Integration and Comparison of Transcriptomic and Proteomic Data for Meningioma


 , , , and
, , , and
Abstract
:Simple Summary
Abstract
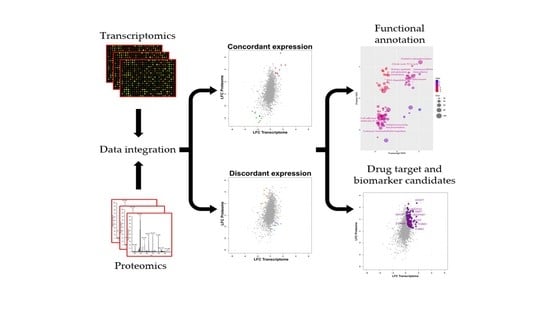
1. Introduction
2. Results
2.1. Integrated Transcriptome–Proteome Analysis of Grade III vs. Grade I Meningioma
2.2. Functional Annotation of Grade III vs. Grade I Meningioma Differential Transcriptome–Proteome Expression
2.3. Validation of Concordant and Discordant Expression in Grade III Meningioma
2.4. Drug–Gene Interaction Analysis
3. Discussion
4. Materials and Methods
4.1. Data Acquisition and Processing
4.2. Functional Enrichment Analyses
4.3. Clinical Material
4.4. Cell Culture and Western Blotting
4.5. Immunohistochemistry
4.6. RNA Isolation and Quantitative PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. WHO Classification of Tumours of the Central Nervous System, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2016; Volume 1, p. 408. [Google Scholar]
- Marosi, C.; Hassler, M.; Roessler, K.; Reni, M.; Sant, M.; Mazza, E.; Vecht, C. Meningioma. Crit. Rev. Oncol. Hematol. 2008, 67, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Apra, C.; Peyre, M.; Kalamarides, M. Current treatment options for meningioma. Expert Rev. Neurother. 2018, 18, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Brodbelt, A.R.; Barclay, M.E.; Greenberg, D.; Williams, M.; Jenkinson, M.D.; Karabatsou, K. The outcome of patients with surgically treated meningioma in England: 1999-2013. A cancer registry data analysis. Br. J. Neurosurg. 2019, 33, 641–647. [Google Scholar] [CrossRef] [Green Version]
- Peyre, M.; Gauchotte, G.; Giry, M.; Froehlich, S.; Pallud, J.; Graillon, T.; Bielle, F.; Cazals-Hatem, D.; Varlet, P.; Figarella-Branger, D.; et al. De novo and secondary anaplastic meningiomas: A study of clinical and histomolecular prognostic factors. Neuro. Oncol. 2018, 20, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Abedalthagafi, M.; Bi, W.L.; Aizer, A.A.; Merrill, P.H.; Brewster, R.; Agarwalla, P.K.; Listewnik, M.L.; Dias-Santagata, D.; Thorner, A.R.; Van Hummelen, P.; et al. Oncogenic PI3K mutations are as common as AKT1 and SMO mutations in meningioma. Neuro. Oncol. 2016, 18, 649–655. [Google Scholar] [CrossRef] [Green Version]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; McKenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avsar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, V.E.; Harmanci, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 2016, 48, 1253–1259. [Google Scholar] [CrossRef] [Green Version]
- Collord, G.; Tarpey, P.; Kurbatova, N.; Martincorena, I.; Moran, S.; Castro, M.; Nagy, T.; Bignell, G.; Maura, F.; Young, M.D.; et al. An integrated genomic analysis of anaplastic meningioma identifies prognostic molecular signatures. Sci. Rep. 2018, 8, 13537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fevre-Montange, M.; Champier, J.; Durand, A.; Wierinckx, A.; Honnorat, J.; Guyotat, J.; Jouvet, A. Microarray gene expression profiling in meningiomas: Differential expression according to grade or histopathological subtype. Int. J. Oncol. 2009, 35, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Mock, A.; Jungk, C.; Sahm, F.; Ull, A.T.; Warta, R.; Lamszus, K.; Gousias, K.; Ketter, R.; Roesch, S.; et al. Transcriptomic analysis of aggressive meningiomas identifies PTTG1 and LEPR as prognostic biomarkers independent of WHO grade. Oncotarget 2016, 7, 14551–14568. [Google Scholar] [CrossRef] [Green Version]
