (-)-epigallocatechin-3-gallate and dacarbazine in mice. J. Cell. Biochem. 2001, 83, 631–642. [Google Scholar] [CrossRef]
- Ryu, B.; Kim, D.S.; Deluca, A.M.; Alani, R.M. Comprehensive expression profiling of tumor cell lines identifies molecular signatures of melanoma progression. PLoS ONE 2007, 2, e594. [Google Scholar] [CrossRef] [PubMed]
- Wilking-Busch, M.J.; Ndiaye, M.A.; Liu, X.; Ahmad, N. RNA interference-mediated knockdown of SIRT1 and/or SIRT2 in melanoma: Identification of downstream targets by large-scale proteomics analysis. J. Proteom. 2018, 170, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xie, Q.R.; Chen, Z.; Lu, S.; Xia, W. Regulation of SIRT2 levels for human non-small cell lung cancer therapy. Lung Cancer 2013, 82, 9–15. [Google Scholar] [CrossRef]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.; Das, J.; Doweyko, A.M.; et al. Discovery of n-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual SRC/ABL kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef]
- Wang, X.D.; Reeves, K.; Luo, F.R.; Xu, L.A.; Lee, F.; Clark, E.; Huang, F. Identification of candidate predictive and surrogate molecular markers for dasatinib in prostate cancer: Rationale for patient selection and efficacy monitoring. Genome Biol. 2007, 8, R255. [Google Scholar] [CrossRef]
- Eustace, A.J.; Crown, J.; Clynes, M.; O’Donovan, N. Preclinical evaluation of dasatinib, a potent SRC kinase inhibitor, in melanoma cell lines. J. Transl. Med. 2008, 6, 53. [Google Scholar] [CrossRef]
- Kluger, H.M.; Dudek, A.Z.; McCann, C.; Ritacco, J.; Southard, N.; Jilaveanu, L.B.; Molinaro, A.; Sznol, M. A phase 2 trial of dasatinib in advanced melanoma. Cancer 2011, 117, 2202–2208. [Google Scholar] [CrossRef]
- Algazi, A.P.; Weber, J.S.; Andrews, S.C.; Urbas, P.; Munster, P.N.; DeConti, R.C.; Hwang, J.; Sondak, V.K.; Messina, J.L.; McCalmont, T.; et al. Phase I clinical trial of the SRC inhibitor dasatinib with dacarbazine in metastatic melanoma. Br. J. Cancer 2012, 106, 85–91. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Infante, J.R.; Daud, A.; Gonzalez, R.; Kefford, R.F.; Sosman, J.; Hamid, O.; Schuchter, L.; Cebon, J.; Ibrahim, N.; et al. Combined BRAF and MEK inhibition in melanoma with BRAF v600 mutations. N. Engl. J. Med. 2012, 367, 1694–1703. [Google Scholar] [CrossRef]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated Re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef]
- Huang, T.S.; Rauth, S.; Das Gupta, T.K. Overexpression of EGF receptor is associated with spontaneous metastases of a human melanoma cell line in nude mice. Anticancer Res. 1996, 16, 3557–3563. [Google Scholar]
- Ma, D.; Niederkorn, J.Y. Role of epidermal growth factor receptor in the metastasis of intraocular melanomas. Invest. Ophthalmol. Vis. Sci. 1998, 39, 1067–1075. [Google Scholar]
- Qiu, L.; Wang, Q.; Di, W.; Jiang, Q.; Schefeller, E.; Derby, S.; Wanebo, H.; Yan, B.; Wan, Y. Transient activation of EGFR/AKT cell survival pathway and expression of survivin contribute to reduced sensitivity of human melanoma cells to betulinic acid. Int. J. Oncol. 2005, 27, 823–830. [Google Scholar]
- Straume, O.; Akslen, L.A. Importance of vascular phenotype by basic fibroblast growth factor, and influence of the angiogenic factors basic fibroblast growth factor/fibroblast growth factor receptor-1 and ephrin-a1/epha2 on melanoma progression. Am. J. Pathol. 2002, 160, 1009–1019. [Google Scholar] [CrossRef]
- Easty, D.J.; Hill, S.P.; Hsu, M.Y.; Fallowfield, M.E.; Florenes, V.A.; Herlyn, M.; Bennett, D.C. Up-regulation of ephrin-a1 during melanoma progression. Int J. Cancer 1999, 84, 494–501. [Google Scholar] [CrossRef]
- Margaryan, N.V.; Strizzi, L.; Abbott, D.E.; Seftor, E.A.; Rao, M.S.; Hendrix, M.J.; Hess, A.R. Epha2 as a promoter of melanoma tumorigenicity. Cancer Biol. Ther. 2009, 8, 279–288. [Google Scholar] [CrossRef]
