The Role of Transient Receptor Potential Melastatin 7 (TRPM7) in Cell Viability: A Potential Target to Suppress Breast Cancer Cell Cycle


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
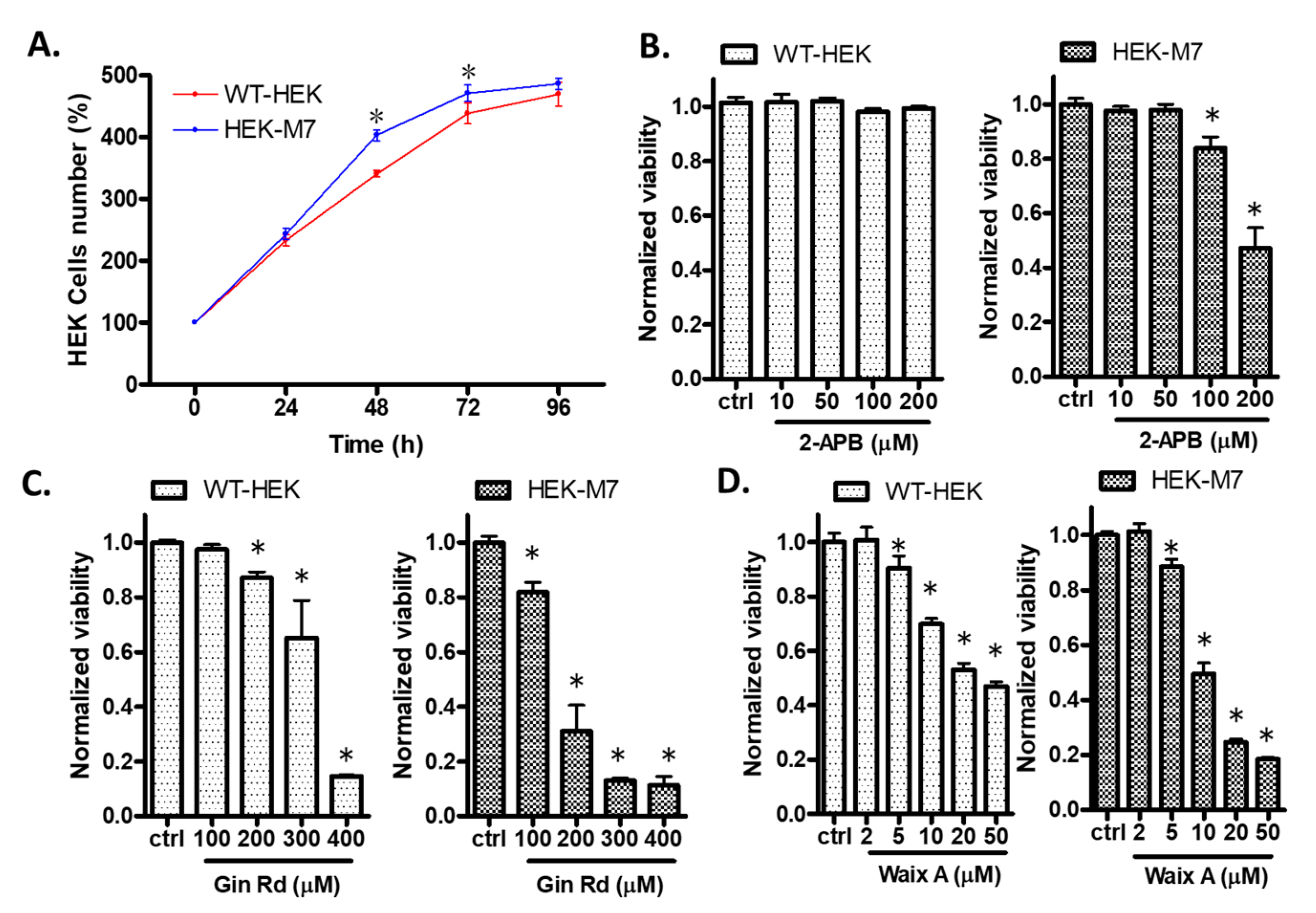
2.1. Effects of TRPM7 Inhibitors on Cell Viability of WT-HEK and HEK-M7
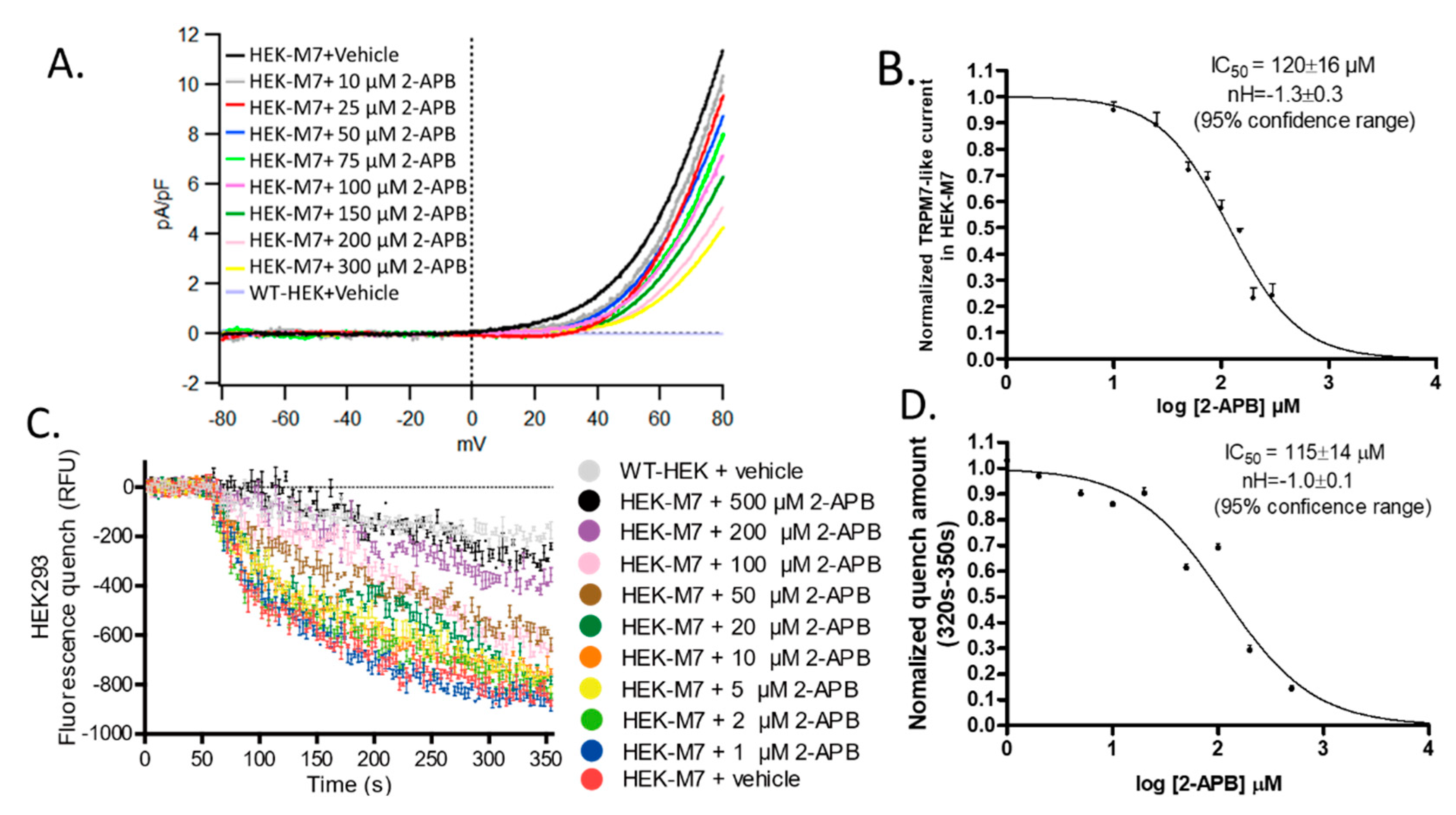
2.2. 2-APB Inhibited TRPM7 Current and Divalent Flux
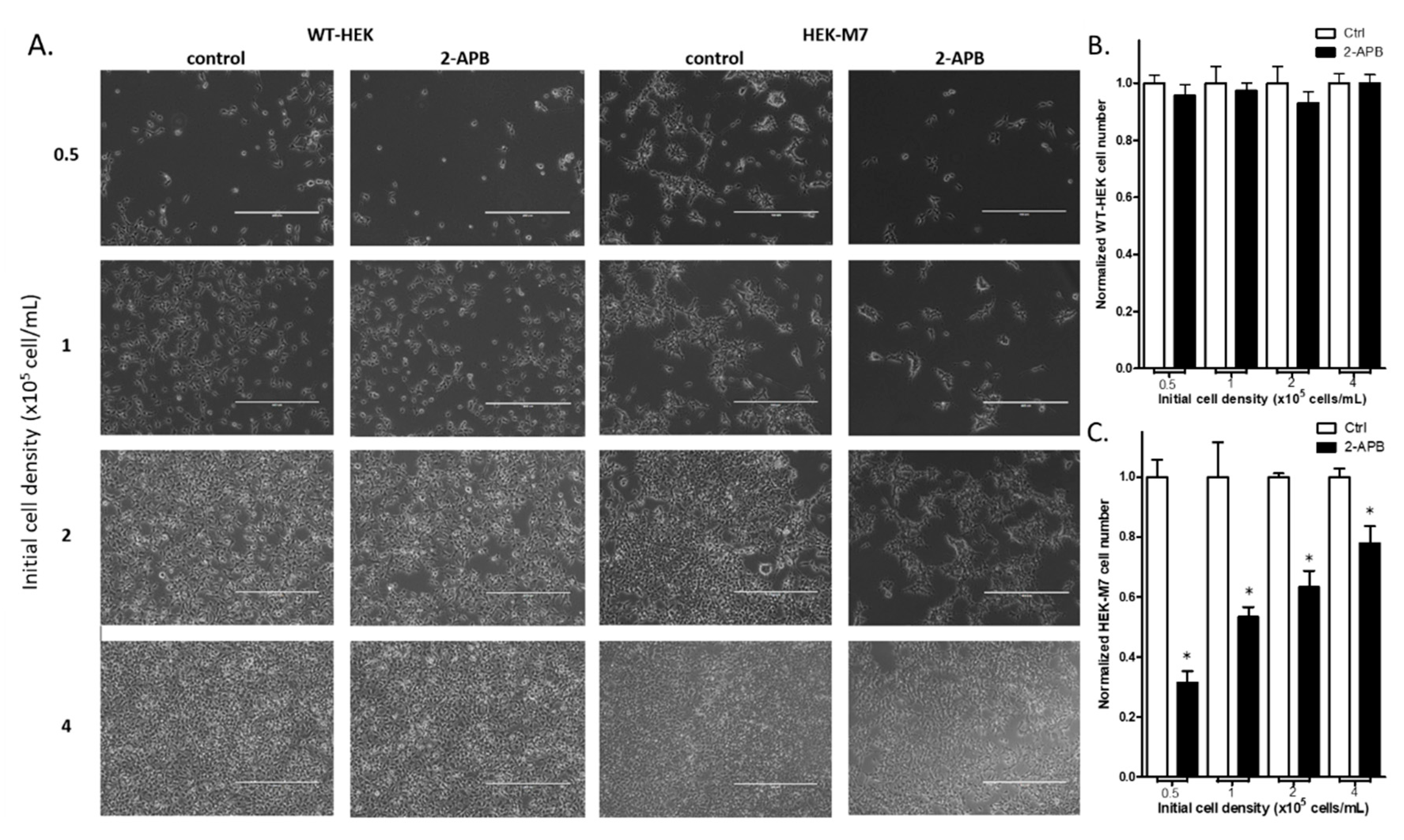
2.3. 2-APB Suppressed the Cell Proliferation in HEK-M7 but not WT-HEK Cells
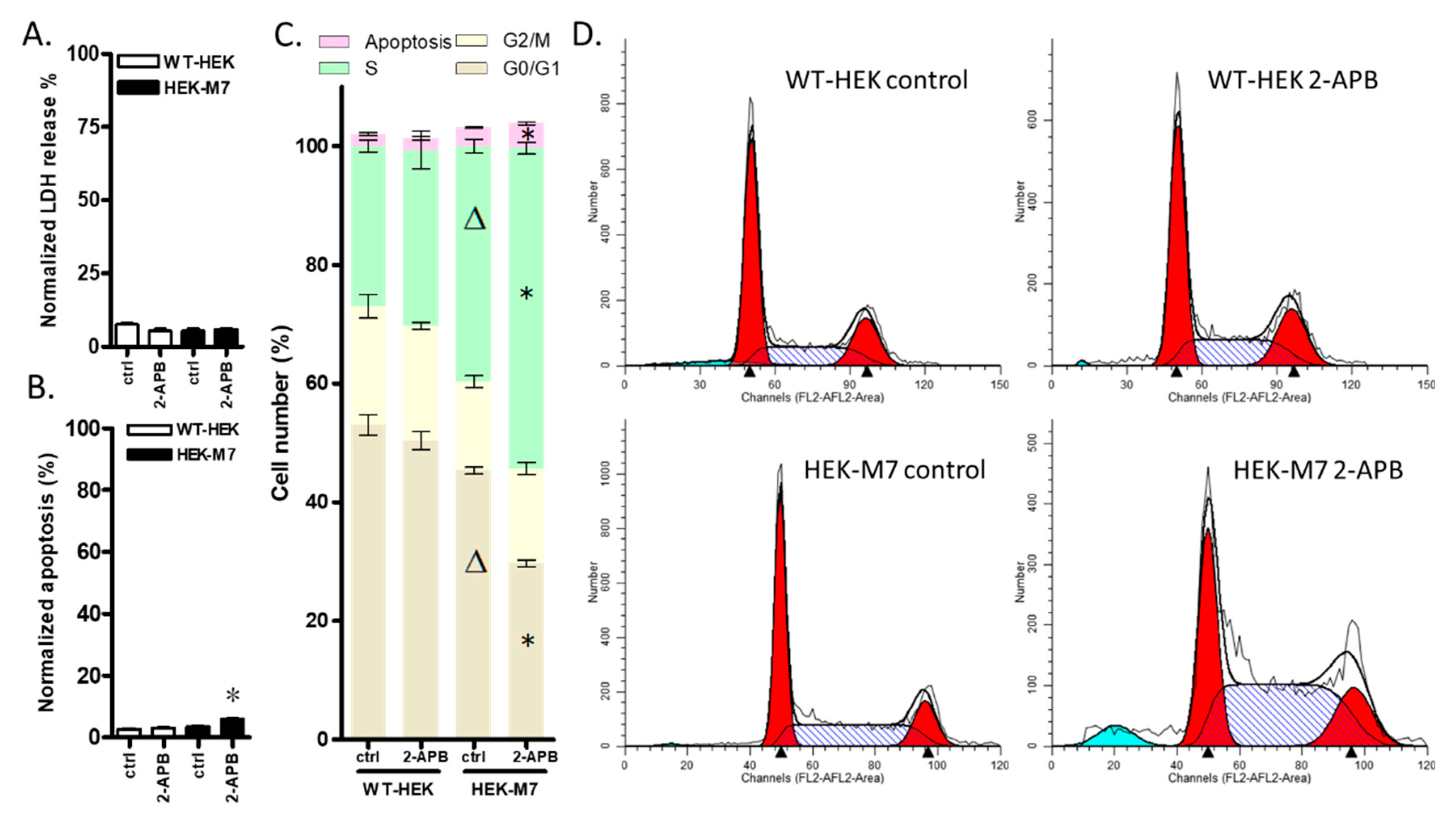
2.4. 2-APB Inhibition of TRPM7 Affected the Cell Cycle but did not Promote Cell Death
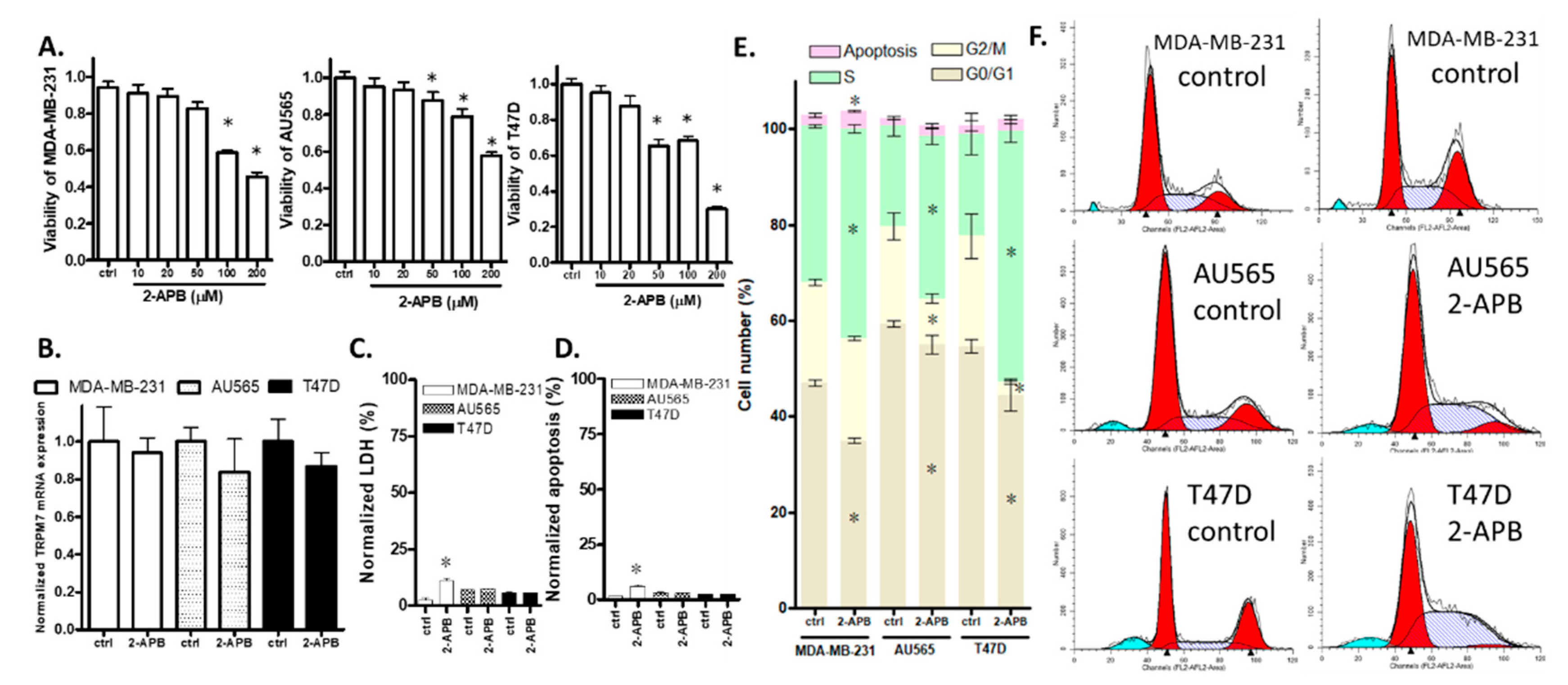
2.5. 2-APB Suppressed Cell Viability in TRPM7-Expressing Breast Cancer Cell Lines
2.6. 2-APB Decreased the Overall Viability of AU565 and T47D by Inhibiting Cell Proliferation Rather Than by Promoting Cell Death
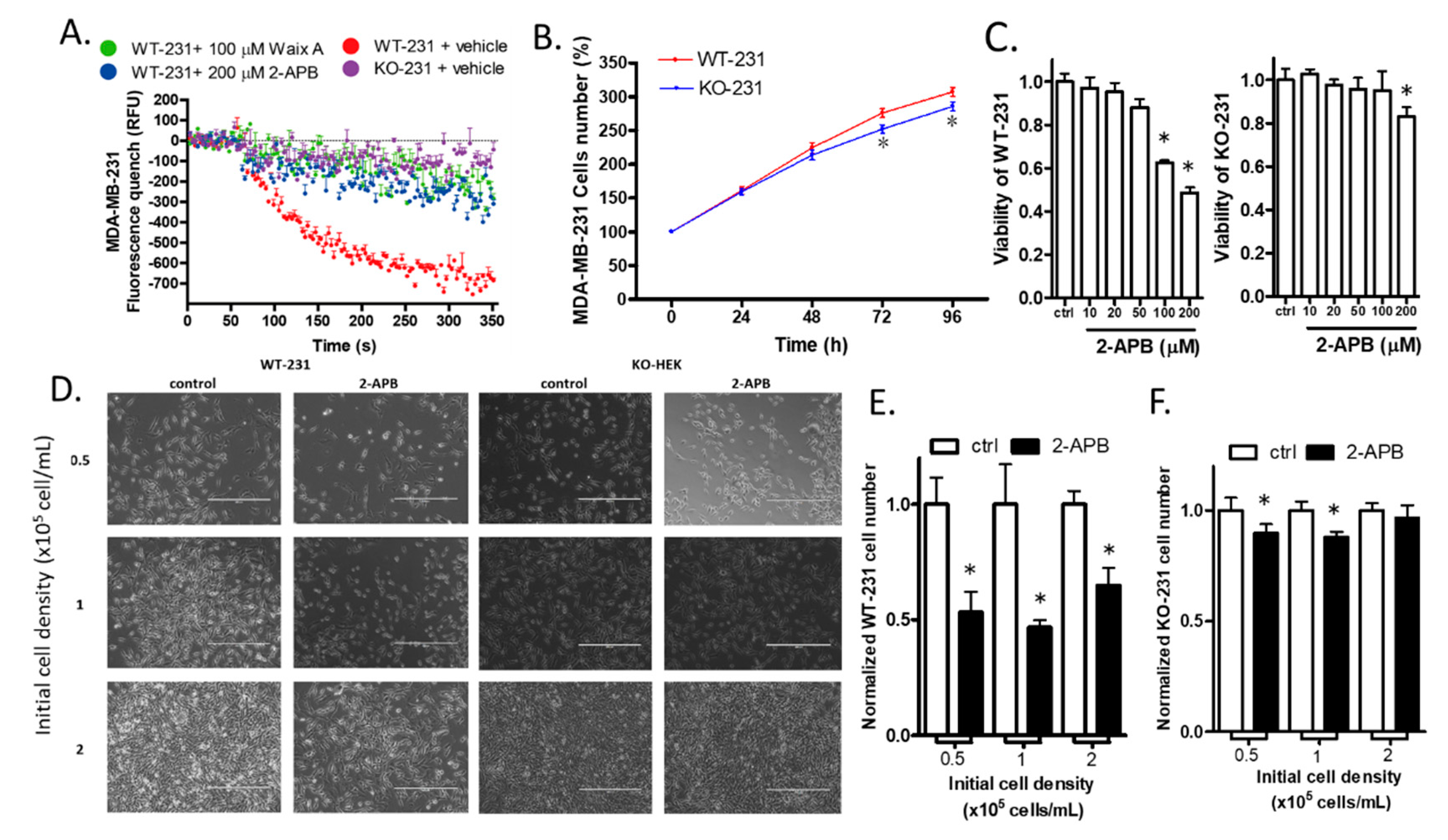
2.7. The Knock-out of TRPM7 Decreased the Divalent Influx in MDA-MB-231
2.8. The Knock-out of TRPM7 Decreased the Proliferation of WT-231 Cells
2.9. 2-APB Selectively Suppressed the Viability of WT-231 Compared with KO-231
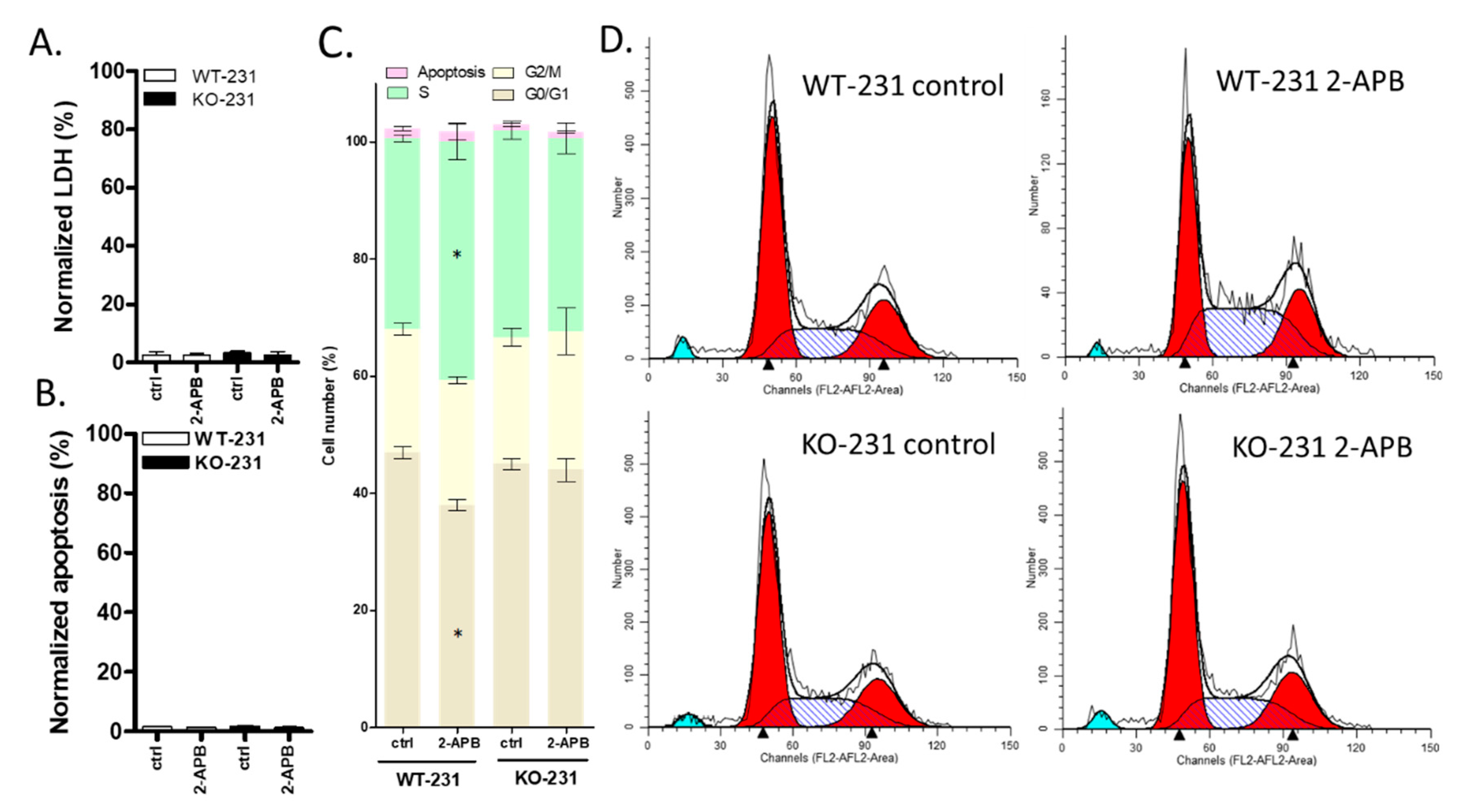
2.10. The Viability of MDA-MB-231 Was Affected by 100 µM 2-APB by Regulating Cell Cycle rather than Cell Death (LDH and Apoptosis) and this Effect was Blocked by TRPM7 Knock-out
2.11. A Second Inhibitor, Gin Rd, Affected MDA-MB-231 Cells Similarly