- Viaene, A.N.; Zhang, B.; Martinez-Lage, M.; Xiang, C.; Tosi, U.; Thawani, J.P.; Gungor, B.; Zhu, Y.; Roccograndi, L.; Zhang, L.; et al. Transcriptome signatures associated with meningioma progression. Acta Neuropathol. Commun. 2019, 7, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, M.A.; Gutmann, D.H.; Peterson, K.; Chicoine, M.R.; Kleinschmidt-DeMasters, B.K.; Brown, H.G.; Perry, A. Molecular characterization of human meningiomas by gene expression profiling using high-density oligonucleotide microarrays. Am. J. Pathol. 2002, 161, 665–672. [Google Scholar] [CrossRef] [Green Version]
- Wrobel, G.; Roerig, P.; Kokocinski, F.; Neben, K.; Hahn, M.; Reifenberger, G.; Lichter, P. Microarray-based gene expression profiling of benign, atypical and anaplastic meningiomas identifies novel genes associated with meningioma progression. Int. J. Cancer 2005, 114, 249–256. [Google Scholar] [CrossRef]
- Diez, P.; Droste, C.; Degano, R.M.; Gonzalez-Munoz, M.; Ibarrola, N.; Perez-Andres, M.; Garin-Muga, A.; Segura, V.; Marko-Varga, G.; LaBaer, J.; et al. Integration of Proteomics and Transcriptomics Data Sets for the Analysis of a Lymphoma B-Cell Line in the Context of the Chromosome-Centric Human Proteome Project. J. Proteome Res. 2015, 14, 3530–3540. [Google Scholar] [CrossRef]
- Latosinska, A.; Makridakis, M.; Frantzi, M.; Borras, D.M.; Janssen, B.; Mullen, W.; Zoidakis, J.; Merseburger, A.S.; Jankowski, V.; Mischak, H.; et al. Integrative analysis of extracellular and intracellular bladder cancer cell line proteome with transcriptome: Improving coverage and validity of -omics findings. Sci. Rep. 2016, 6, 25619. [Google Scholar] [CrossRef]
- Schwanhausser, B.; Busse, D.; Li, N.; Dittmar, G.; Schuchhardt, J.; Wolf, J.; Chen, W.; Selbach, M. Global quantification of mammalian gene expression control. Nature 2011, 473, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, J.; Ferluga, S.; Sharma, V.; Futschik, M.; Hilton, D.A.; Adams, C.L.; Lasonder, E.; Hanemann, C.O. Proteomic analysis discovers the differential expression of novel proteins and phosphoproteins in meningioma including NEK9, HK2 and SET and deregulation of RNA metabolism. EBioMedicine 2019, 40, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Parada, C.A.; Osbun, J.; Kaur, S.; Yakkioui, Y.; Shi, M.; Pan, C.; Busald, T.; Karasozen, Y.; Gonzalez-Cuyar, L.F.; Rostomily, R.; et al. Kinome and phosphoproteome of high-grade meningiomas reveal AKAP12 as a central regulator of aggressiveness and its possible role in progression. Sci. Rep. 2018, 8, 2098. [Google Scholar] [CrossRef] [Green Version]
