Evolution of Mutational Landscape and Tumor Immune-Microenvironment in Liver Oligo-Metastatic Colorectal Cancer

 , ,
, ,  , , , ,
, , , ,  , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
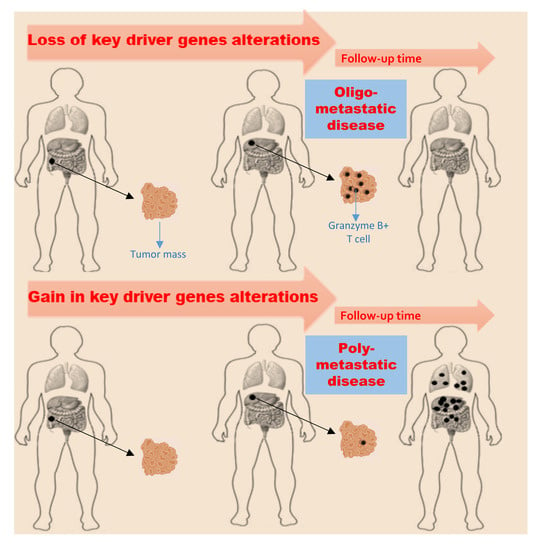
1. Introduction
2. Results
2.1. Characteristics of Patients
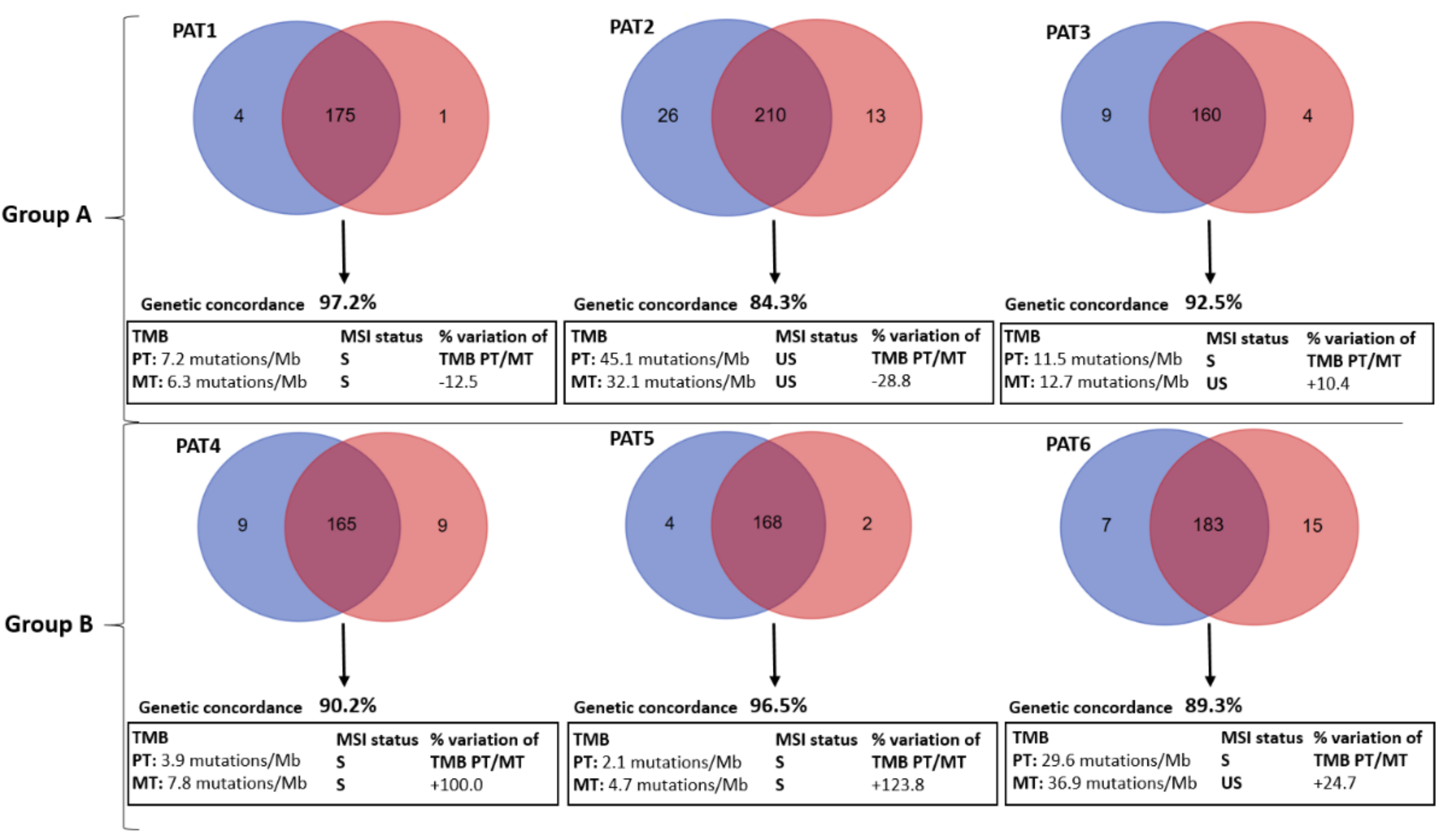
2.2. Mutational Concordance, Tumor Mutation Burden (TMB), Microsatellite (MSI) Status and Mutational Signatures
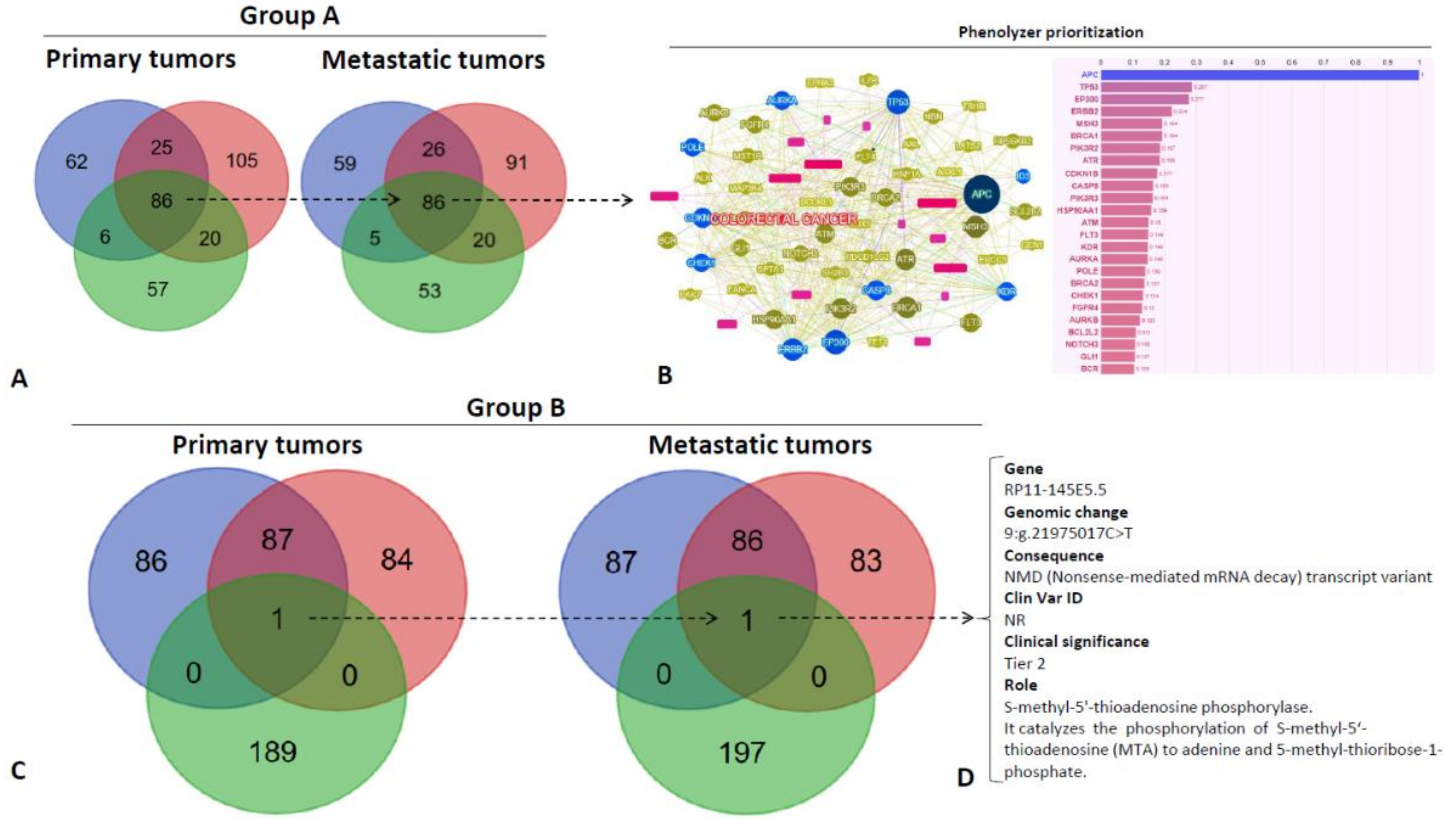
2.3. Overall Genes Evolution
2.4. Key Driver Genes Evolution
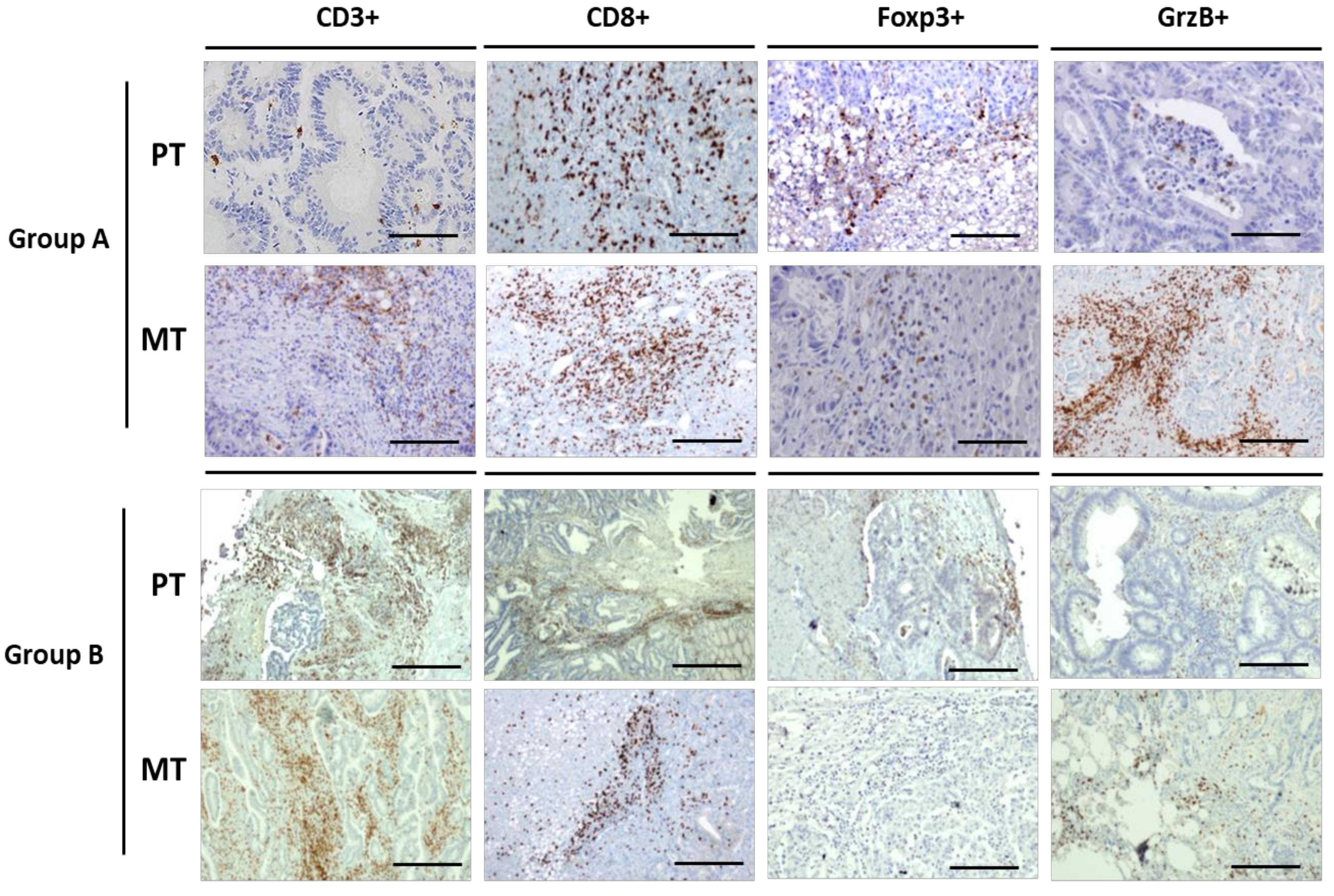
2.5. Tumor Microenvironment Evolution
3. Discussion
4. Materials and Methods
4.1. Tumor Specimens and Sequencing
4.2. Tumor Mutational Burden (TMB), Microsatellite Instability (MSI) and Mutational Profiles
4.3. Tumor-Infiltrating Lymphocytes Analysis
4.4. Biostatistical Analysis and Data Presentation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Xie, R.; Wei, R.; Cai, C.; Bi, D.; Yin, D.; Liu, H.; Zheng, J.; Zhang, Y.; Song, F.; et al. Integrin α5 subunit is required for the tumor supportive role of fibroblasts in colorectal adenocarcinoma and serves as a potential stroma prognostic marker. Mol. Oncol. 2019, 13, 2697–2714. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, S.; Kunze, P.; Hampel, C.; Eckstein, M.; Bramsen, J.B.; Muenzner, J.K.; Carlé, B.; Ndreshkjana, B.; Kemenes, S.; Gasparini, P.; et al. DAPK1 loss triggers tumor invasion in colorectal tumor cells. Cell Death Dis. 2019, 10, 895. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Feng, X.; Fang, Y.; Li, Y.; Li, Z.; Zhan, Y.; Li, M.; Li, G.; Ding, Y.; Deng, H. POTEE drives colorectal cancer development via regulating SPHK1/p65 signaling. Cell Death Dis. 2019, 10, 863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordgaard, C.; Doll, S.; Matos, A.L.D.S.A.; Høeberg, M.; Kazi, J.U.; Friis, S.; Stenvang, J.; Rönnstrand, L.; Mann, M.; Moreira, J.M.A. inhibitor 1 (TIMP-1) promotes receptor tyrosine kinase c-Kit signaling in colorectal cancer. Mol. Oncol. 2019, 13, 2646–2662. [Google Scholar] [CrossRef] [PubMed]
- DiPrima, M.; Wang, D.; Tröster, A.; Maric, A.; Terrades-Garcia, N.; Ha, T.; Kwak, H.; Sánchez-Martín, D.; Kudlinzki, D.; Schwalbe, H.; et al. Identification of Eph receptor signaling as a regulator of autophagy and a therapeutic target in colorectal carcinoma. Mol. Oncol. 2019, 13, 2441–2459. [Google Scholar] [CrossRef]