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Drugs and Treatments
4.3. MTT Assay
4.4. Cell Counting
4.5. Patch-Clamp Recording
4.6. Fura-2AM-Based Quench Assay
4.7. Quantitative Real Time-PCR
4.8. LDH Assay
4.9. Apoptosis Assay
4.10. Cell Cycle Flow Cytometry
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parekh, A.B.; Penner, R. Store depletion and calcium influx. Physiol. Rev. 1997, 77, 901–930. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Capacitative calcium entry. Biochem. J. 1995, 312, 1. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Bird, G.; Hajnóczky, G.; Robb-Gaspers, L.; Putney, J., Jr. Spatial and temporal aspects of cellular calcium signaling. FASEB J. 1996, 10, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, I.S.; Delling, M.; Clapham, D.E. An introduction to TRP channels. Annu. Rev. Physiol. 2006, 68, 619–647. [Google Scholar] [CrossRef] [Green Version]
- Fleig, A.; Penner, R. The TRPM ion channel subfamily: Molecular, biophysical and functional features. Trends Pharmacol. Sci. 2004, 25, 633–639. [Google Scholar] [CrossRef]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517. [Google Scholar] [CrossRef]
- Gautier, M.; Perrière, M.; Monet, M.; Vanlaeys, A.; Korichneva, I.; Dhennin-Duthille, I.; Ouadid-Ahidouch, H. Recent advances in oncogenic roles of the TRPM7 chanzyme. Curr. Med. Chem. 2016, 23, 4092–4107. [Google Scholar] [CrossRef]