- Misra, B.B.; Langefeld, C.D.; Olivier, M.; Cox, L.A. Integrated Omics: Tools, Advances, and Future Approaches. J. Mol. Endocrinol. 2019, 62, R21–R45. [Google Scholar] [CrossRef] [Green Version]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA methylation-based classification and grading system for meningioma: A multicentre, retrospective analysis. Lancet Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Harmanci, A.S.; Youngblood, M.W.; Clark, V.E.; Coskun, S.; Henegariu, O.; Duran, D.; Erson-Omay, E.Z.; Kaulen, L.D.; Lee, T.I.; Abraham, B.J.; et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat. Commun. 2018, 9, 16215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paramasivam, N.; Hubschmann, D.; Toprak, U.H.; Ishaque, N.; Neidert, M.; Schrimpf, D.; Stichel, D.; Reuss, D.; Sievers, P.; Reinhardt, A.; et al. Mutational patterns and regulatory networks in epigenetic subgroups of meningioma. Acta Neuropathol. 2019, 138, 295–308. [Google Scholar] [CrossRef]
- Vasudevan, H.N.; Braunstein, S.E.; Phillips, J.J.; Pekmezci, M.; Tomlin, B.A.; Wu, A.; Reis, G.F.; Magill, S.T.; Zhang, J.; Feng, F.Y.; et al. Comprehensive Molecular Profiling Identifies FOXM1 as a Key Transcription Factor for Meningioma Proliferation. Cell Rep. 2018, 22, 3672–3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menghi, F.; Orzan, F.N.; Eoli, M.; Farinotti, M.; Maderna, E.; Pisati, F.; Bianchessi, D.; Valletta, L.; Lodrini, S.; Galli, G.; et al. DNA microarray analysis identifies CKS2 and LEPR as potential markers of meningioma recurrence. Oncologist 2011, 16, 1440–1450. [Google Scholar] [CrossRef] [Green Version]
- Miller, R., Jr.; DeCandio, M.L.; Dixon-Mah, Y.; Giglio, P.; Vandergrift, W.A., 3rd; Banik, N.L.; Patel, S.J.; Varma, A.K.; Das, A. Molecular Targets and Treatment of Meningioma. J. Neurol. Neurosurg. 2014, 1, 1. [Google Scholar]
- Farrah, T.; Deutsch, E.W.; Omenn, G.S.; Campbell, D.S.; Sun, Z.; Bletz, J.A.; Mallick, P.; Katz, J.E.; Malmstrom, J.; Ossola, R.; et al. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol. Cell. Proteom. 2011, 10, M110.006353. [Google Scholar] [CrossRef] [Green Version]
- Cotto, K.C.; Wagner, A.H.; Feng, Y.Y.; Kiwala, S.; Coffman, A.C.; Spies, G.; Wollam, A.; Spies, N.C.; Griffith, O.L.; Griffith, M. DGIdb 3.0: A redesign and expansion of the drug-gene interaction database. Nucleic Acids Res. 2018, 46, D1068–D1073. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, E.; Fagerberg, L.; Klevebring, D.; Matic, I.; Geiger, T.; Cox, J.; Algenas, C.; Lundeberg, J.; Mann, M.; Uhlen, M. Defining the transcriptome and proteome in three functionally different human cell lines. Mol. Syst. Biol. 2010, 6, 450. [Google Scholar] [CrossRef]
- Vogel, C.; Abreu Rde, S.; Ko, D.; Le, S.Y.; Shapiro, B.A.; Burns, S.C.; Sandhu, D.; Boutz, D.R.; Marcotte, E.M.; Penalva, L.O. Sequence signatures and mRNA concentration can explain two-thirds of protein abundance variation in a human cell line. Mol. Syst. Biol. 2010, 6, 400. [Google Scholar] [CrossRef]
- Dahal Lamichane, B.; Jung, S.Y.; Yun, J.; Kang, S.; Kim, D.Y.; Lamichane, S.; Kim, Y.J.; Park, J.H.; Jang, W.B.; Ji, S.T.; et al. AGR2 is a target of canonical Wnt/beta-catenin signaling and is important for stemness maintenance in colorectal cancer stem cells. Biochem. Biophys. Res. Commun. 2019, 515, 600–606. [Google Scholar] [CrossRef]