- Vatandoust, S.; Price, T.J.; Karapetis, C.S. Colorectal cancer: Metastases to a single organ. World J. Gastroenterol. 2015, 21, 11767–11776. [Google Scholar] [CrossRef]
- Chan, H.T.; Nagayama, S.; Chin, Y.M.; Otaki, M.; Hayashi, R.; Kiyotani, K.; Fukunaga, Y.; Ueno, M.; Nakamura, Y.; Low, S.-K. Clinical significance of clonal hematopoiesis in the interpretation of blood liquid biopsy. Mol. Oncol. 2020, 14, 1719–1730. [Google Scholar] [CrossRef]
- Rusthoven, K.E.; Kavanagh, B.D.; Burri, S.H.; Chen, C.; Cardenes, H.; Chidel, M.A.; Pugh, T.J.; Kane, M.; Gaspar, L.E.; Schefter, T.E. Multi -Institutional Phase I/II Trial of Stereotactic Body Radiation Therapy for Lung Metastases. J. Clin. Oncol. 2009, 27, 1579–1584. [Google Scholar] [CrossRef]
- Hellman, S.; Weichselbaum, R.R. Oligometastases. J. Clin. Oncol. 1995, 13, 8–10. [Google Scholar] [CrossRef]
- Niibe, Y.; Chang, J.Y.; Onishi, H.; Salama, J.; Hiraki, T.; Yamashita, H. Oligometastases/oligo-recurrence of lung cancer. J. Clin. Oncol. 2014, 13, 8–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Withers, H.R.; Lee, S.P. Modeling growth kinetics and statistical distribution of oligometastases. Semin. Radiat. Oncol. 2016, 16, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Lussier, Y.A.; Khodarev, N.N.; Regan, K.; Corbin, K.; Li, H.; Ganai, S.; Khan, S.A.; Gnerlich, J.; Darga, T.E.; Fan, H.; et al. Oligo -and polymetastatic progression in lungmetastasis(es) patients is associated with specific MicroRNAs. PLoS ONE 2012, 7, e50141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastos, P.; Gomes, T.; Ribeiro, L. Catechol-O-Methyltransferase (COMT): An Updateon Its Role in Cancer, Neurological and Cardiovascular Diseases. Rev. Physiol. Biochem. Pharmacol. 2017, 173, 1–39. [Google Scholar] [PubMed]
- Balzan, S.; Lubrano, V. LOX-1 receptor: A potential link in atherosclerosis and cancer. Life Sci. 2018, 198, 79–86. [Google Scholar] [CrossRef]
- Ouyang, L.; Zhang, K.; Chen, J.; Wang, J.; Huang, H. Roles of platelet derived growth factor in vascular calcification. J. Cell. Physiol. 2018, 233, 2804–2814. [Google Scholar] [CrossRef]