- Yee, N. Role of TRPM7 in cancer: Potential as molecular biomarker and therapeutic target. Pharmaceuticals 2017, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.-L.; Kong, C.-Z.; Zhang, Z.; Li, Z.-L.; Bi, J.-B.; Liu, X.-K. TRPM7 is overexpressed in bladder cancer and promotes proliferation, migration, invasion and tumor growth. Oncol. Rep. 2017, 38, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J. Involvement of transient receptor potential melastatin 7 channels in sophorae radix-induced apoptosis in cancer cells: Sophorae radix and TRPM7. J. Pharmacopunct. 2012, 15, 31. [Google Scholar] [CrossRef]
- Jiang, J.; Li, M.-H.; Inoue, K.; Chu, X.-P.; Seeds, J.; Xiong, Z.-G. Transient Receptor Potential Melastatin 7–like Current in Human Head and Neck Carcinoma Cells: Role in Cell Proliferation. Cancer Res. 2007, 67, 10929–10938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteilh-Zoller, M.K.; Hermosura, M.C.; Nadler, M.J.; Scharenberg, A.M.; Penner, R.; Fleig, A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J. Gen. Physiol. 2003, 121, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, C.; Perraud, A.-L.; Johnson, C.O.; Inabe, K.; Smith, M.K.; Penner, R.; Kurosaki, T.; Fleig, A.; Scharenberg, A.M. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell 2003, 114, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Selvaraj, S.; Varma, A.; Derry, S.; Sahmoun, A.E.; Singh, B.B. Increase in serum Ca2+/Mg2+ ratio promotes proliferation of prostate cancer cells by activating TRPM7 channels. J. Biol. Chem. 2013, 288, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Nadler, M.J.; Hermosura, M.C.; Inabe, K.; Perraud, A.-L.; Zhu, Q.; Stokes, A.J.; Kurosaki, T.; Kinet, J.-P.; Penner, R.; Scharenberg, A.M. LTRPC7 is a Mg·ATP-regulated divalent cation channel required for cell viability. Nature 2001, 411, 590. [Google Scholar] [CrossRef]
- Chokshi, R.; Fruasaha, P.; Kozak, J.A. 2-aminoethyl diphenyl borinate (2-APB) inhibits TRPM7 channels through an intracellular acidification mechanism. Channels 2012, 6, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J. Involvement of melastatin type transient receptor potential 7 channels in ginsenoside Rd-induced apoptosis in gastric and breast cancer cells. J. Ginseng Res. 2013, 37, 201. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J.; Nam, J.H.; Kwon, Y.K.; So, I.; Kim, S.J. The role of waixenicin a as transient receptor potential melastatin 7 blocker. Basic Clin. Pharmacol. Toxicol. 2013, 112, 83–89. [Google Scholar] [CrossRef]
- Aarts, M.; Iihara, K.; Wei, W.L.; Xiong, Z.G.; Arundine, M.; Cerwinski, W.; MacDonald, J.F.; Tymianski, M. A key role for TRPM7 channels in anoxic neuronal death. Cell 2003, 115, 863–877. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.C.; Xie, J.; Zhang, Z.; Su, L.T.; Yue, L.; Runnels, L.W. Blockade of TRPM7 channel activity and cell death by inhibitors of 5-lipoxygenase. PLoS ONE 2010, 5, e11161. [Google Scholar] [CrossRef] [Green Version]
