Differential Depletion of Bone Marrow Resident B-ALL after Systemic Administration of Endosomal TLR Agonists

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Direct and Indirect Cytotoxic Effects of Endosomal TLR Agonists on Mouse Primary B-ALL Cells In Vitro
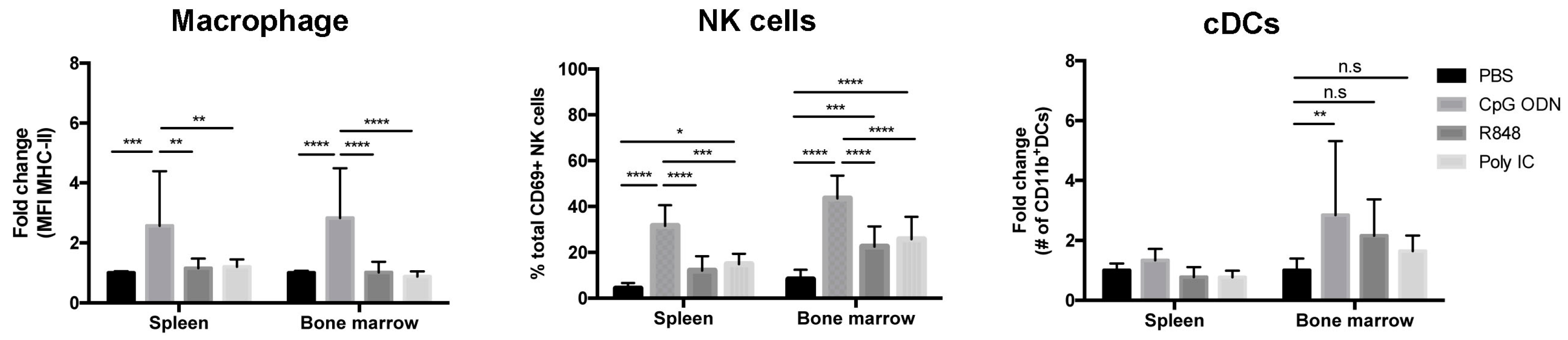
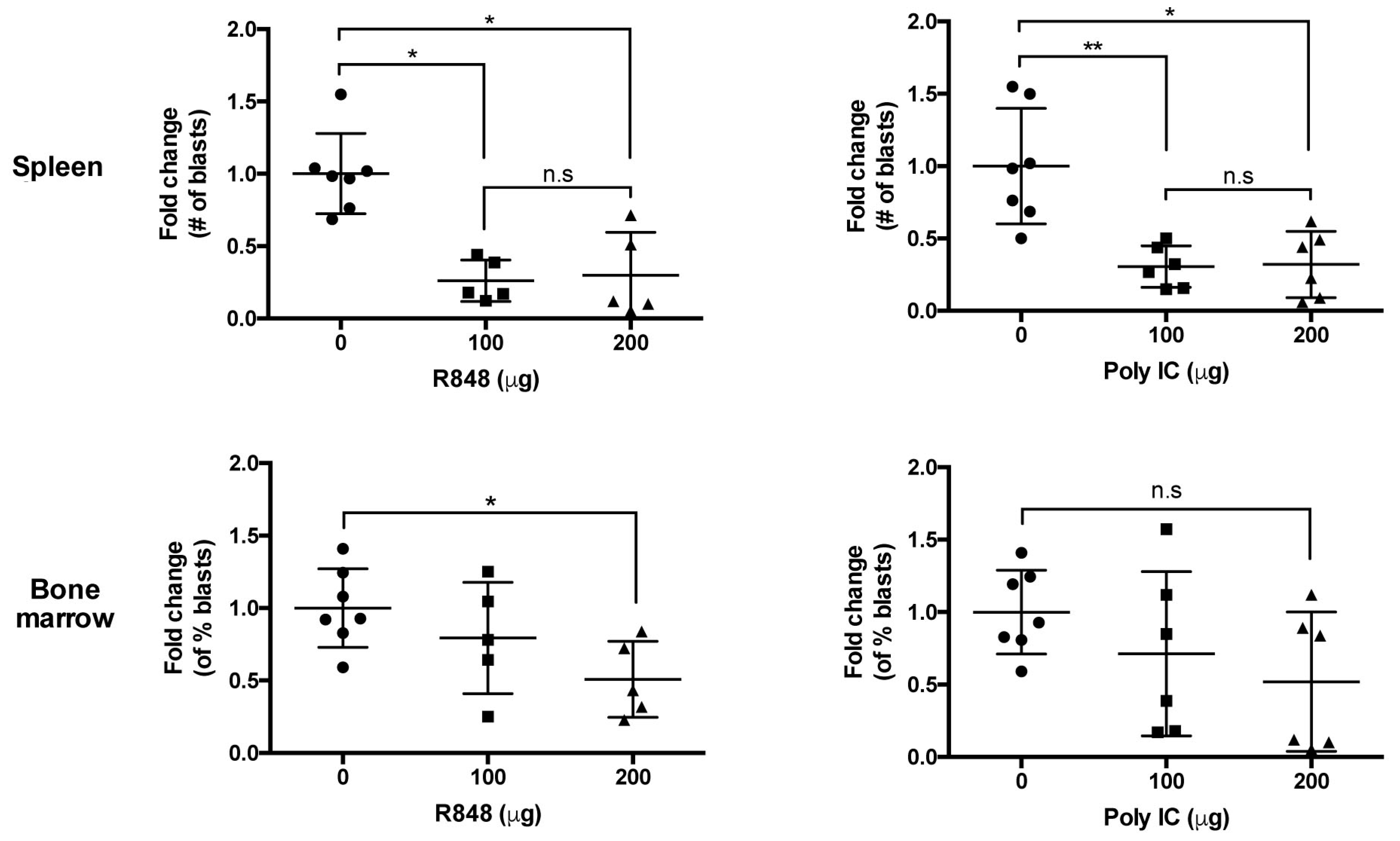