- Ottaiano, A.; De Divitiis, C.; Capozzi, M.; Guetz, G.D.; Pisano, C.; Pignata, S.; Tafuto, S. Obesity and Cancer: Biological Links and Treatment Implications. Curr. Cancer Drug Targets 2018, 18, 231–238. [Google Scholar] [CrossRef]
- Nechama, M.; Kwon, J.; Wei, S.; Kyi, A.T.; Welner, R.S.; Ben-Dov, I.Z. The IL-33-PIN1-IRAK-M axis is critical for type 2 immunity inIL-33-induced allergic airway inflammation. Nat. Commun. 2018, 9, 1603. [Google Scholar] [CrossRef] [Green Version]
- Marenholz, I.; Esparza-Gordillo, J.; Rüschendorf, F.; Bauerfeind, A.; Strachan, D.P.; Spycher, B.D.; Ege, M. Meta-analysis identifies seven susceptibility loci involved in the atopic march. Nat. Commun. 2015, 6, 8804. [Google Scholar] [CrossRef] [Green Version]
- Kozłowska, R.; Bożek, A.; Jarząb, J. Association between cancer and allergies. Allergy Asthma Clin. Immunol. 2016, 12, 39. [Google Scholar] [CrossRef] [Green Version]
- Chou, W.-Y.; Lai, P.-Y.; Hu, J.-M.; Hsu, C.-H.; Chen, Y.-C.; Tian, Y.-F.; You, S.-L.; Hsiao, C.-W.; Chou, Y.-C.; Sun, C.-A. Association between atopic dermatitis and colorectal cancer risk: A nationwide cohort study. Medicine (Baltimore) 2020, 99, e18530. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Garlanda, C.; Borrello, M.G.; Sica, A.; Mantovani, A. Pathways connecting inflammation and cancer. Curr. Opin. Genet. Dev. 2008, 18, 3–10. [Google Scholar] [CrossRef]
- Borrello, M.G.; Degl’Innocenti, D.; Pierotti, M.A. Inflammation and cancer: The oncogene-driven connection. Cancer Lett. 2008, 267, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Marazzi, I.; Greenbaum, B.D.; Low, D.H.P.; Guccione, E. Chromatin dependencies in cancer and inflammation. Nat. Rev. Mol. Cell Biol. 2008, 19, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Scala, S.; Circelli, L.; Lombardi, A.; Scala, S.; Martucci, N.; Galon, J.; Buonanno, M.; Scognamiglio, G.; Botti, G.; Hermitte, F.; et al. Genetic trajectory and immune microenvironment of lung-specific oligometastatic colorectal cancer. Cell Death Dis. 2020, 11, 275. [Google Scholar]
- Wang, Q.; Shi, Y.-L.; Zhou, K.; Wang, L.-L.; Yan, Z.-X.; Liu, Y.-L.; Xu, L.-L.; Zhao, S.-W.; Chu, H.-L.; Shi, T.-T.; et al. PIK3CA mutations confer resistance to first-line chemotherapy in colorectal cancer. Cell Death Dis. 2018, 9, 739. [Google Scholar] [CrossRef]