- Neve, R.M.; Chin, K.; Fridlyand, J.; Yeh, J.; Baehner, F.L.; Fevr, T.; Clark, L.; Bayani, N.; Coppe, J.P.; Tong, F.; et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006, 10, 515–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.; Miller, K.; Jemal, A. Cancer statistics (2018). CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.F.; Li, C.W.; Kang, B.X.; Gao, G.; Li, C.; Zhang, Z.M. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhennin-Duthille, I.; Gautier, M.; Faouzi, M.; Guilbert, A.; Brevet, M.; Vaudry, D.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: Correlation with pathological parameters. Cell. Physiol. Biochem. 2011, 28, 813–822. [Google Scholar] [CrossRef]
- Hantute-Ghesquier, A.; Haustrate, A.; Prevarskaya, N. TRPM family channels in cancer. Pharmaceuticals 2018, 11, 58. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Lee, H.C.; Yoon, S.Y.; Lykke-Hartmann, K.; Fissore, R.A.; Carvacho, I. TRPV3 channels mediate Ca2+ influx induced by 2-APB in mouse eggs. Cell Calcium 2016, 59, 21–31. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, L.; Zhang, X.; Bai, J.; Shi, M.; Zhao, G. Ginsenoside-Rd attenuates TRPM7 and ASIC1a but promotes ASIC2a expression in rats after focal cerebral ischemia. Neurol. Sci. 2012, 33, 1125–1131. [Google Scholar] [CrossRef]
