From Conventional Therapies to Immunotherapy: Melanoma Treatment in Review

 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Melanoma—Epidemiology and Prevalence
3. Conventional Cancer Therapies
3.1. Excisional Surgery
3.2. Chemotherapy
- Dacarbazine (DTIC): Approved by the FDA in 1975 for treatment of melanoma, it is an alkylating agent. Like every other chemotherapeutic drug, it is not highly selective for cancer cells over healthy cells, and the high number of clinical trials which have been carried out have reported a modest anti-tumor efficacy. Despite this, dacarbazine remains one of the first-line treatments for metastatic melanoma [32].
- Temozolomide: Despite being considered an analogue of dacarbazine, it has been studied because it has the advantage of oral administration, which is usually more versatile for the patient. Furthermore, temozolomide can reach the central nervous system and, since brain is one of the most common sites for melanoma to metastasize, this represents a crucial point for advanced melanoma treatment [32].
3.3. Targeted Therapies
- BRAF inhibitors: Since BRAF is the most frequently mutated oncogene in melanoma [33], its inhibitors have shown promising results in several clinical trials, with rapid regression of metastasis and positive responses in 50–60% melanoma patients [32,34]. The first drug belonging to this class that has been approved for melanoma is vemurafenib, a selective inhibitor of V600-mutant BRAF [33]. In a randomized phase III clinical trial (BRIM3), vemurafenib showed an objective response rate (ORR) of 48% versus 5% for dacarbazine, and a median progression-free survival (PFS) of 5.3 months versus 1.6 months for dacarbazine [33,35]. The second BRAF inhibitor came soon after the first one, with similar promising results [33]. Toxicities associated with this class of therapeutic agents include rash, arthralgia, fatigue, fever (for dabrafenib only) and photosensitivity (for vemurafenib only), but also the development of secondary non-melanoma cutaneous lesions, such as squamous-cell carcinoma [36,37].