2.2. Organ-Specific Depletion of Primary B-ALL Cells by Endosomal TLR-Induced Innate Immune Responses
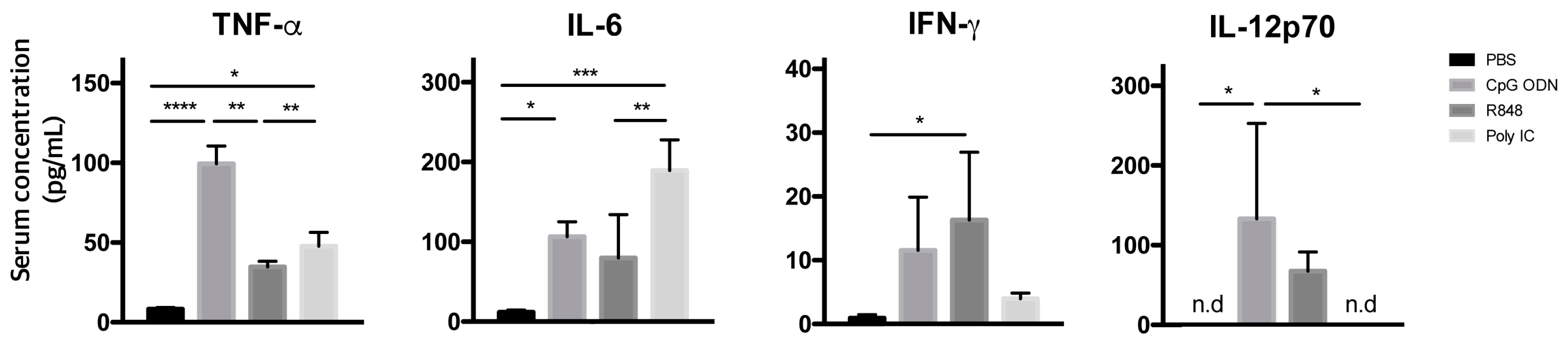
2.3. Immunostimulatory Effects of Endosomal TLR Agonists in Mice Bearing Primary B-ALL
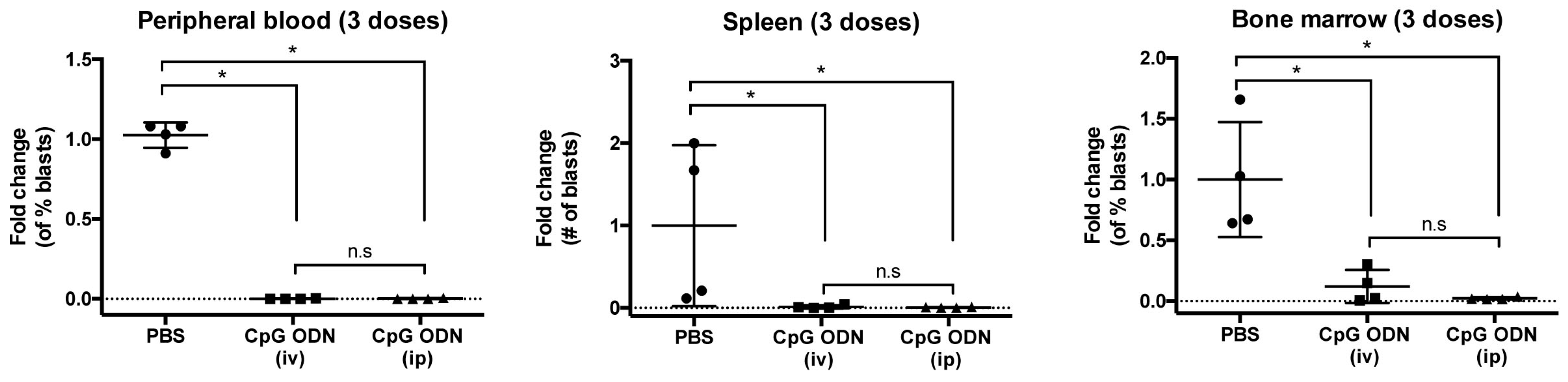
2.4. Systemic Administration of TLR Agonists Achieves Durable Control of Primary B-ALL Progression In Vivo
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Mice
4.3. Cells
4.4. PCR
- Tlr3F—TCGGATTCTTGGTTTCAAGG; Tlr3R—TTTCGGCTTCTTTTGATGCT;
- Tlr7F—GGAGCTCTGTCCTTGAGTGG; Tlr7R—CAAGGCATGTCCTAGGTGGT;
- Tlr8F—GGCACAACTCCCTTGTGATT; Tlr8R—CATTTGGGTGCTGTTGTTTG;
- Tlr9F—TCGCTTTGTGGACTTGTCAG; Tlr9R—GGCTCAGGCTAAGACACTGG.