- Parri, M.; Taddei, M.L.; Bianchini, F.; Calorini, L.; Chiarugi, P. Epha2 reexpression prompts invasion of melanoma cells shifting from mesenchymal to amoeboid-like motility style. Cancer Res. 2009, 69, 2072–2081. [Google Scholar] [CrossRef]
- Udayakumar, D.; Zhang, G.; Ji, Z.; Njauw, C.N.; Mroz, P.; Tsao, H. Epha2 is a critical oncogene in melanoma. Oncogene 2011, 30, 4921–4929. [Google Scholar] [CrossRef]
- Sussmuth, S.D.; Haider, S.; Landwehrmeyer, G.B.; Farmer, R.; Frost, C.; Tripepi, G.; Andersen, C.A.; Di Bacco, M.; Lamanna, C.; Diodato, E.; et al. An exploratory double-blind, randomized clinical trial with selisistat, a sirt1 inhibitor, in patients with huntington’s disease. Br. J. Clin. Pharmacol. 2015, 79, 465–476. [Google Scholar] [CrossRef]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenet. 2016, 8, 61. [Google Scholar] [CrossRef]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Olah, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective SIRT2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [PubMed]
- Karas’, K.; Sałkowska, A.; Sobalska-Kwapis, M.; Walczak-Drzewiecka, A.; Strapagiel, D.; Dastych, J.; Bachorz, R.A. and Ratajewski, M. Digoxin, an overlooked agonist of RORγ/RORγT. Front. Pharmacol. 2019, 9, 1460. [Google Scholar] [CrossRef]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. Panther version 11: Expanded annotation data from gene ontology and reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017, 45, D183–D189. [Google Scholar] [CrossRef]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef]
- Repetto, G.; del Peso, A.; Zurita, J.L. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat. Protoc. 2008, 3, 1125–1131. [Google Scholar] [CrossRef]
- Ratajewski, M.; Walczak-Drzewiecka, A.; Salkowska, A.; Dastych, J. Aflatoxins upregulate CYP3A4 mRNA expression in a process that involves the pxr transcription factor. Toxicol. Lett. 2011, 205, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.C.; Christensen, M.E.; Waterhouse, N.J. Measuring survival of adherent cells with the colony-forming assay. Cold Spring Harb. Protoc. 2016, 2016, 8. [Google Scholar] [CrossRef]
- Guzman, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. Colonyarea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Protein | Effect | Average Fold Change |
|---|---|---|---|
| Integrins | |||
| ITGA1 | integrin subunit alpha 1 | ↓ | 0.462 |
| ITGA3 | integrin subunit alpha 3 | ↑ | 2.173 |
| ITGA4 | integrin subunit alpha 4 | ↓ | 0.556 |
| ITGA5 | integrin subunit alpha 5 | ↑ | 1.588 |
| ITGA6 | integrin subunit alpha 6 | ↑ | 2.221 |
| ITGA9 | integrin subunit alpha 9 | ↑ | 2.498 |
| ITGA11 | integrin subunit alpha 11 | ↓ | 0.551 |
| ITGAE | integrin subunit alpha E | ↓ | 0.540 |
| ITGAL | integrin subunit alpha L | ↓ | 0.540 |
| Cadherins | |||
| CDH2 | cadherin 2 | ↑ | 1.770 |
| CDH19 | cadherin 19 | ↑ | 4.203 |
| Selectins | |||
| SELL | Selectin L | ↑ | 8.589 |
| Syndecans | |||
| SDC1 | syndecan 1 | ↓ | 0.300 |
| SDC2 | syndecan 2 | ↑ | 4.304 |
| Immunoglobulin-like Cell Adhesion Molecules | |||
| NCAM1 | neural cell adhesion molecule | ↑ | 21.328 |
| Receptor tyrosine kinases | |||
| EGFR | epidermal growth factor receptor | ↓ | 0.456 |
| IGF1R | insulin like growth factor 1 receptor | ↑ | 1.653 |
| IGFLR1 | IGF like family receptor 1 | ↓ | 0.647 |
| FGFR1 | fibroblast growth factor receptor 1 | ↑ | 4.052 |
| NGFR | nerve growth factor receptor | ↓ | 0.579 |
| EPHA2 | EPH receptor A2 | ↓ | 0.477 |
| EPHA3 | EPH receptor A3 | ↑ | 1.646 |
| EPHA5 | EPH receptor A3 | ↓ | 0.421 |
| EPHB3 | EPH receptor B3 | ↑ | 2.767 |
| AXL | AXL receptor tyrosine kinase | ↓ | 0.592 |
| ROR1 | receptor tyrosine kinase like orphan receptor 1 | ↑ | 2.547 |
| MAP/ERK pathway | |||
| GRB14 | growth factor receptor bound protein 14 | ↑ | 3.678 |
| RASAL1 | RAS protein activator like 1 | ↑ | 7.833 |
| RASD2 | RASD family member 2 | ↑ | 3.716 |