- MEK inhibitors: The development of MEK inhibitors became a priority after the success with BRAF-inhibitors, and it was led by the acknowledgement that BRAF signaling is dependent on MEK1/2 downstream activation [33,38]. Trametinib belongs to this class of new targeted therapies [32], and represents the first drug of its class to be approved by the FDA as a single agent, since in the phase III METRIC clinical trial it showed an ORR of 22% and a median PFS of 4.8 months [39]. Aside from the use of MEK inhibitors to target BRAF-mutated melanomas, there is also preclinical evidence that indicates vulnerability to MEK inhibitors in a not insignificant number of melanomas which do not present BRAF V600 mutations, called wild-type BRAF melanomas (especially in NRAS-Q61-mutant tumors), and also in BRAF/NRAS wild-type melanomas, together with melanomas harboring non-V600 BRAF mutations [33,40].
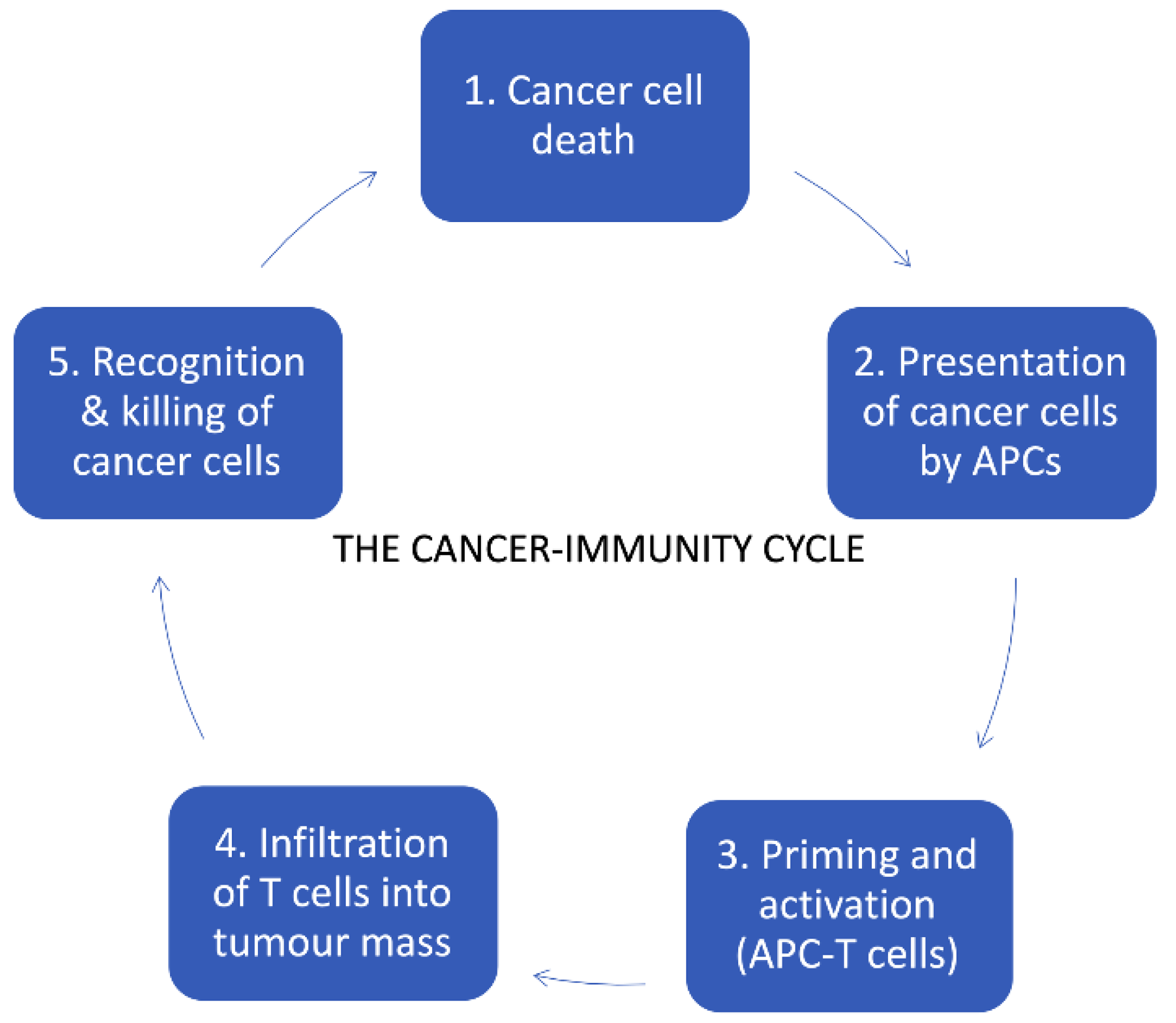
4. Cancer Immunotherapy
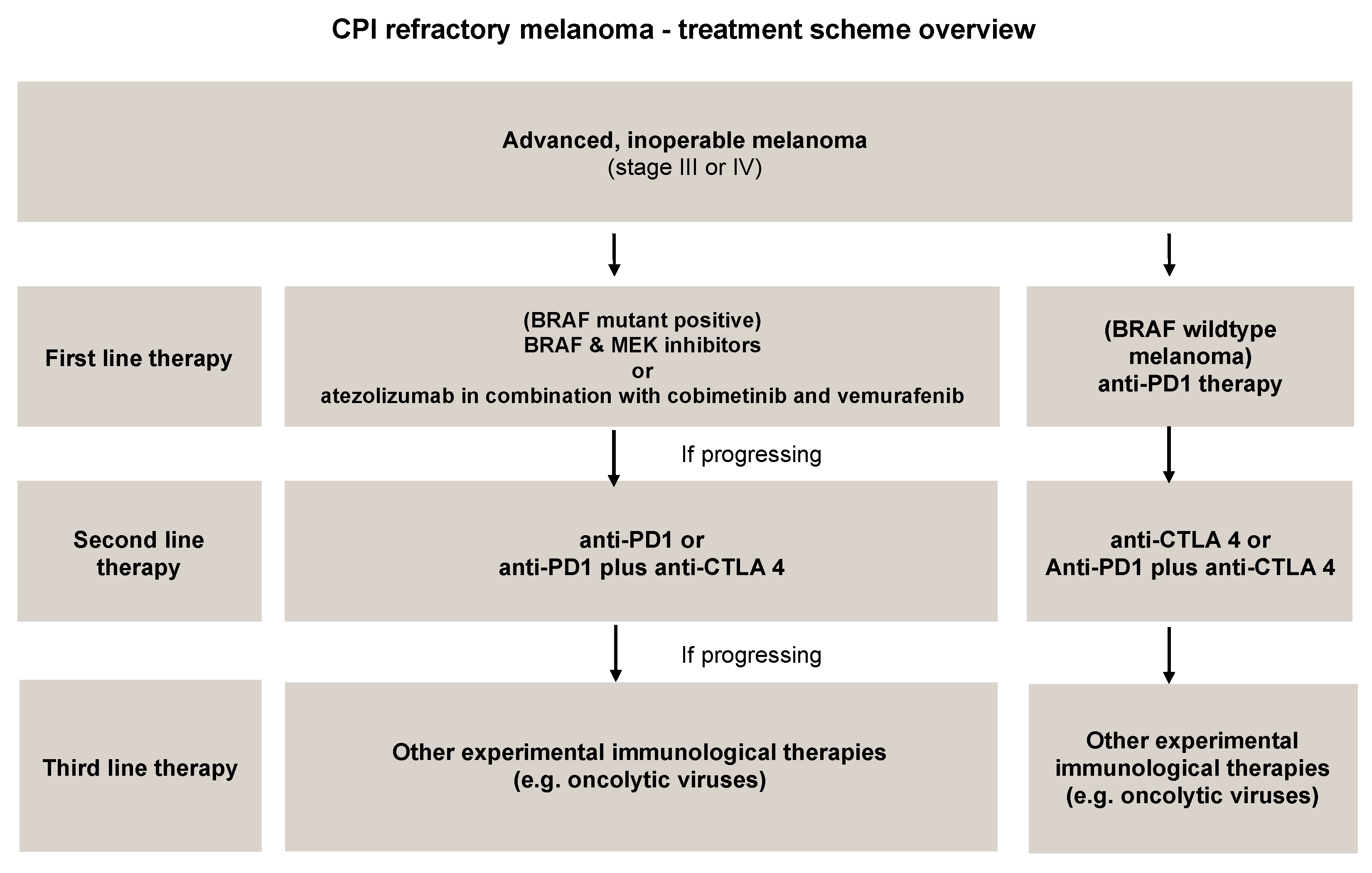
4.1. Immune Checkpoint Inhibitors (ICIs)
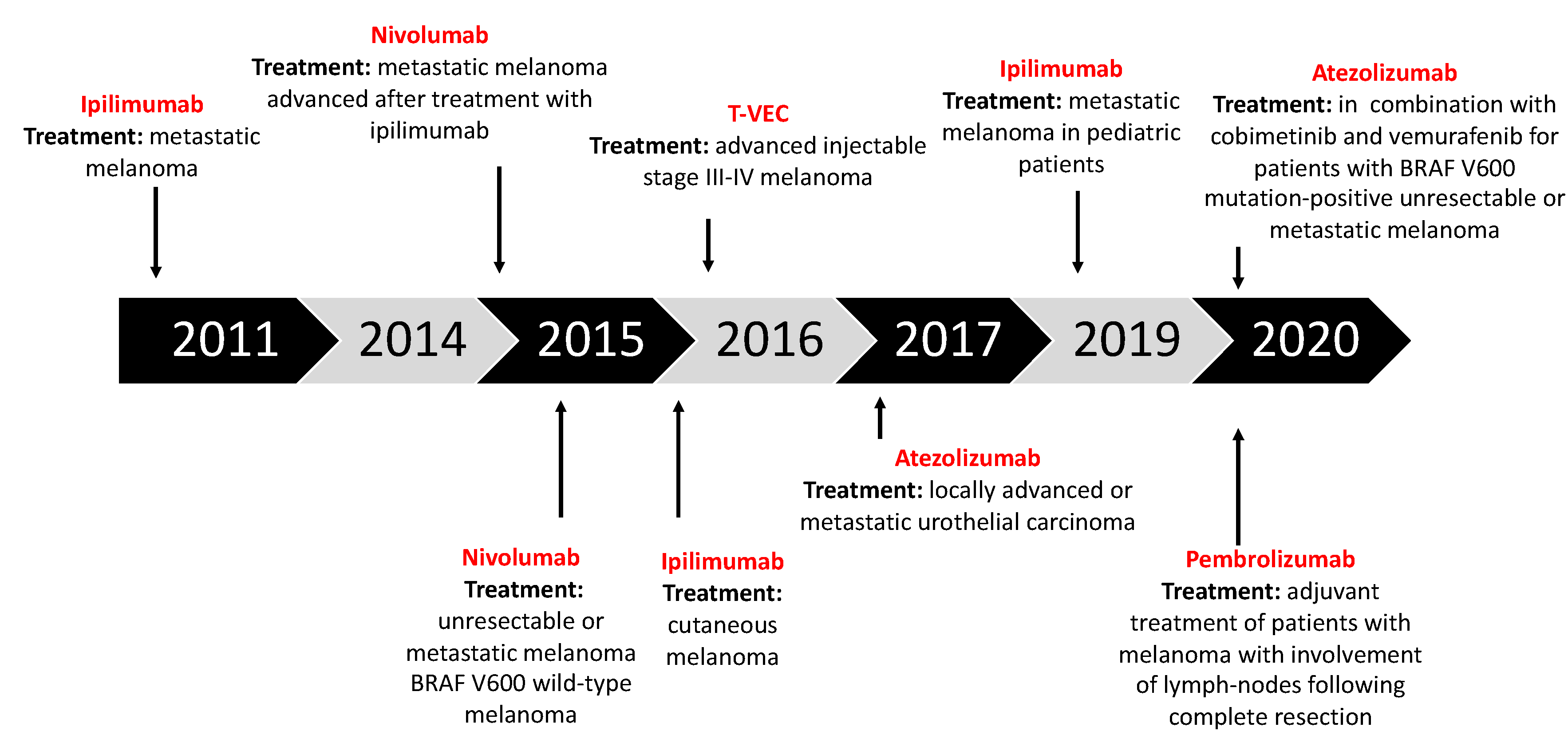
- Ipilimumab (anti-CTLA-4): Gained regulatory approval by the FDA to treat melanoma after a series of phase III clinical trials (CA184-002 as a single agent, CA184-024 in combination with dacarbazine). The tumor responses according to the Response Criteria in Solid Tumors (RECIST) criteria varied from 5.7% to 11.0% in the anti-CTLA-4 treatment arms. The median overall survival (OS) was improved to 10 months for the anti-CTLA-4 monotherapy arm as compared to 6.4 months for the peptide vaccine-alone arm (HR 0.68; p < 0.001 [58], CA184-002, NCT00094653). The five-year survival rate was 18.2% (95% CI, 13.6% to 23.4%) for patients treated with anti-CTLA-4 + dacarbazine vs. 8.8% (95% CI, 5.7% to 12.8%) for patients treated with placebo plus dacarbazine (p = 0.002, CA184-024, NCT00324155) [59]. Toxicity associated with ipilimumab includes immune-related symptoms such as dermatitis, colitis, diarrhea and, less commonly, hepatitis, uveitis and hypophysitis [60].
- Pembrolizumab and nivolumab (anti-PD1): After the ipilimumab proof of concept that a checkpoint blockade could actually be an effective strategy to treat melanoma, pembrolizumab and nivolumab were investigated for the same indication, even if (or maybe especially because) they are selective for another receptor which is usually expressed on immune T cell surface—PD-1. Phase III clinical trial reported the median overall survival which has not been reached in the nivolumab-plus-ipilimumab group and was 37.6 months in the nivolumab group, as compared with 19.9 months in the ipilimumab group (hazard ratio for death with nivolumab plus ipilimumab vs. ipilimumab, 0.55 [p < 0.001]; hazard ratio for death with nivolumab vs. ipilimumab, 0.65 [p < 0.001]). The overall survival rate at 3 years was 58% in the nivolumab-plus-ipilimumab group and 52% in the nivolumab group, as compared with 34% in the ipilimumab group (NCT01844505) [33,61,62,63].