- Sung, H.Y.; Choi, E.N.; Lyu, D.; Park, A.K.; Ju, W.; Ahn, J.H. Aberrant hypomethylation-mediated AGR2 overexpression induces an aggressive phenotype in ovarian cancer cells. Oncol. Rep. 2014, 32, 815–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, I.; Baeesa, S.; Bangash, M.; Schulten, H.J.; Alghamdi, F.; Qashqari, H.; Madkhali, N.; Carracedo, A.; Saka, M.; Jamal, A.; et al. Pleomorphism and drug resistant cancer stem cells are characteristic of aggressive primary meningioma cell lines. Cancer Cell Int. 2017, 17, 72. [Google Scholar] [CrossRef] [Green Version]
- Kagawa, Y.; Umaru, B.A.; Ariful, I.; Shil, S.K.; Miyazaki, H.; Yamamoto, Y.; Ogata, M.; Owada, Y. Role of FABP7 in tumor cell signaling. Adv. Biol. Regul. 2019, 71, 206–218. [Google Scholar] [CrossRef]
- Jiang, J.; Lin, C.; Liu, N.; Zhang, Z.; Sun, Y.; Fang, X.; Qi, J. The expression of fatty acid metabolism-associated proteins is correlated with the prognosis of meningiomas. APMIS 2013, 121, 997–1003. [Google Scholar] [CrossRef]
- Panagopoulos, A.T.; Lancellotti, C.L.; Veiga, J.C.; de Aguiar, P.H.; Colquhoun, A. Expression of cell adhesion proteins and proteins related to angiogenesis and fatty acid metabolism in benign, atypical, and anaplastic meningiomas. J. Neurooncol. 2008, 89, 73–87. [Google Scholar] [CrossRef]
- Berger, W.T.; Ralph, B.P.; Kaczocha, M.; Sun, J.; Balius, T.E.; Rizzo, R.C.; Haj-Dahmane, S.; Ojima, I.; Deutsch, D.G. Targeting fatty acid binding protein (FABP) anandamide transporters—A novel strategy for development of anti-inflammatory and anti-nociceptive drugs. PLoS ONE 2012, 7, e50968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczocha, M.; Rebecchi, M.J.; Ralph, B.P.; Teng, Y.H.; Berger, W.T.; Galbavy, W.; Elmes, M.W.; Glaser, S.T.; Wang, L.; Rizzo, R.C.; et al. Inhibition of fatty acid binding proteins elevates brain anandamide levels and produces analgesia. PLoS ONE 2014, 9, e94200. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.Z.; Graham, K.; Glubrecht, D.D.; Lai, R.; Mackey, J.R.; Godbout, R. A fatty acid-binding protein 7/RXRbeta pathway enhances survival and proliferation in triple-negative breast cancer. J. Pathol. 2012, 228, 310–321. [Google Scholar] [CrossRef]
- Slipicevic, A.; Jorgensen, K.; Skrede, M.; Rosnes, A.K.; Troen, G.; Davidson, B.; Florenes, V.A. The fatty acid binding protein 7 (FABP7) is involved in proliferation and invasion of melanoma cells. BMC Cancer 2008, 8, 276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Shi, J.; Qin, J.; Jin, G.; Han, X.; Li, H. Brain lipid binding protein mediates the proliferation of human glioblastoma cells by regulating ERK1/2 signaling pathway in vitro. Vitr. Cell. Dev. Biol. Anim. 2018, 54, 156–162. [Google Scholar] [CrossRef]
- Tolle, A.; Krause, H.; Miller, K.; Jung, K.; Stephan, C. Importance of braintype fatty acid binding protein for cell-biological processes in human renal carcinoma cells. Oncol. Rep. 2011, 25, 1307–1312. [Google Scholar] [CrossRef] [Green Version]
- Timpl, R.; Sasaki, T.; Kostka, G.; Chu, M.L. Fibulins: A versatile family of extracellular matrix proteins. Nat. Rev. Mol. Cell Biol. 2003, 4, 479–489. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, J.; Yin, H.B.; Liu, Y.F.; Liu, J.H. Fibulin-1 functions as a prognostic factor in lung adenocarcinoma. Jpn. J. Clin. Oncol. 2015, 45, 854–859. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Yao, C.; Li, P.; Feng, Y.; Wang, F.; Liu, Y.F.; Guo, Y.B.; Mao, Q.S.; Xue, W.J. Low expression of fibulin-1 correlates with unfavorable prognosis in gastric cancer. Tumour Biol. 2016, 37, 9399–9410. [Google Scholar] [CrossRef] [PubMed]