4.5. Adoptive Transfer Experiments
4.6. Flow Cytometry
4.7. In Vivo Bioluminescence Imaging
4.8. Serum Cytokine Analysis
4.9. Statistical Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A






References
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Bhojwani, D.; Pui, C.H. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013, 14, e205–e217. [Google Scholar] [CrossRef]
- Gaynon, P.S.; Desai, A.A.; Bostrom, B.C.; Hutchinson, R.J.; Lange, B.J.; Nachman, J.B.; Reaman, G.H.; Sather, H.N.; Steinherz, P.G.; Trigg, M.E.; et al. Early Response to Therapy and Outcome in Childhood. Cancer 1997, 80, 1717–1726. [Google Scholar] [CrossRef]
- Lauten, M.; Möricke, A.; Beier, R.; Zimmermann, M.; Stanulla, M.; Meissner, B.; Odenwald, E.; Attarbaschi, A.; Niemeyer, C.; Niggli, F.; et al. Prediction of outcome by early bone marrow response in childhood acute lymphoblastic leukemia treated in the ALL-BFM 95 trial: Differential effects in precursor B-cell and T-cell leukemia. Haematologica 2012, 97, 1048–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowitz, M.J.; Devidas, M.; Hunger, S.P.; Bowman, W.P.; Carroll, A.J.; Carroll, W.L.; Linda, S.; Martin, P.L.; Pullen, D.J.; Viswanatha, D.; et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: A Children’ s Oncology Group study. Blood 2008, 111, 5477–5485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conter, V.; Bartram, C.R.; Valsecchi, M.G.; Panzer-gru, R.; Arico, M.; Zimmermann, M.; Mann, G.; Rossi, G. De.; Stanulla, M.; Locatelli, F.; et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood 2010, 115, 3206–3214. [Google Scholar] [CrossRef]
- Berry, D.A.; Zhou, S.; Higley, H.; Mukundan, L.; Fu, S.; Reaman, G.H.; Wood, B.L.; Kelloff, G.J.; Jessup, J.M.; Radich, J.P. Association of Minimal Residual Disease With Clinical Outcome in Pediatric and Adult Acute Lymphoblastic Leukemia: A Meta-analysis. JAMA Oncol. 2017, 3, e170580. [Google Scholar] [CrossRef]
- Rabin, K.R.; Gramatges, M.M.; Borowitz, M.J.; Palla, S.L.; Shi, X.; Margolin, J.F.; Zweidler-Mckay, P.A. Absolute lymphocyte counts refine minimal residual disease-based risk stratification in childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2012, 59, 468–474. [Google Scholar] [CrossRef] [Green Version]
- Rubnitz, J.E.; Campbell, P.; Zhou, Y.; Sandlund, J.T.; Jeha, S.; Ribeiro, R.C.; Inaba, H.; Bhojwani, D.; Relling, M.V.; Howard, S.C.; et al. Prognostic impact of absolute lymphocyte counts at the end of remission induction in childhood acute lymphoblastic leukemia. Cancer 2013, 119, 2061–2066. [Google Scholar] [CrossRef] [Green Version]
- Hatzipantelis, E.; Pana, Z.D.; Vlachou, M.; Papageorgiou, T.; Tragiannidis, A.; Athanassiadou, F. Peripheral blood lymphocyte recovery and overall survival in pediatric acute lymphoblastic leukemia. Pediatr. Blood Cancer 2014, 61, 181–183. [Google Scholar] [CrossRef]
- Gupta, A.; Kapoor, G.; Jain, S.; Bajpai, R. Absolute Lymphocyte Count Recovery Independently Predicts Outcome in Childhood Acute Lymphoblastic Leukemia: Experience from a Tertiary Care Cancer Center of a Developing Country. J. Pediatr. Hematol. Oncol. 2015, 37, e143–e149. [Google Scholar] [CrossRef] [PubMed]
- Hirase, S.; Hasegawa, D.; Takahashi, H.; Moriwaki, K.; Saito, A.; Kozaki, A.; Ishida, T.; Yanai, T.; Kawasaki, K.; Yamamoto, N. Absolute lymphocyte count at the end of induction therapy is a prognostic factor in childhood acute lymphoblastic leukemia. Int. J. Hematol. 2015, 102, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Rolf, N.; Smolen, K.K.; Kariminia, A.; Velenosi, A.; Fidanza, M.; Strahlendorf, C.; Seif, A.E.; Reid, G.S. Absolute lymphocyte counts at end-of-induction correlate with distinct immune cell compartments in pediatric B cell precursor acute lymphoblastic leukemia. Cancer Immune Immunother. 2018, 67, 225–236. [Google Scholar]