4.2. Oncolytic Virotherapy
- Toll-like receptors (TLRs): This pathway is activated by pathogen-associated molecular patterns (PAMPs), which consist of elements of viral capsid, DNA, RNA and viral proteins. These elements are recognized by TLRs, and they stimulate the innate immune system through a variety of signaling factors (MYD88, TRIF, IRF7, IRF3, NF-kβ), leading to the release of pro-inflammatory cytokines and local type I interferon (IFN-I) [82,83].
- RIG-1 pathway: This pathway is activated by the detection of viral dsDNA and uses some of the same factors exploited by the TLRs pathway, such as IRF3/7. It leads to the release of IFN-I [6].
- IFN-I pathway: This is activated by the local production of type I interferon. After IFN-I binds to its receptor, IFNR, a cascade of signals is triggered and, through the JAK-STAT pathway, it leads to the upregulation of cell-cycle regulators such as PKR and IRF7. These two factors are important in order to contain viral spread because they induce abortive apoptosis, which blocks the replicative cycle of viruses before the viral progeny is ready to be released [82].
5. Combinatorial Approaches with OVs in Melanoma Treatments
5.1. OVs with Immune Checkpoint Inhibitors
5.2. OVs with Chemotherapeutic Agents—Future Prospects
- Use OVs as adjuvant to chemotherapy, which is the most clinically relevant approach, since chemotherapeutic agents represent the cornerstone of almost every cancer standard-of-care therapy [78].
- Use chemotherapeutic drugs to counteract or inhibit factors that limit the effectiveness of oncolytic virotherapy such as large tumor size, poor vasculature, elevated interstitial pressure and other physical barriers [112].
- Many chemotherapeutic agents induce apoptosis in cancer cells, while OVs need actively dividing cells to complete their replicative cycle successfully;
- Other chemotherapeutic drugs target angiogenetic mechanisms to impair tumor expansion, but this would also affect viral trafficking inside the tumor mass;
- The immune modulation exerted by some chemotherapeutic drugs could dampen the antitumor immune response triggered by OVs.