| RASEF | RAS and EF-hand domain containing | ↑ | 2.321 |
| RASGEF1C | RasGEF domain family member 1C | ↓ | 0.195 |
| RASGRP1 | RAS guanyl releasing protein 1 | ↑ | 6.635 |
| RASGRP3 | RAS guanyl releasing protein 3 | ↓ | 0.417 |
| RASSF2 | Ras association domain family member 2 | ↑ | 2.186 |
| RASSF4 | Ras association domain family member 4 | ↓ | 0.594 |
| RASSF5 | Ras association domain family member 5 | ↑ | 2.972 |
| MAP2K3 | mitogen-activated protein kinase kinase 3 | ↓ | 0.593 |
| MAP2K6 | mitogen-activated protein kinase kinase 6 | ↑ | 2.309 |
| MAP3K4 | mitogen-activated protein kinase kinase kinase 4 | ↓ | 0.598 |
| MAP3K14 | mitogen-activated protein kinase kinase kinase 14 | ↓ | 0.585 |
| MAP3K21 | mitogen-activated protein kinase kinase kinase 21 | ↑ | 3.521 |
| Other protein kinases | |||
| BTK | tyrosine-protein kinase BTK | ↓ | 0.342 |
| GAK | cyclin G-assiciated kinase | ↓ | 0.583 |
| LYN | tyrosine protein kinase LYN | ↓ | 0.275 |
| Cell cycle regulators | |||
| CDK5R1 | cyclin dependent kinase 5 regulatory subunit 1 | ↓ | 0.653 |
| CDK6 | Cyclin-dependent kinase 6 | ↑ | 1.660 |
| CDKL2 | cyclin dependent kinase like 2 | ↑ | 2.001 |
| CDKN1A | Cyclin-dependent kinase inhibitor 1A (P21WAF) | ↓ | 0.615 |
| CDKN2B | cyclin dependent kinase inhibitor 2B | ↑ | 4.107 |
| CDKN2C | cyclin dependent kinase inhibitor 2C | ↓ | 0.546 |
| CDKN2D | cyclin dependent kinase inhibitor 2D | ↓ | 0.587 |
| Gene Symbol | Protein | Effect | Average Fold Change |
|---|---|---|---|
| Integrins | |||
| ITGA1 | integrin subunit alpha 1 | ↓ | 0.051 |
| ITGBL1 | integrin subunit beta like 1 | ↓ | 0.227 |
| Cadherins | |||
| CDH12 | cadherin 12 | ↑ | 8.313 |
| Syndecans | |||
| SDC2 | syndecan 2 | ↓ | 0.452 |
| Immunoglobulin-like Cell Adhesion Molecules | |||
| NCAM1 | neural cell adhesion molecule | ↓ | 0.572 |
| ICAM5 | intercellular adhesion molecule 5 | ↓ | 0.449 |
| Receptor tyrosine kinases | |||
| EGFR | epidermal growth factor receptor | ↓ | 0.195 |
| FGFR1 | fibroblast growth factor receptor 1 | ↑ | 2.005 |
| EPHA2 | EPH receptor A2 | ↓ | 0.423 |
| EPHB1 | EPH receptor B1 | ↓ | 0.360 |
| AXL | AXL receptor tyrosine kinase | ↑ | 2.086 |
| DDR1 | Discoidin domain receptor family, member 1 | ↓ | 0.576 |
| MAP/ERK pathway | |||
| GRB14 | growth factor receptor bound protein 14 | ↓ | 0.234 |
| RASL10B | RAS like family 10 member B | ↑ | 2.220 |
| MAP3K9 | mitogen-activated protein kinase kinase kinase 9 | ↓ | 0.638 |
| MAPK8IP1 | mitogen-activated protein kinase 8 interacting protein 1 | ↓ | 0.342 |
| MAPK8IP2 | mitogen-activated protein kinase 8 interacting protein 2 | ↓ | 0.587 |
| Cell cycle regulators | |||
| CDK14 | Cyclin-dependent kinase 14 | ↓ | 0.207 |
| CDK18 | Cyclin-dependent kinase 18 | ↑ | 1.536 |
| CDK20 | Cyclin-dependent kinase 20 | ↓ | 0.073 |
| CDKN2D | cyclin dependent kinase inhibitor 2D | ↓ | 0.643 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karwaciak, I.; Sałkowska, A.; Karaś, K.; Sobalska-Kwapis, M.; Walczak-Drzewiecka, A.; Pułaski, Ł.; Strapagiel, D.; Dastych, J.; Ratajewski, M. SIRT2 Contributes to the Resistance of Melanoma Cells to the Multikinase Inhibitor Dasatinib. Cancers 2019, 11, 673. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11050673
Karwaciak I, Sałkowska A, Karaś K, Sobalska-Kwapis M, Walczak-Drzewiecka A, Pułaski Ł, Strapagiel D, Dastych J, Ratajewski M. SIRT2 Contributes to the Resistance of Melanoma Cells to the Multikinase Inhibitor Dasatinib. Cancers. 2019; 11(5):673. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11050673
Chicago/Turabian StyleKarwaciak, Iwona, Anna Sałkowska, Kaja Karaś, Marta Sobalska-Kwapis, Aurelia Walczak-Drzewiecka, Łukasz Pułaski, Dominik Strapagiel, Jarosław Dastych, and Marcin Ratajewski. 2019. "SIRT2 Contributes to the Resistance of Melanoma Cells to the Multikinase Inhibitor Dasatinib" Cancers 11, no. 5: 673. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11050673