- Rakoff-Nahoum, S.; Medzhitov, R. Toll-like receptors and cancer. Nat. Rev. Cancer 2009, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tacken, P.J.; Zeelenberg, I.S.; Cruz, L.J.; van Hout-Kuijer, M.A.; van de Glind, G.; Fokkink, R.G.; Lambeck, A.J.; Figdor, C.G. Targeted delivery of Toll-like receptor ligands to human and mouse dendritic cells strongly enhances adjuvanticity. Blood 2011, 118, 6836–6844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Galluzzi, L.; Smyth, M.; Kroemer, G. Mechanism of Action of Conventional and Targeted Anticancer Therapies: Reinstating Immunosurveillance. Immunity 2013, 39, 74–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iribarren, K.; Bloy, N.; Buqué, A.; Cremer, I.; Eggermont, A.; Fridman, W.H.; Fucikova, J.; Galon, J.; Špíšek, R.; Zitvogel, L.; et al. Trial Watch: Immunostimulation with Toll-like receptor agonists in cancer therapy. Oncoimmunology 2015, 5, e1088631. [Google Scholar] [CrossRef] [Green Version]
- Sagiv-Barfi, I.; Czerwinski, D.K.; Levy, S.; Alam, I.S.; Mayer, A.T.; Gambhir, S.S.; Levy, R. Eradication of spontaneous malignancy by local immunotherapy. Sci. Transl. Med. 2018, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Campos, J.; Gallotta, M.; Gong, M.; Crain, C.; Naik, E.; Coffman, R.L.; Guiducci, C. Intratumoral injection of a CpG oligonucleotide reverts resistance to PD-1 blockade by expanding multifunctional CD8+ T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7240–E7249. [Google Scholar] [CrossRef] [Green Version]
- Sato-Kaneko, F.; Yao, S.; Ahmadi, A.; Zhang, S.S.; Hosoya, T.; Kaneda, M.M.; Varner, J.A.; Pu, M.; Messer, K.S.; Guiducci, C.; et al. Combination immunotherapy with TLR agonists and checkpoint inhibitors suppresses head and neck cancer. JCI Insight 2017, 2, 1–18. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, A.A.; Schultze, J.L.; Boussiotis, V.A.; Freeman, G.J.; Seamon, M.J.; Laszlo, S.; Billet, A.; Sallan, S.E.; Gribben, J.G.; Nadler, L.M. Pre-B acute lymphoblastic leukemia cells may induce T-cell anergy to alloantigen. Blood 1996, 88, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Alessandri, A.J.; Reid, G.S.D.; Bader, S.A.; Massing, B.G.; Sorensen, P.H.B.; Schultz, K.R. ETV6 (TEL)-AML1 pre-B acute lymphoblastic leukaemia cells are associated with a distinct antigen-presenting phenotype. Br. J. Haematol. 2002, 116, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Reid, G.S.D.; She, K.; Terrett, L.; Food, M.R.; Trudeau, J.D.; Schultz, K.R. CpG stimulation of precursor B-lineage acute lymphoblastic leukemia induces a distinct change in costimulatory molecule expression and shifts allogeneic T cells toward a Th1 response. Blood 2005, 105, 3641–3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corthals, S.L.; Wynne, K.; She, K.; Shimizu, H.; Curman, D.; Garbutt, K.; Reid, G.S.D. Differential immune effects mediated by Toll-like receptors stimulation in precursor B-cell acute lymphoblastic leukaemia. Br. J. Haematol. 2006, 132, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Trudeau, J.D.; Teachey, D.T.; Fish, J.D.; Grupp, S.A.; Schultz, K.R.; Reid, G.S.D. In vivo control of acute lymphoblastic leukemia by immunostimulatory CpG oligonucleotides. Blood 2007, 109, 2008–2013. [Google Scholar] [CrossRef] [PubMed]