5.3. OVs with Radiotherapy—Future Prospects
- Radiotherapy may reduce the internal pressure within the tumor mass, making it easier for the OV to penetrate it and work properly.
- Some OVs, such as vesicular stomatitis virus (VSV) or HSV, are able to preferentially target Ras-mutated cancer cells (Ras is one of the driver mutations in melanoma). Since Ras mutations in cancer cells are associated with resistance to radiotherapy, OVs which can target these cells will exert a complementary therapeutic effect to radiotherapy.
- Infection of melanoma cells by OVs will lead to a release of cytokines like TNFα or TRAIL, which can sensitize tumor cells to radiation therapy.
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Loeb, K.R.; Loeb, L.A. Significance of multiple mutations in cancer. Carcinogenesis 2000, 21, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Calì, B.; Molon, B.; Viola, A. Tuning cancer fate: The unremitting role of host immunity. Open Biol. 2020, 7, 170006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The Immune System in Cancer Pathogenesis: Potential Therapeutic Approaches. J. Immunol. Res. 2016, 2016, 4273943. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gun, S.Y.; Lee, S.W.L.; Sieow, J.L.; Wong, S.C. Targeting immune cells for cancer therapy. Redox Biol. 2019, 25, 101174. [Google Scholar] [CrossRef]
- Elion, D.L.; Jacobson, M.E.; Hicks, D.J.; Rahman, B.; Sanchez, V.; Gonzales-Ericsson, P.I.; Fedorova, O.; Pyle, A.M.; Wilson, J.T.; Cook, R.S. Therapeutically Active RIG-I Agonist Induces Immunogenic Tumor Cell Killing in Breast Cancers. Cancer Res. 2018, 78, 6183–6195. [Google Scholar] [CrossRef] [Green Version]
- Lakshmi Narendra, B.; Eshvendar Reddy, K.; Shantikumar, S.; Ramakrishna, S. Immune system: A double-edged sword in cancer. Inflamm. Res. 2013, 62, 823–834. [Google Scholar] [CrossRef]
- Swann, J.B.; Smyth, M.J. Immune surveillance of tumors. J. Clin. Investig. 2007, 117, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Chow, M.T.; Möller, A.; Smyth, M.J. Inflammation and immune surveillance in cancer. Semin. Cancer Biol. 2012, 22, 23–32. [Google Scholar] [CrossRef]
- Goldszmid, R.S.; Dzutsev, A.; Trinchieri, G. Host Immune Response to Infection and Cancer: Unexpected Commonalities. Cell Host Microbe 2014, 15, 295–305. [Google Scholar] [CrossRef] [Green Version]
- Corrales, L.; Matson, V.; Flood, B.; Spranger, S.; Gajewski, T.F. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 2017, 27, 96–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamora, A.E.; Crawford, J.C.; Thomas, P.G. Hitting the Target: How T Cells Detect and Eliminate Tumors. J. Immunol. 2018, 200, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Durgeau, A.; Virk, Y.; Corgnac, S.; Mami-Chouaib, F. Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 2018, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.; Wang, J.; Sahoo, A. T-cell tolerance in cancer. Immunotherapy 2013, 5, 513–531. [Google Scholar] [CrossRef] [Green Version]
- Makkouk, A.; Weiner, G.J. Cancer Immunotherapy and Breaking Immune Tolerance: New Approaches to an Old Challenge. Cancer Res. 2015, 75, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen vaccine: An emerging tumor immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [Green Version]
- Uong, A.; Zon, L.I. Melanocytes in development and cancer. J. Cell. Physiol. 2010, 222, 38–41. [Google Scholar] [CrossRef] [Green Version]
- Bastian, B.C. The Molecular Pathology of Melanoma: An Integrated Taxonomy of Melanocytic Neoplasia. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 239–271. [Google Scholar] [CrossRef] [Green Version]
- Balois, T.; Chatelain, C.; Ben Amar, M. Patterns in melanocytic lesions: Impact of the geometry on growth and transport inside the epidermis. J. R. Soc. Interface 2014, 11, 20140339. [Google Scholar] [CrossRef]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, W.H.; Farma, J.M. Cutaneous Melanoma: Etiology and Therapy; Codon Publication: Wishart, Australia, 2018; ISBN 9780994438140. [Google Scholar]
- De Fabo, E.C.; Noonan, F.P.; Fears, T.; Merlino, G. Ultraviolet B but not Ultraviolet a Radiation Initiates Melanoma. Cancer Res. 2004, 64, 6372–6376. [Google Scholar] [CrossRef] [Green Version]
- Gandini, S.; Sera, F.; Cattaruzza, M.S.; Pasquini, P.; Picconi, O.; Boyle, P.; Melchi, C.F. Meta-analysis of risk factors for cutaneous melanoma: II. Sun exposure. Eur. J. Cancer 2005, 41, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.; Lee, A.; Smith, S.D. The Safety and Efficacy of Narrow Band Ultraviolet B Treatment in Dermatology: A Review. Am. J. Clin. Dermatol. 2015, 16, 501–531. [Google Scholar] [CrossRef]
- Berlin, N.L.; Cartmel, B.; Leffell, D.J.; Bale, A.E.; Mayne, S.T.; Ferrucci, L.M. Family history of skin cancer is associated with early-onset basal cell carcinoma independent of MC1R genotype. Cancer Epidemiol. 2015, 39, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Bossen, K.S.; Sørensen, H.T. The impact of comorbidity on cancer survival: A review. Clin. Epidemiol. 2013, 5, 3–29. [Google Scholar]
- Zbytek, B.; Carlson, J.A.; Granese, J.; Ross, J.; Mihm, M.; Slominski, A. Current concepts of metastasis in melanoma. Expert Rev. Dermatol. 2008, 3, 569–585. [Google Scholar] [CrossRef] [Green Version]