- Kanda, M.; Nomoto, S.; Okamura, Y.; Hayashi, M.; Hishida, M.; Fujii, T.; Nishikawa, Y.; Sugimoto, H.; Takeda, S.; Nakao, A. Promoter hypermethylation of fibulin 1 gene is associated with tumor progression in hepatocellular carcinoma. Mol. Carcinog. 2011, 50, 571–579. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, J.; Li, H.; Guan, W.; Xia, D.; Yu, G.; Xiao, H.; Lang, B.; Ma, X.; Liu, J.; et al. Fibulin-1 is down-regulated through promoter hypermethylation and suppresses renal cell carcinoma progression. J. Urol. 2013, 190, 291–301. [Google Scholar] [CrossRef]
- Kalamarides, M.; Stemmer-Rachamimov, A.O.; Niwa-Kawakita, M.; Chareyre, F.; Taranchon, E.; Han, Z.Y.; Martinelli, C.; Lusis, E.A.; Hegedus, B.; Gutmann, D.H.; et al. Identification of a progenitor cell of origin capable of generating diverse meningioma histological subtypes. Oncogene 2011, 30, 2333–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chua, S., Jr. Leptin Function and Regulation. Compr. Physiol. 2017, 8, 351–369. [Google Scholar] [CrossRef]
- Han, G.; Li, Y.; Cao, Y.; Yue, Z.; Zhang, Y.; Wang, L.; Liu, J. Overexpression of leptin receptor in human glioblastoma: Correlation with vasculogenic mimicry and poor prognosis. Oncotarget 2017, 8, 58163–58171. [Google Scholar] [CrossRef] [Green Version]
- Vuletic, M.S.; Milosevic, V.S.; Jancic, S.A.; Zujovic, J.T.; Krstic, M.S.; Vukmirovic, F.C. Clinical significance of Leptin receptor (LEPR) and Endoglin (CD105) expressions in colorectal adenocarcinoma. J. BU ON 2019, 24, 2448–2457. [Google Scholar]
- Fan, Y.L.; Li, X.Q. Expression of leptin and its receptor in thyroid carcinoma: Distinctive prognostic significance in different subtypes. Clin. Endocrinol. 2015, 83, 261–267. [Google Scholar] [CrossRef]
- Osorio, C.F.; Souza, D.B.; Gallo, C.B.; Costa, W.S.; Sampaio, F.J. Leptin and leptin receptor expressions in prostate tumors may predict disease aggressiveness? Acta Cir. Bras. 2014, 29(Suppl. 3), 44–48. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.S.; Tsai, K.B.; Chung, Y.F.; Chan, T.F.; Yeh, Y.T.; Tsai, L.Y.; Su, J.H. Aberrant expression and possible involvement of the leptin receptor in endometrial cancer. Gynecol. Oncol. 2004, 92, 769–775. [Google Scholar] [CrossRef]
- Kumar, D.; Bansal, G.; Narang, A.; Basak, T.; Abbas, T.; Dash, D. Integrating transcriptome and proteome profiling: Strategies and applications. Proteomics 2016, 16, 2533–2544. [Google Scholar] [CrossRef]
- Maier, T.; Guell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [Green Version]
- Parks, S.K.; Pouyssegur, J. The Na(+)/HCO3(-) Co-Transporter SLC4A4 Plays a Role in Growth and Migration of Colon and Breast Cancer Cells. J. Cell. Physiol. 2015, 230, 1954–1963. [Google Scholar] [CrossRef]