- Seif, A.E.; Barrett, D.M.; Milone, M.; Brown, V.I.; Grupp, S.A.; Reid, G.S.D. Long-term protection from syngeneic acute lymphoblastic leukemia by CpG ODN-mediated stimulation of innate and adaptive immune responses. Blood 2009, 114, 2459–2466. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Malkin, D.; Tabori, U.; Shlien, A.; Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; et al. Comprehensive Analysis of Hypermutation in Human Cancer. Cell 2017, 171, 1042–1056. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Wasserman, R.; Zeng, X.X.; Hardy, R.R. The evolution of B precursor leukemia in the Emu-ret mouse. Blood 1998, 92, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.X.; Zhang, H.; Hardy, R.R.; Wasserman, R. The fetal origin of B-precursor leukemia in the E-mu-ret mouse. Blood 1998, 92, 3529–3536. [Google Scholar] [CrossRef] [PubMed]
- Barrett, D.M.; Seif, A.E.; Carpenito, C.; Teachey, D.T.; Fish, J.D.; June, C.H.; Grupp, S.A.; Reid, G.S.D. Noninvasive bioluminescent imaging of primary patient acute lymphoblastic leukemia: A strategy for preclinical modeling. Blood 2011, 118, 112–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duque-Afonso, J.; Feng, J.; Scherer, F.; Lin, C.H.; Wong, S.H.K.; Wang, Z.; Iwasaki, M.; Cleary, M.L. Comparative genomics reveals multistep pathogenesis of E2A-PBX1 acute lymphoblastic leukemia. J. Clin. Invest. 2015, 125, 3667–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pui, C.-H.; Carroll, W.L.; Meshinchi, S.; Arceci, R.J. Biology, risk stratification, and therapy of pediatric acute leukemias: An update. J. Clin. Oncol. 2011, 29, 551–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunger, S.P.; Lu, X.; Devidas, M.; Camitta, B.M.; Gaynon, P.S.; Winick, N.J.; Reaman, G.H.; Carroll, W.L. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the children’s oncology group. J. Clin. Oncol. 2012, 30, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Zhang, X. Activation of Murine Macrophages. Curr. Protoc. Immunol. 2008, 83, 14.2.1–14.2.10. [Google Scholar] [CrossRef]
- Zanoni, I.; Foti, M.; Ricciardi-Castagnoli, P.; Granucci, F. TLR-Dependent Activation Stimuli Associated with Th1 Responses Confer NK Cell Stimulatory Capacity to Mouse Dendritic Cells. J. Immunol. 2005, 175, 286–292. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Ishii, K.; Nguyen, S.; Su, P.P.; Burk, C.R.; Kim, B.H.; Duncan, B.B.; Tarun, S.; Shah, N.N.; Kohler, M.E.; et al. Murine Pre-B cell ALL induces T cell dysfunction not fully reversed by introduction of a chimeric antigen receptor. Blood 2018, 132, 1899–1910. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Lee, J.H.; Mattei, J.J.; Barrett, D.M.; van den Elzen, P.; Grupp, S.A.; Reid, G.S.D.; Seif, A.E. Generation of a multi-antigen-directed immune response for durable control of acute lymphoblastic leukemia. Leukemia 2017, 32, 539–542. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, S.; Fotovati, A.; Duque-Afonso, J.; Cleary, M.L.; van den Elzen, P.; Seif, A.E.; Reid, G.S.D. Differential Depletion of Bone Marrow Resident B-ALL after Systemic Administration of Endosomal TLR Agonists. Cancers 2020, 12, 169. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12010169
Jo S, Fotovati A, Duque-Afonso J, Cleary ML, van den Elzen P, Seif AE, Reid GSD. Differential Depletion of Bone Marrow Resident B-ALL after Systemic Administration of Endosomal TLR Agonists. Cancers. 2020; 12(1):169. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12010169
Chicago/Turabian StyleJo, Sumin, Abbas Fotovati, Jesus Duque-Afonso, Michael L. Cleary, Peter van den Elzen, Alix E. Seif, and Gregor S.D. Reid. 2020. "Differential Depletion of Bone Marrow Resident B-ALL after Systemic Administration of Endosomal TLR Agonists" Cancers 12, no. 1: 169. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12010169