- Jang, S.; Hong, M.; Shin, M.K.; Kim, B.C.; Shin, H.-S.; Jang, S.J.; Hong, S.-M.; Kim, J.; Chun, S.-M.; Kim, T.-I.; et al. KRAS and PIK3CA mutations in colorectal adenocarcinomas correlate with aggressive histological features and behavior. Hum. Pathol. 2017, 65, 21–30. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Salz, T.; Zajac-Kaye, M.; Liao, D.; Huang, S.; Qiu, Y. Overexpression of histone deacetylases in cancer cells is controlled by interplay of transcription factors and epigenetic modulators. FASEB J. 2014, 4, 4265–4279. [Google Scholar] [CrossRef] [Green Version]
- Iyer, N.G.; Ozdag, H. Caldas C p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.T.; Demenais, F.; Iles, M.M.; Harland, M.; Taylor, J.C.; Corda, E.; Randerson-Moor, J.; Aitken, J.F.; Avril, M.-F.; Azizi, E.; et al. Genome-wide association study identifies three loci associated with melanoma risk. Nat. Genet. 2009, 8, 920–925. [Google Scholar] [CrossRef] [PubMed]
- Stevens, A.P.; Spangler, B.; Wallner, S.; Kreutz, M.; Dettmer, K.; Oefner, P.J.; Bosserhoff, A.-K. Direct and tumor microenvironment mediated influences of 5′-deoxy-5′-(methylthio)adenosine on tumor progression of malignant melanoma. J. Cell. Biochem. 2009, 106, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Lu, K.; Zhu, S.; Li, W.; Sun, S. Characterization of methylthioadenosin phosphorylase (MTAP) expression in colorectal cancer. Artif. Cells Nanomed. Biotechnol. 2018, 46, 2082–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snezhkina, A.V.; Krasnov, G.S.; Lipatova, A.V.; Sadritdinova, A.F.; Kardymon, O.L.; Fedorova, M.S.; Melnikova, N.V.; Stepanov, O.A.; Zaretsky, A.R.; Kaprin, A.D.; et al. The Dysregulation of Polyamine Metabolism in Colorectal Cancer Is Associated with Overexpression of c-Myc and C/EBPβ rather than Enterotoxigenic Bacteroides fragilis Infection. Oxid. Med. Cell Longev. 2016, 2016, 353560. [Google Scholar] [CrossRef] [Green Version]
- Ishaque, N.; Abba, M.L.; Hauser, C.; Patil, N.; Paramasivam, N.; Huebschmann, D.; Leupold, J.H.; Balasubramanian, G.P.; Kleinheinz, K.; Toprak, U.H.; et al. Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nat. Commun. 2018, 9, 4782. [Google Scholar] [CrossRef] [Green Version]