- Mani, C.; Tripathi, K.; Luan, S.; Clark, D.W.; Andrews, J.F.; Vindigni, A.; Thomas, G.; Palle, K. The multifunctional protein PACS-1 is required for HDAC2- and HDAC3-dependent chromatin maturation and genomic stability. Oncogene 2020, 39, 2583–2596. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, J.; Rothzerg, E.; Wu, X.; Xu, H.; Zhu, S.; Xu, J. Molecular structure and the role of high-temperature requirement protein 1 in skeletal disorders and cancers. Cell Prolif. 2020, 53, e12746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onder, E.; Arikok, A.T.; Seckin, H.; Alper, M. Decrease in serine protease HtrA1 expression correlates with grade and recurrence in meningiomas. Adv. Med. Sci. 2015, 60, 139–143. [Google Scholar] [CrossRef]
- Klose, R.; Adam, M.G.; Weis, E.M.; Moll, I.; Wustehube-Lausch, J.; Tetzlaff, F.; Oka, C.; Ehrmann, M.; Fischer, A. Inactivation of the serine protease HTRA1 inhibits tumor growth by deregulating angiogenesis. Oncogene 2018, 37, 4260–4272. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Klumperman, J. Lysosome biogenesis and lysosomal membrane proteins: Trafficking meets function. Nat. Rev. Mol. Cell Biol. 2009, 10, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, W.; Zhang, R.; Liu, J.; Yu, J.; Wu, X.; Xu, Y.; Ma, M.; Huang, J. High expression of LAMP2 predicts poor prognosis in patients with esophageal squamous cell carcinoma. Cancer Biomark. 2017, 19, 305–311. [Google Scholar] [CrossRef]
- Morell, C.; Bort, A.; Vara-Ciruelos, D.; Ramos-Torres, A.; Altamirano-Dimas, M.; Diaz-Laviada, I.; Rodriguez-Henche, N. Up-Regulated Expression of LAMP2 and Autophagy Activity during Neuroendocrine Differentiation of Prostate Cancer LNCaP Cells. PLoS ONE 2016, 11, e0162977. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, O.; Wang, W.C.; Lotan, R.; Fukuda, M. Differential glycosylation and cell surface expression of lysosomal membrane glycoproteins in sublines of a human colon cancer exhibiting distinct metastatic potentials. J. Biol. Chem. 1992, 267, 5700–5711. [Google Scholar]
- Gabriele, C.; Cantiello, F.; Nicastri, A.; Crocerossa, F.; Russo, G.I.; Cicione, A.; Vartolomei, M.D.; Ferro, M.; Morgia, G.; Lucarelli, G.; et al. High-throughput detection of low abundance sialylated glycoproteins in human serum by TiO2 enrichment and targeted LC-MS/MS analysis: Application to a prostate cancer sample set. Anal. Bioanal. Chem. 2019, 411, 755–763. [Google Scholar] [CrossRef]
- Surace, E.I.; Lusis, E.; Murakami, Y.; Scheithauer, B.W.; Perry, A.; Gutmann, D.H. Loss of tumor suppressor in lung cancer-1 (TSLC1) expression in meningioma correlates with increased malignancy grade and reduced patient survival. J. Neuropathol. Exp. Neurol. 2004, 63, 1015–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.L.; Chen, G.Q.; Li, Z.Y.; Wang, B.R. Function and histopathology of a cell adhesion molecule TSLC1 in cancer. Cancer Investig. 2011, 29, 107–112. [Google Scholar] [CrossRef]
- Marconi, G.D.; Gallorini, M.; Carradori, S.; Guglielmi, P.; Cataldi, A.; Zara, S. The Up-Regulation of Oxidative Stress as a Potential Mechanism of Novel MAO-B Inhibitors for Glioblastoma Treatment. Molecules 2019, 24, 2005. [Google Scholar] [CrossRef] [Green Version]
- Sharpe, M.A.; Livingston, A.D.; Gist, T.L.; Ghosh, P.; Han, J.; Baskin, D.S. Successful Treatment of Intracranial Glioblastoma Xenografts With a Monoamine Oxidase B-Activated Pro-Drug. EBioMedicine 2015, 2, 1122–1132. [Google Scholar] [CrossRef] [Green Version]