- Harries, M.; Malvehy, J.; Lebbe, C.; Heron, L.; Amelio, J.; Szabo, Z.; Schadendorf, D. Treatment patterns of advanced malignant melanoma (stage III–IV)—A review of current standards in Europe. Eur. J. Cancer 2016, 60, 179–189. [Google Scholar] [CrossRef]
- Perera, E.; Gnaneswaran, N.; Jennens, R.; Sinclair, R. Malignant Melanoma. Healthcare 2014, 2, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Mishra, H.; Mishra, P.K.; Ekielski, A.; Jaggi, M.; Iqbal, Z.; Talegaonkar, S. Melanoma treatment: From conventional to nanotechnology. J. Cancer Res. Clin. Oncol. 2018, 144, 2283–2302. [Google Scholar] [CrossRef] [PubMed]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [Green Version]
- Menzies, A.M.; Long, G.V. Review Systemic treatment for BRAF-mutant melanoma: Where do we go next? Lancet Oncol. 2014, 15, e371–e381. [Google Scholar] [CrossRef]
- Ravnan, M.C.; Matalka, M.S. Vemurafenib in Patients with BRAF V600E Mutation–Positive Advanced Melanoma. Clin. Ther. 2012, 34, 1474–1486. [Google Scholar] [CrossRef] [PubMed]
- Hauschild, A.; Grob, J.-J.; Demidov, L.V.; Jouary, T.; Gutzmer, R.; Millward, M.; Rutkowski, P.; Blank, C.U.; Miller, W.H.; Kaempgen, E.; et al. Dabrafenib in BRAF-mutated metastatic melanoma: A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012, 380, 358–365. [Google Scholar] [CrossRef]
- Daud, A.; Tsai, K. Management of Treatment-Related Adverse Events with Agents Targeting the MAPK Pathway in Patients with Metastatic Melanoma. Oncologist 2017, 22, 823–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, J.N.; Wang, T.; Cohen, M.S. BRAF and MEK Inhibitors: Use and Resistance in BRAF-Mutated Cancers. Drugs 2018, 78, 549–566. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved Survival with Vemurafenib in Melanoma with BRAF V600E Mutation. N. Engl. J. Med. 2011, 364, 2507–2523. [Google Scholar] [CrossRef] [Green Version]
- Kudchadkar, R.; Paraiso, K.H.T.; Smalley, K.S.M. Targeting Mutant BRAF in Melanoma: Current Status and Future Development of Combination Therapy Strategies. Cancer J. 2012, 18, 124. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Gonzalez, R.; Pavlick, A.; Hamid, O.; Gajewski, T.F.; Daud, A.; Flaherty, L.; Logan, T.; Chmielowski, B.; Lewis, K.; et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAFV600-mutated melanoma: A phase 1b study. Lancet Oncol. 2014, 15, 954–965. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Zappasodi, R.; Merghoub, T.; Wolchok, J.D. Emerging Concepts for Immune Checkpoint Blockade-Based Combination Therapies. Cancer Cell 2018, 33, 581–598. [Google Scholar] [CrossRef] [Green Version]
- Kuryk, L.; Møller, A.-S.W.; Jaderberg, M. Abscopal effect when combining oncolytic adenovirus and checkpoint inhibitor in a humanized NOG mouse model of melanoma. J. Med. Virol. 2019, 91, 1702–1706. [Google Scholar] [CrossRef] [Green Version]
- Kuryk, L.; Møller, A.S.W.; Jaderberg, M. Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. Oncoimmunology 2019, 8, 1–11. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Freeman, G.J. The B7–CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Wang, S.; He, Z.; Wang, X.; Li, H.; Liu, X.-S. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. eLife 2019, 8, e49020. [Google Scholar] [CrossRef]
- Cowey, C.L.; Liu, F.X.; Black-Shinn, J.; Stevinson, K.; Boyd, M.; Frytak, J.R.; Ebbinghaus, S.W. Pembrolizumab Utilization and Outcomes for Advanced Melanoma in US Community Oncology Practices. J. Immunother. 2018, 41, 86. [Google Scholar] [CrossRef] [Green Version]
- Deeks, E.D. Pembrolizumab: A Review in Advanced Melanoma. Drugs 2016, 76, 375–386. [Google Scholar] [CrossRef]
- Aris, M.; Barrio, M.M. Combining Immunotherapy with Oncogene-Targeted Therapy: A New Road for Melanoma Treatment. Front. Immunol. 2015, 6, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining Immune Checkpoint Inhibitors: Established and Emerging Targets and Strategies to Improve Outcomes in Melanoma. Front. Immunol. 2019, 10, 453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer, L.; Apuri, S.; Eroglu, Z.; Kottschade, L.A.; Forschner, A.; Gutzmer, R.; Schlaak, M.; Heinzerling, L.; Krackhardt, A.M.; Loquai, C.; et al. Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur. J. Cancer 2017, 75, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Constantin Kirchberger, M.; Moreira, A.; Erdmann, M.; Schuler, G.; Heinzerling, L. Real world experience in low-dose ipilimumab in combination with PD-1 blockade in advanced melanoma patients. Oncotarget 2018, 9, 28903. [Google Scholar] [CrossRef] [PubMed]
- Seliger, B. Basis of PD1/PD-L1 Therapies. J. Clin. Med. 2019, 8, 2168. [Google Scholar] [CrossRef] [Green Version]
- Coit, D.G.; Andtbacka, R.; Bichakjian, C.K.; Dilawari, R.A.; DiMaio, D.; Guild, V.; Halpern, A.C.; Hodi, F.S.; Kashani-Sabet, M.; Lange, J.R.; et al. Melanoma: Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2009, 7, 250–275. [Google Scholar] [CrossRef] [Green Version]
- Rogiers, A.; Boekhout, A.; Schwarze, J.K.; Awada, G.; Blank, C.U.; Neyns, B. Long-Term Survival, Quality of Life, and Psychosocial Outcomes in Advanced Melanoma Patients Treated with Immune Checkpoint Inhibitors. J. Oncol. 2019, 2019, 5269062. [Google Scholar] [CrossRef]