- Braxton, D.R.; Zhang, R.; Morrissette, J.D.; Aiza-Bonilla, A.L.; Furth, E.E. Clinicopathogenomic analysis of mismatch repair proficient colorectal adenocarcinoma uncovers novel prognostic subgroups with differing patterns of genetic evolution. Int. J. Cancer 2016, 139, 1546–1556. [Google Scholar] [CrossRef]
- Van Helden, E.J.; Angus, L.; van der Houven van Oordt, W.M.; Heideman, D.A.M.; Boon, E.; Van Es, S.C.; Radema, S.A.; Van Herpen, C.M.L.; De Groot, D.J.A.; De Vries, E.G.E.; et al. RAS and BRAF mutations in cell-free DNA are predictive for outcome of cetuximab monotherapy in patients with tissue-tested RAS wild-type advanced colorectal cancer. Mol. Oncol. 2019, 13, 2361–2374. [Google Scholar] [CrossRef] [Green Version]
- Elez, E.; Chianese, C.; Sanz-García, E.; Martinelli, E.; Noguerido, A.; Mancuso, F.M.; Caratù, G.; Matito, J.; Grasselli, J.; Cardone, C.; et al. Impact of circulating tumor DNA mutant allele fraction on prognosis in RAS-mutant metastatic colorectal cancer. Mol. Oncol. 2019, 13, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Elez, E.; Chianese, C.; Sanz-García, E.; Martinelli, E.; Noguerido, A.; Mancuso, F.M.; Caratù, G.; Matito, J.; Grasselli, J.; Cardone, C.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar]
- Cortes-Ciriano, I.; Lee, S.; Park, W.Y.; Kim, T.M.; Park, P.J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef] [Green Version]
- Mazzaschi, G.; Madeddu, D.; Falco, A.; Bocchialini, G.; Goldoni, M.; Sogni, F.; Armani, G.; Lagrasta, C.A.; Lorusso, B.; Mangiaracina, C.; et al. Low PD-1 Expression in Cytotoxic CD8(+) Tumor-Infiltrating Lymphocytes Confers an Immune-Privileged Tissue Microenvironment in NSCLC with a Prognostic and Predictive Value. Clin. Cancer Res. 2018, 24, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.-S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus Immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transorfm. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.M. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer: A Joint Consensus Recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [Green Version]
- Dumont, P.; Leu, J.I.; Della Pietra, A.C.; George, D.L.; Murphy, M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 2003, 33, 357–365. [Google Scholar] [CrossRef]