- Becker, R.A.; Chambers, J.M.; Reeve Wilks, A. The New S Language: A Programming Environment for Data Analysis and Graphics; Wadsworth & Brooks/Cole Advanced Books & Software: Monterey, CA, USA, 1988; p. 702. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Hilton, D.A.; Shivane, A.; Kirk, L.; Bassiri, K.; Enki, D.G.; Hanemann, C.O. Activation of multiple growth factor signalling pathways is frequent in meningiomas. Neuropathology 2016, 36, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Protein Name | LFC Transcript | Rank Transcript | LFC Protein | Rank Protein | Average Rank |
|---|---|---|---|---|---|---|
| MBP | Myelin basic protein | −1.21 | 3521 | 2.05 | 3234 | 3377.5 |
| PACS1 | Phosphofurin acidic cluster sorting protein 1 | −0.51 | 3218 | 3.34 | 3534 | 3376 |
| APP | Amyloid-beta precursor protein | −0.77 | 3395 | 2.26 | 3318 | 3356.5 |
| DSP | Desmoplakin | −1.43 | 3544 | 1.48 | 2938 | 3241 |
| FCGBP | IgGFc-binding protein | −0.46 | 3156 | 2.11 | 3264 | 3210 |
| LAMP2 | Lysosome-associated membrane glycoprotein 2 | −0.71 | 3369 | 1.64 | 3043 | 3206 |
| BOLA2B | BolA-like protein 2 | −0.28 | 2823 | 3.30 | 3532 | 3177.5 |
| PTPRF | Receptor-type tyrosine-protein phosphatase F | −1.03 | 3487 | 1.33 | 2828 | 3157.5 |
| DCN | Decorin | −0.77 | 3400 | 1.39 | 2875 | 3137.5 |
| AK3 | GTP:AMP phosphotransferase AK3, mitochondrial | −0.51 | 3219 | 1.58 | 3006 | 3112.5 |
| HTRA1 | Serine protease HTRA1 | −1.47 | 3550 | 1.12 | 2670 | 3110 |
| OGN | Mimecan | −0.89 | 3445 | 1.19 | 2731 | 3088 |
| GOLIM4 | Golgi integral membrane protein 4 | −0.73 | 3379 | 1.25 | 2771 | 3075 |
| LAMB2 | Laminin subunit beta-2 | −0.64 | 3331 | 1.29 | 2799 | 3065 |
| ITM2B | Integral membrane protein 2B | −0.63 | 3319 | 1.24 | 2766 | 3042.5 |
| SDHB | Succinate dehydrogenase [ubiquinone] iron-sulfur subunit, mitochondrial | −0.31 | 2870 | 1.90 | 3175 | 3022.5 |
| CST3 | Cystatin-C | −0.38 | 3034 | 1.51 | 2962 | 2998 |
| LTBP4 | Latent-transforming growth factor beta-binding protein 4 | −0.53 | 3239 | 1.21 | 2745 | 2992 |
| PPP3CA | Serine/threonine-protein phosphatase 2B catalytic subunit alpha isoform | −0.54 | 3245 | 1.15 | 2694 | 2969.5 |
| HEXA | Beta-hexosaminidase subunit alpha | −0.27 | 2765 | 1.87 | 3163 | 2964 |
| PCSK1N | ProSAAS | −0.83 | 3426 | 0.91 | 2483 | 2954.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dunn, J.; Lenis, V.P.; Hilton, D.A.; Warta, R.; Herold-Mende, C.; Hanemann, C.O.; Futschik, M.E. Integration and Comparison of Transcriptomic and Proteomic Data for Meningioma. Cancers 2020, 12, 3270. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113270
Dunn J, Lenis VP, Hilton DA, Warta R, Herold-Mende C, Hanemann CO, Futschik ME. Integration and Comparison of Transcriptomic and Proteomic Data for Meningioma. Cancers. 2020; 12(11):3270. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113270
Chicago/Turabian StyleDunn, Jemma, Vasileios P. Lenis, David A. Hilton, Rolf Warta, Christel Herold-Mende, C. Oliver Hanemann, and Matthias E. Futschik. 2020. "Integration and Comparison of Transcriptomic and Proteomic Data for Meningioma" Cancers 12, no. 11: 3270. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113270