- Maio, M.; Grob, J.-J.; Aamdal, S.; Bondarenko, I.; Robert, C.; Thomas, L.; Garbe, C.; Chiarion-Sileni, V.; Testori, A.; Chen, T.-T.; et al. Five-Year Survival Rates for Treatment-Naive Patients with Advanced Melanoma Who Received Ipilimumab Plus Dacarbazine in a Phase III Trial. J. Clin. Oncol. 2015, 33, 1191–1196. [Google Scholar] [CrossRef]
- Savoia, P.; Astrua, C.; Fava, P. Ipilimumab (Anti-Ctla-4 Mab) in the treatment of metastatic melanoma: Effectiveness and toxicity management. Hum. Vaccin. Immunother. 2016, 12, 1092–1101. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, Durable Tumor Remission, and Long-Term Safety in Patients With Advanced Melanoma Receiving Nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Jessurun, C.A.C.; Vos, J.A.M.; Limpens, J.; Luiten, R.M. Biomarkers for Response of Melanoma Patients to Immune Checkpoint Inhibitors: A Systematic Review. Front. Oncol. 2017, 7, 233. [Google Scholar] [CrossRef] [PubMed]
- Kuryk, L.; Møller, A.S.W. Chimeric oncolytic Ad5/3 virus replicates and lyses ovarian cancer cells through desmoglein-2 cell entry receptor. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuryk, L.; Møller, A.-S.W.; Jaderberg, M. Quantification and functional evaluation of CD40L production from the adenovirus vector ONCOS-401. Cancer Gene Ther. 2018. [Google Scholar] [CrossRef]
- Kuryk, L.; Møller, A.-S.; Vuolanto, A.; Pesonen, S.; Garofalo, M.; Cerullo, V.; Jaderberg, M. Optimization of Early Steps in Oncolytic Adenovirus ONCOS-401 Production in T-175 and HYPERFlasks. Int. J. Mol. Sci. 2019, 20, 621. [Google Scholar] [CrossRef] [Green Version]
- Capasso, C.; Magarkar, A.; Cervera-carrascon, V.; Fusciello, M.; Feola, S.; Muller, M. ORIGINAL RESEARCH A novel in silico framework to improve MHC-I epitopes and break the tolerance to melanoma. Oncoimmunology 2017, 6, e1319028. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Villa, A.; Crescenti, D.; Marzagalli, M.; Kuryk, L.; Limonta, P.; Mazzaferro, V.; Ciana, P. Heterologous and cross-species tropism of cancer- derived extracellular vesicles. Theranostics 2019, 9, 5681. [Google Scholar] [CrossRef]
- Garofalo, M.; Villa, A.; Rizzi, N.; Kuryk, L.; Mazzaferro, V.; Ciana, P. Systemic Administration and Targeted Delivery of Immunogenic Oncolytic Adenovirus Encapsulated in Extracellular Vesicles for Cancer Therapies. Viruses 2018, 10, 558. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Villa, A.; Rizzi, N.; Kuryk, L.; Rinner, B.; Cerullo, V.; Yliperttula, M. Extracellular vesicles enhance the targeted delivery of immunogenic oncolytic adenovirus and paclitaxel in immunocompetent mice. J. Control. Release 2019, 294, 165–175. [Google Scholar] [CrossRef]
- Jhawar, S.R.; Thandoni, A.; Bommareddy, P.K.; Hassan, S.; Kohlhapp, F.J.; Goyal, S.; Schenkel, J.M.; Silk, A.W.; Zloza, A. Oncolytic Viruses—Natural and Genetically Engineered Cancer Immunotherapies. Front. Oncol. 2017, 7, 202. [Google Scholar] [CrossRef] [Green Version]
- Kuryk, Ł.; Wieczorek, M.; Diedrich, S.; Böttcher, S.; Witek, A.; Litwińska, B. Genetic analysis of poliovirus strains isolated from sewage in Poland. J. Med. Virol. 2014, 86, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Ciąćka, A.; Witek, A.; Kuryk, Ł.; Żuk-Wasek, A. Environmental Surveillance of Non-polio Enteroviruses in Poland, 2011. Food Environ. Virol. 2015, 7, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Pesonen, S.; Kangasniemi, L.; Hemminki, A. Oncolytic Adenoviruses for the Treatment of Human Cancer: Focus on Translational and Clinical Data. Mol. Pharm. 2011, 8, 12–28. [Google Scholar] [CrossRef]
- Garofalo, M.; Saari, H.; Somersalo, P.; Crescenti, D.; Kuryk, L.; Aksela, L.; Capasso, C.; Madetoja, M.; Koskinen, K.; Oksanen, T.; et al. Antitumor effect of oncolytic virus and paclitaxel encapsulated in extracellular vesicles for lung cancer treatment. J. Control. Release 2018, 283, 223–234. [Google Scholar] [CrossRef]
- Kuryk, L.; Møller, A.-S.W.; Garofalo, M.; Cerullo, V.; Pesonen, S.; Alemany, R.; Jaderberg, M. Anti-tumor specific T-cell responses induced by oncolytic adenovirus ONCOS-102 in peritoneal mesothelioma mouse model. J. Med. Virol. 2018, 1669–1673. [Google Scholar] [CrossRef] [Green Version]
- Capasso, C.; Hirvinen, M.; Garofalo, M.; Romaniuk, D.; Kuryk, L.; Sarvela, T.; Vitale, A.; Antopolsky, M.; Magarkar, A.; Viitala, T.; et al. Oncolytic adenoviruses coated with MHC-I tumor epitopes increase the antitumor immunity and efficacy against melanoma. Oncoimmunology 2016, 5, e1105429. [Google Scholar] [CrossRef] [Green Version]