- Jeong, B.S.; Hu, W.; Belyi, V.; Rabadan, R.; Levine, A.J. Differential levels of transcription of p53-regulated genes by the arginine/proline polymorphism: p53 with arginine at codon 72 favors apoptosis. FASEB J. 2010, 24, 1347–1353. [Google Scholar] [CrossRef]
- Bank, L. p53 polymorphic variants at codon 72 exert different effects on cell cycle progression. Int. J. Cancer 2004, 108, 196–199. [Google Scholar]
- Furihata, M.; Takeuchi, T.; Matsumoto, M.; Kurabayashi, A.; Ohtsuki, Y.; Terao, N.; Kuwahara, M.; Shuin, T. p53 mutation arising in Arg72 allele in the tumorigenesis and development of carcinoma of the urinary tract. Clin. Cancer Res. 2002, 8, 1192–1195. [Google Scholar]
- Basu, S.; Gnanapradeepan, K.; Barnoud, T.; Kung, C.-P.; Tavecchio, M.; Scott, J.; Watters, A.; Chen, Q.; Kossenkov, A.V.; Murphy, M.E. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1α. Genes Dev. 2018, 32, 230–243. [Google Scholar] [CrossRef]
- Yang, H.; Robinson, P.N.; Wang, K. Phenolyzer: Phenotype-based prioritization of candidate genes for human diseases. Nat. Methods 2015, 12, 841–843. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient ID | Year of Diagnosis | Age | Ge | BMI (Kg/m2) | Histology, Pathologic Stage at Diagnosis *; Grading (G) in PT and MT | Tumor Side, and Type of Surgery | Ad CT | Sites of Metastases Presentation | TTM (m) |
|---|---|---|---|---|---|---|---|---|---|
| Group A | |||||||||
| PAT1 | 2014 April | 76 | F | 19.5 | Adenocarcinoma, pT3pN1aM1a pV0pR0; G2, G2 | Left, combined left hemicolectomy and segment 3 pR0 metastasectomy. | No | None | NR |
| PAT2 | 2017 March | 60 | M | 19.8 | Adenocarcinoma, pT3pN0M1a pV1pR0; G2, G3 | Right, combined right hemicolectomy and segment 5 pR0 metastasectomy. | Capox | None | NR |
| PAT3 | 2017 January | 81 | M | 21.0 | Adenocarcinoma, pT2pN1bM1apV0pR0; G2, G3 | Left, combined left hemicolectomy and segment 6 pR0 metastasectomy. | Cape | None | NR |
| Group B | |||||||||
| PAT4 | 2014 October | 81 | M | 18.2 | Adenocarcinoma, pT3pN1bM1a pV1pR0; G2, G3 | Right, combined right hemicolectomy and segment 5 pR0 metastasectomy. | Cape | Liver, peritoneum | 6.0 |
| PAT5 | 2018 March | 61 | F | 19.0 | Adenocarcinoma, pT2pN2bM1a pV1pR0; G2, G2 | Right, combined right hemicolectomy and segment 8 pR0 metastasectomy. | Capox | Liver, lungs, peritoneum | 3.5 |