- Huang, J.; Furuya, H.; Faouzi, M.; Zhang, Z.; Monteilh-Zoller, M.; Kawabata, K.G.; Horgen, F.D.; Kawamori, T.; Penner, R.; Fleig, A. Inhibition of TRPM7 suppresses cell proliferation of colon adenocarcinoma in vitro and induces hypomagnesemia in vivo without affecting azoxymethane-induced early colon cancer in mice. Cell Commun. Signal. 2017, 15, 30. [Google Scholar] [CrossRef]
- Zierler, S.; Yao, G.; Zhang, Z.; Kuo, W.C.; Pörzgen, P.; Penner, R.; Horgen, F.D.; Fleig, A. Waixenicin A inhibits cell proliferation through magnesium-dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J. Biol. Chem. 2011, 286, 39328–39335. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Jiang, J.; Yue, L. Functional characterization of homo-and heteromeric channel kinases TRPM6 and TRPM7. J. Gen. Physiol. 2006, 127, 525–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, L.T.; Agapito, M.A.; Li, M.; Simonson, W.T.; Huttenlocher, A.; Habas, R.; Yue, L.; Runnels, L.W. TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain. J. Biol. Chem. 2006, 281, 11260–11270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, N.; Ichimura, A.; Takei, D.; Sakaguchi, R.; Kitani, A.; Nagaoka, R.; Tomizawa, M.; Miyazaki, Y.; Miyachi, H.; Numata, T. TRPM7 channels mediate spontaneous Ca2+ fluctuations in growth plate chondrocytes that promote bone development. Sci. Signal. 2019, 12, eaaw4847. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Zhang, W.; Shi, J.; Jiang, S. TGF-β1-induced airway smooth muscle cell proliferation involves TRPM7-dependent calcium influx via TGFβR/SMAD3 (Revision). Mol. Immunol. 2018, 103, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Hong, W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer cell 2008, 13, 188–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gérard, C.; Goldbeter, A. The balance between cell cycle arrest and cell proliferation: Control by the extracellular matrix and by contact inhibition. Interface Focus 2014, 4, 20130075. [Google Scholar] [CrossRef] [Green Version]
- Ribatti, D. A revisited concept: Contact inhibition of growth. From cell biology to malignancy. Exp. Cell Res. 2017, 359, 17–19. [Google Scholar] [CrossRef]
- Faouzi, M.; Kilch, T.; Horgen, F.D.; Fleig, A.; Penner, R. The TRPM7 channel kinase regulates store-operated calcium entry. J. Physiol. 2017, 595, 3165–3180. [Google Scholar] [CrossRef] [Green Version]
- Cao, R.; Meng, Z.; Liu, T.; Wang, G.; Qian, G.; Cao, T.; Guan, X.; Dan, H.; Xiao, Y.; Wang, X. Decreased TRPM7 inhibits activities and induces apoptosis of bladder cancer cells via ERK1/2 pathway. Oncotarget 2016, 7, 72941. [Google Scholar] [CrossRef] [Green Version]
- Pietenpol, J.; Stewart, Z. Cell cycle checkpoint signaling: Cell cycle arrest versus apoptosis. Toxicology 2002, 181, 475–481. [Google Scholar] [CrossRef]
