Modulation of de Novo Lipogenesis Improves Response to Enzalutamide Treatment in Prostate Cancer

 , and
, and 
Abstract
:Simple Summary
Abstract
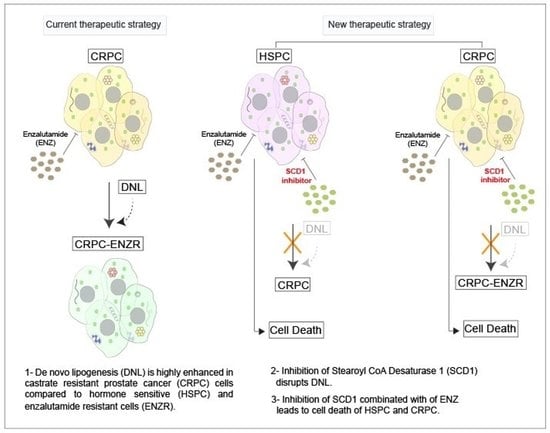
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
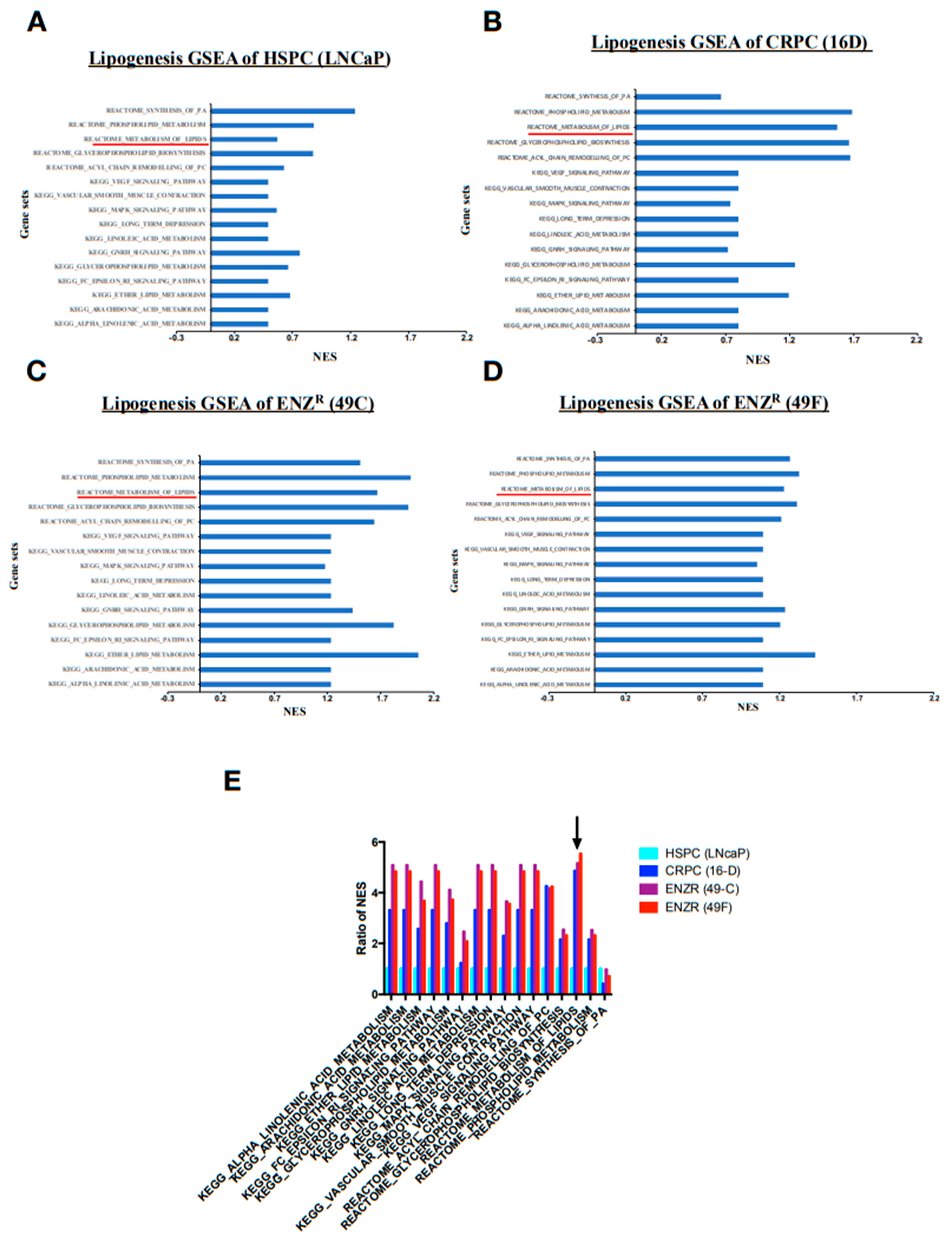