| PAT6 | 2013 March | 27 | M | 18.9 | Adenocarcinoma, pT3pN1bM1a pV1pR0; G2, G3 | Left, combined left hemicolectomy and segment 8 pR0 metastasectomy. | Capox | Liver | 7.5 |
| Patient ID | First-Line CT | PFS1 (m) | No. of Cycles; Duration of CT (Months) | Second-Line CT | PFS2 (m) | No. of Cycles; Duration of CT (Months) | Third-Line CT | PFS3 (m) | No. of Cycles; Duration of CT (m) | Vital Status | Survival Time from Diagnosis (m) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Group A | |||||||||||
| PAT1 | None | 0.0 | 0; 0.0 | None | 0.0 | 0; 0.0 | None | 0.0 | 0; 0.0 | AWD | NR |
| PAT2 | None | 0.0 | 0; 0.0 | None | 0.0 | 0; 0.0 | None | 0.0 | 0; 0.0 | AWD | NR |
| PAT3 | None | 0.0 | 0; 0.0 | None | 0.0 | 0; 0.0 | None | 0.0 | 0; 0.0 | AWD | NR |
| Group B | |||||||||||
| PAT4 | FU/Beva | 11.0 | 20; 10.5 | Rego | 5.5 | 5; 5.0 | None | 0.0 | 0; 0.0 | D | 18.0 |
| PAT5 | Folfox/Beva | 3.0 | 6; 3.0 | Folfiri | 2.0 | 4; 2.0 | None | 0.0 | 0; 0.0 | D | 5.5 |
| PAT6 | Folfiri/Pani | 9.5 | 17; 9.0 | Folfox/Beva | 9.0 | 16; 8.0 | Folfiri Rech | 2.0 | 2; 4.0 | D | 21.0 |
| Groups | Patient | PT Mutated Genes | MT Mutated Genes |
|---|---|---|---|
| Group A | PAT1 | APC (p.R554TER), TP53 (p.R273H, p.P72R*), BRCA2 (p.Y1655TER). | APC (p.R554TER), TP53 (p.P72R, p.R273H), BRCA2 (p.Y1655TER). |
| PAT2 | APC (p.1450TER), TP53 (p.K382TER, p.P72R), KRAS (p.A146T), SMAD4 (p.R361H). | APC (p.1450TER), TP53 (p.K382TER, p.P72R). | |
| PAT3 | APC (p.S587TER), TP53 (p.V197E, p.V173A, p.P72R), KRAS (p.G13D), KRAS (p.G12D). | APC (p.S587TER), TP53 (p.V197E, p.V173A, p.P72R), KRAS (p.G13D). | |
| Group B | PAT4 | APC (p.Q236TER), TP53 (p.C135F, p.P72R). | APC (p.Q236TER), TP53 (p.C135F, p.P72R), KRAS (p.G12C), PIK3CA (p.E545K). |
| PAT5 | APC (p.R876TER, p.M1413TER), TP53 (p.R273H, p.P72R), NRAS (p.Q61K) | APC (p.R876TER, p.M1413TER), TP53 (p.R273H, p.P72R), NRAS (p.Q61K), PIK3CA (p.M1043I), SMAD4 (p.Q256TER). | |
| PAT6 | APC (p.H298TER, p.K581TER), TP53 (p.P72R). | APC (p.H298TER, p.K581TER), TP53 (p.P72R), PIK3CA (p.R88Q), BRAF (p.P403L). |
| Patients | Primary Tumors | Metastatic Tumors | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD3+ | CD8+ | Foxp3 | GrzB | CD3+ | CD8+ | Foxp3 | GrzB | |||||||||
| TC | IM | TC | IM | TC | IM | TC | IM | TC | IM | TC | IM | TC | IM | TC | IM | |