- Yee, N.S.; Zhou, W.; Liang, I.C. Transient receptor potential ion channel Trpm7 regulates exocrine pancreatic epithelial proliferation by Mg2+-sensitive Socs3a signaling in development and cancer. Dis. Models Mech. 2011, 4, 240–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Xiao, C.; Liu, H.; Huang, Y.; Dilger, J.P.; Lin, J. Effects of local anesthetics on breast cancer cell viability and migration. BMC Cancer 2018, 18, 666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Ji, X.; Yang, X.; Zhang, X. Cell type-and density-dependent effect of 1 T static magnetic field on cell proliferation. Oncotarget 2017, 8, 13126. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Dilger, J.P.; Lin, J. The Role of Transient Receptor Potential Melastatin 7 (TRPM7) in Cell Viability: A Potential Target to Suppress Breast Cancer Cell Cycle. Cancers 2020, 12, 131. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12010131
Liu H, Dilger JP, Lin J. The Role of Transient Receptor Potential Melastatin 7 (TRPM7) in Cell Viability: A Potential Target to Suppress Breast Cancer Cell Cycle. Cancers. 2020; 12(1):131. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12010131
Chicago/Turabian StyleLiu, Hengrui, James P. Dilger, and Jun Lin. 2020. "The Role of Transient Receptor Potential Melastatin 7 (TRPM7) in Cell Viability: A Potential Target to Suppress Breast Cancer Cell Cycle" Cancers 12, no. 1: 131. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12010131