- Kuryk, L.; Haavisto, E.; Garofalo, M.; Capasso, C.; Hirvinen, M.; Pesonen, S.; Ranki, T.; Vassilev, L.; Cerullo, V. Synergistic anti-tumor efficacy of immunogenic adenovirus ONCOS-102 (Ad5/3-D24-GM-CSF) and standard of care chemotherapy in preclinical mesothelioma model. Int. J. Cancer 2016, 139, 1883–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuryk, L.; Vassilev, L.; Ranki, T.; Hemminki, A.; Karioja-Kallio, A.; Levälampi, O.; Vuolanto, A.; Cerullo, V.; Pesonen, S. Toxicological and bio-distribution profile of a GM-CSF-expressing, double-targeted, chimeric oncolytic adenovirus ONCOS-102—Support for clinical studies on advanced cancer treatment. PLoS ONE 2017, 12, e0182715. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Iovine, B.; Kuryk, L.; Capasso, C.; Hirvinen, M.; Vitale, A.; Yliperttula, M.; Bevilacqua, M.A.; Cerullo, V. Oncolytic Adenovirus Loaded with L-carnosine as Novel Strategy to Enhance the Antitumor Activity. Mol. Cancer Ther. 2016, 15, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Hirvinen, M.; Capasso, C.; Guse, K.; Garofalo, M.; Vitale, A.; Ahonen, M.; Kuryk, L.; Vähä-Koskela, M.; Hemminki, A.; Fortino, V.; et al. Expression of DAI by an oncolytic vaccinia virus boosts the immunogenicity of the virus and enhances antitumor immunity. Mol. Ther. Oncolytics 2016, 3, 16002. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Chen, X.; Ye, K.; Yao, Y.; Li, Y. Application potential of toll-like receptors in cancer immunotherapy: Systematic review. Medicine 2016, 95, e3951. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef] [Green Version]
- Vähä-Koskela, M.J.V.; Heikkilä, J.E.; Hinkkanen, A.E. Oncolytic viruses in cancer therapy. Cancer Lett. 2007, 254, 178–216. [Google Scholar] [CrossRef]
- Coit, D.G.; Thompson, J.A.; Algazi, A.; Andtbacka, R.; Bichakjian, C.K.; Carson, W.E.; Daniels, G.A.; DiMaio, D.; Ernstoff, M.; Fields, R.C.; et al. Melanoma, Version 2.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2016, 14, 450–473. [Google Scholar] [CrossRef]
- Antohe, M.; Nedelcu, R.I.; Nichita, L.; Popp, C.G.; Cioplea, M.; Brinzea, A.; Hodorogea, A.; Calinescu, A.; Balaban, M.; Ion, D.A.; et al. Tumor infiltrating lymphocytes: The regulator of melanoma evolution (Review). Oncol. Lett. 2019, 17, 4155–4161. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Corrigan, P.A.; Beaulieu, C.; Patel, R.B.; Lowe, D.K. Talimogene Laherparepvec: An Oncolytic Virus Therapy for Melanoma. Ann. Pharmacother. 2017, 51, 675–681. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, C.; Doepker, M.P.; Zager, J.S. Talimogene laherparepvec: Overview, combination therapy and current practices. Melanoma Manag. 2016, 3, 267–272. [Google Scholar] [CrossRef]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Sivanandam, V.; Larocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncolytics 2019, 13, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervera-Carrascon, V.; Siurala, M.; Santos, J.M.; Havunen, R.; Tähtinen, S.; Karell, P.; Sorsa, S.; Kanerva, A.; Hemminki, A. TNFa and IL-2 armed adenoviruses enable complete responses by anti-PD-1 checkpoint blockade. Oncoimmunology 2018, 7, e1412902. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer 2019, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hromic-jahjefendic, A.; Lundstrom, K. Viral Vector-Based Melanoma Gene Therapy. Biomedicines 2020, 8, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larocca, C.A.; LeBoeuf, N.R.; Silk, A.W.; Kaufman, H.L. An Update on the Role of Talimogene Laherparepvec (T-VEC) in the Treatment of Melanoma: Best Practices and Future Directions. Am. J. Clin. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Funchain, P.; Song, J.M.; Rayman, P.; Tannenbaum, C.; Ko, J.; Mcnamara, M.; Marcela Diaz-Montero, C.; Gastman, B. Talimogene Laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: A case series. J. Immunother. Cancer 2018, 6, 36. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Imbert-Fernandez, Y.; Telang, S.; Baum, M.; Ranjan, S.; Fraig, M.; Batty, N. Potential clinical and immunotherapeutic utility of talimogene laherparepvec for patients with melanoma after disease progression on immune checkpoint inhibitors and BRAF inhibitors. Melanoma Res. 2018, 28, 250. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.I.; Curti, B.D.; Hallmeyer, S.; Feng, Z.; Paustian, C.; Bifulco, C.; Fox, B.; Grose, M.; Shafren, D. Phase II calm extension study: Coxsackievirus A21 delivered intratumorally to patients with advanced melanoma induces immune-cell infiltration in the tumor microenvironment. J. Immunother. Cancer 2015, 3, 343. [Google Scholar] [CrossRef] [Green Version]
- Curti, B.; Richards, J.; Hallmeyer, S.; Faries, M.; Andtbacka, R.; Daniels, G.; Grose, M.; Shafren, D.R. Abstract CT114: The MITCI (Phase 1b) study: A novel immunotherapy combination of intralesional Coxsackievirus A21 and systemic ipilimumab in advanced melanoma patients with or without previous immune checkpoint therapy treatment. Cancer Res. 2017, 77, CT114. [Google Scholar] [CrossRef]
- Schmid, P.; Cruz, C.; Braiteh, F.S.; Eder, J.P.; Tolaney, S.; Kuter, I.; Nanda, R.; Chung, C.; Cassier, P.; Delord, J.-P.; et al. Abstract 2986: Atezolizumab in metastatic TNBC (mTNBC): Long-term clinical outcomes and biomarker analyses. Cancer Res. 2017, 77, 2986. [Google Scholar] [CrossRef]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimäki, A.; et al. Phase I study with ONCOS-102 for the treatment of solid tumors—An evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, M.; Armstrong, E.J.L.; Jennings, V.; Foo, S.; Crespo-Rodriguez, E.; Bozhanova, G.; Patin, E.C.; McLaughlin, M.; Mansfield, D.; Baker, G.; et al. Combination therapy with oncolytic viruses and immune checkpoint inhibitors. Expert Opin. Biol. Ther. 2020, 20, 635–652. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.; Peng, K.W.; Russell, S.J.; Diaz, R.M. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs 2019, 33, 485–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchini, A.; Daeffler, L.; Pozdeev, V.I.; Angelova, A.; Rommelaere, J. Immune Conversion of Tumor Microenvironment by Oncolytic Viruses: The Protoparvovirus H-1PV Case Study. Front. Immunol. 2019, 10, 1848. [Google Scholar] [CrossRef]