| PAT1 | 600 ± 15.5 | 2000 ± 21.2 | 50 ± 5.5 | 150 ± 11.1 | 67 ± 5.4 | 40 ± 4.2 | 32 ± 2.7 | 45 ± 3.3 | 200 ± 9.9 | 1500 ± 24.9 | 50 ± 2.3 | 120 ± 8.7 | 20 ± 2.1 | 40 ± 3.6 | 74 ± 6.8 | 30 ± 2.2 |
| PAT2 | 589 ± 13.2 | 305 ± 14.5 | 50 ± 10.6 | 150 ± 12.2 | 48 ± 4.8. | 9 ± 0.8 | 30 ± 2.2 | 40 ± 3.2 | 200 ± 11.6 | 500 ± 21.4 | 70 ± 5.8 | 100 ± 12.7 | 50 ± 5.1 | 45 ± 2.9 | 44 ± 3.3 | 25 ± 3.2 |
| PAT3 | 400 ± 9.5 | 500 ± 23.2 | 80 ± 6.2 | 200 ± 8.4 | 40 ± 3.3 | 40 ± 3.9 | 25 ± 2.1 | 30 ± 2.6 | 530 ± 19.2 | 2015 ± 23.8 | 78 ± 6.8 | 100 ± 8.8 | 10 ± 0.7 | 10 ± 1.4 | 58 ± 3.6 | 45 ± 4.8 |
| PAT4 | 800 ± 17.4 | 500 ± 20.1 | 60 ± 5.4 | 700 ± 22.5 | 55 ± 4.2 | 22 ± 4.1 | 22 ± 1.6 | 27 ± 1.9 | 200 ± 12.1 | 2014 ± 20.7 | 154 ± 10.2 | 110 ± 11.6 | 10 ± 0.8 | 35 ± 2.6 | 33 ± 2.2 | 50 ± 3.7 |
| PAT5 | 820 ± 19.7 | 1500 ± 17.2 | 600 ± 26.2 | 500 ± 18.4 | 30 ± 3.3 | 20 ± 2.9 | 120 ± 13.3 | 110 ± 13.9 | 130 ± 8.2 | 1610 ± 19.8 | 30 ± 2.8 | 60 ± 6.3 | 11 ± 0.7 | 15 ± 1.4 | 24 ± 2.6 | 35 ± 2.7 |
| PAT6 | 2000 ± 23.4 | 1040 ± 16.1 | 100 ± 7.4 | 400 ± 12.3 | 30 ± 4.2 | 30 ± 3.3 | 25 ± 1.6 | 42 ± 2.9 | 310 ± 12.1 | 3000 ± 20.7 | 100 ± 10.3 | 530 ± 21.6 | 28 ± 1.7 | 30 ± 2.6 | 26 ± 2.4 | 42 ± 3.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottaiano, A.; Caraglia, M.; Di Mauro, A.; Botti, G.; Lombardi, A.; Galon, J.; Luce, A.; D’Amore, L.; Perri, F.; Santorsola, M.; et al. Evolution of Mutational Landscape and Tumor Immune-Microenvironment in Liver Oligo-Metastatic Colorectal Cancer. Cancers 2020, 12, 3073. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103073
Ottaiano A, Caraglia M, Di Mauro A, Botti G, Lombardi A, Galon J, Luce A, D’Amore L, Perri F, Santorsola M, et al. Evolution of Mutational Landscape and Tumor Immune-Microenvironment in Liver Oligo-Metastatic Colorectal Cancer. Cancers. 2020; 12(10):3073. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103073
Chicago/Turabian StyleOttaiano, Alessandro, Michele Caraglia, Annabella Di Mauro, Gerardo Botti, Angela Lombardi, Jerome Galon, Amalia Luce, Luigi D’Amore, Francesco Perri, Mariachiara Santorsola, and et al. 2020. "Evolution of Mutational Landscape and Tumor Immune-Microenvironment in Liver Oligo-Metastatic Colorectal Cancer" Cancers 12, no. 10: 3073. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12103073