- Doloff, J.C.; Waxman, D.J. Dual E1A oncolytic adenovirus: Targeting tumor heterogeneity with two independent cancer-specific promoter elements, DF3/MUC1 and hTERT. Cancer Gene Ther. 2011, 18, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized Oncolytic Virotherapy Overcomes Systemic Tumor Resistance to Immune Checkpoint Blockade Immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [Green Version]
- Woller, N.; Gürlevik, E.; Fleischmann-Mundt, B.; Schumacher, A.; Knocke, S.; Kloos, A.M.; Saborowski, M.; Geffers, R.; Manns, M.P.; Wirth, T.C.; et al. Viral Infection of Tumors Overcomes Resistance to PD-1-immunotherapy by Broadening Neoantigenome-directed T-cell Responses. Mol. Ther. 2015, 23, 1630–1640. [Google Scholar] [CrossRef] [Green Version]
- Kruger, S.; Ilmer, M.; Kobold, S.; Cadilha, B.L.; Endres, S.; Ormanns, S.; Schuebbe, G.; Renz, B.W.; D’Haese, J.G.; Schloesser, H.; et al. Advances in cancer immunotherapy 2019—Latest trends. J. Exp. Clin. Cancer Res. 2019, 38, 268. [Google Scholar] [CrossRef] [Green Version]
- Kaunitz, G.J.; Cottrell, T.R.; Lilo, M.; Muthappan, V.; Esandrio, J.; Berry, S.; Xu, H.; Ogurtsova, A.; Anders, R.A.; Fischer, A.H.; et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Investig. 2017, 97, 1063–1071. [Google Scholar] [CrossRef] [Green Version]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef] [Green Version]
- Cerullo, V.; Vähä-Koskela, M.; Hemminki, A. Oncolytic adenoviruses: A potent form of tumor immunovirotherapy. Oncoimmunology 2012, 1, 979–981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, A.; Ho, L.; Wan, Y. Chemotherapy and Oncolytic Virotherapy: Advanced Tactics in the War against Cancer. Front. Oncol. 2014, 4, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirin, C.; Mainka, A.; Hesse, A.; Nettelbeck, D.M. Combining adenoviral oncolysis with temozolomide improves cell killing of melanoma cells. Int. J. Cancer 2007, 2807, 2801–2807. [Google Scholar] [CrossRef] [PubMed]
- Adusumilli, P.S.; Stiles, B.M.; Chan, M.-K.; Chou, T.-C.; Wong, R.J.; Rusch, V.W.; Fong, Y. Radiation Therapy Potentiates Effective Oncolytic Viral Therapy in the Treatment of Lung Cancer. Ann. Thorac. Surg. 2005, 80, 409–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adusumilli, P.S.; Chan, M.-K.; Hezel, M.; Yu, Z.; Stiles, B.M.; Chou, T.-C.; Rusch, V.W.; Fong, Y. Radiation-Induced Cellular DNA Damage Repair Response Enhances Viral Gene Therapy Efficacy in the Treatment of Malignant Pleural Mesothelioma. Ann. Surg. Oncol. 2007, 14, 258–269. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef]
- Zhang, T.; Suryawanshi, Y.R.; Woyczesczyk, H.M.; Essani, K. Targeting Melanoma with Cancer-Killing Viruses. Open Virol. J. 2017, 11, 28–47. [Google Scholar] [CrossRef] [Green Version]
- Noser, J.A.; Mael, A.A.; Sakuma, R.; Ohmine, S.; Marcato, P.; Lee, P.W.K.; Ikeda, Y. The RAS/Raf1/MEK/ERK Signaling Pathway Facilitates VSV-mediated Oncolysis: Implication for the Defective Interferon Response in Cancer Cells. Mol. Ther. 2007, 15, 1531–1536. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| OV | Checkpoint Inhibitor | Indication | Response Data | ClinicalTrials.gov ID |
|---|---|---|---|---|
| T-VEC | Ipilimumab | Melanoma | ORR 39% (comb.) vs. 18% (ipi alone) | NCT01740297 |
| T-VEC | Pembrolizumab | Stage IIIB–IV melanoma | 48% ORR | NCT02263508 |
| T-VEC | Pembrolizumab | Stage III–IV melanoma | N/A | NCT02965716 |
| HF-10 | Ipilimumab | Melanoma | N/A | NCT031530085 |
| HF-10 | Ipilimumab | Melanoma | BORR 24% (at 24 weeks); median PFS 19 months; median OS 21.8 months | NCT02272855 |
| HF-10 | Nivolumab | Stage IIIB, IIIC, IVM1a melanoma | N/A | NCT03259425 |
| CAVATAK | Ipilimumab | Uveal melanoma with liver metastasis | N/A | NCT03408587 |
| CAVATAK | Pembrolizumab | Melanoma | N/A | NCT02565992 |
| ONCOS-102 | Pembrolizumab | Advanced or unresectable melanoma | N/A | NCT03003676 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuryk, L.; Bertinato, L.; Staniszewska, M.; Pancer, K.; Wieczorek, M.; Salmaso, S.; Caliceti, P.; Garofalo, M. From Conventional Therapies to Immunotherapy: Melanoma Treatment in Review. Cancers 2020, 12, 3057. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103057
Kuryk L, Bertinato L, Staniszewska M, Pancer K, Wieczorek M, Salmaso S, Caliceti P, Garofalo M. From Conventional Therapies to Immunotherapy: Melanoma Treatment in Review. Cancers. 2020; 12(10):3057. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103057
Chicago/Turabian StyleKuryk, Lukasz, Laura Bertinato, Monika Staniszewska, Katarzyna Pancer, Magdalena Wieczorek, Stefano Salmaso, Paolo Caliceti, and Mariangela Garofalo. 2020. "From Conventional Therapies to Immunotherapy: Melanoma Treatment in Review" Cancers 12, no. 10: 3057. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103057