2.1. Lipid Synthesis Is Increased as Prostate Cancer Progresses from Hormone Sensitive to Castrate Resistant to Enzalutamide Resistant State
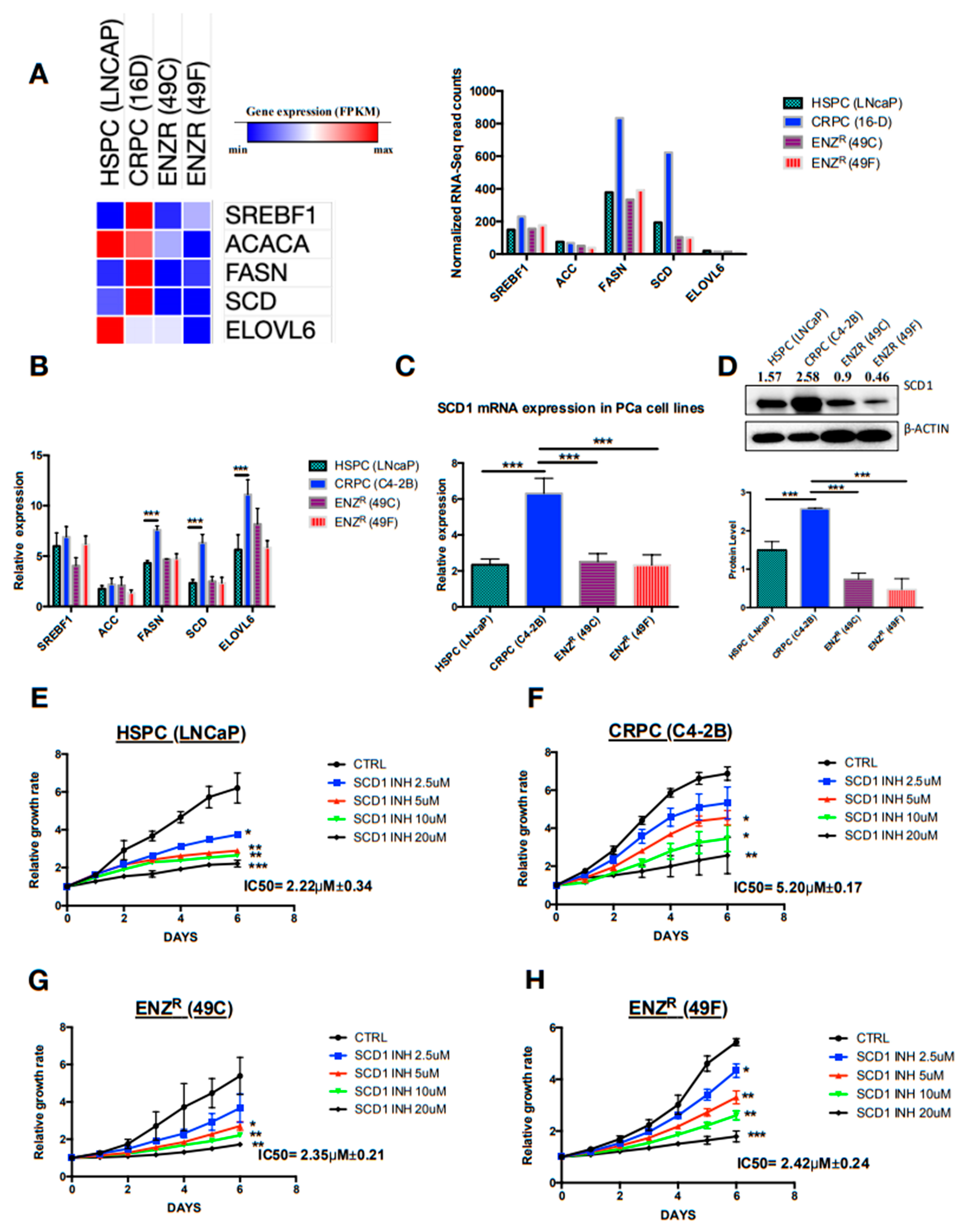
2.2. De Novo Lipogenesis Is Increased in CRPC and ENZR Cell Lines and This Modulation Affects Survival of Prostate Cancer Cell Lines
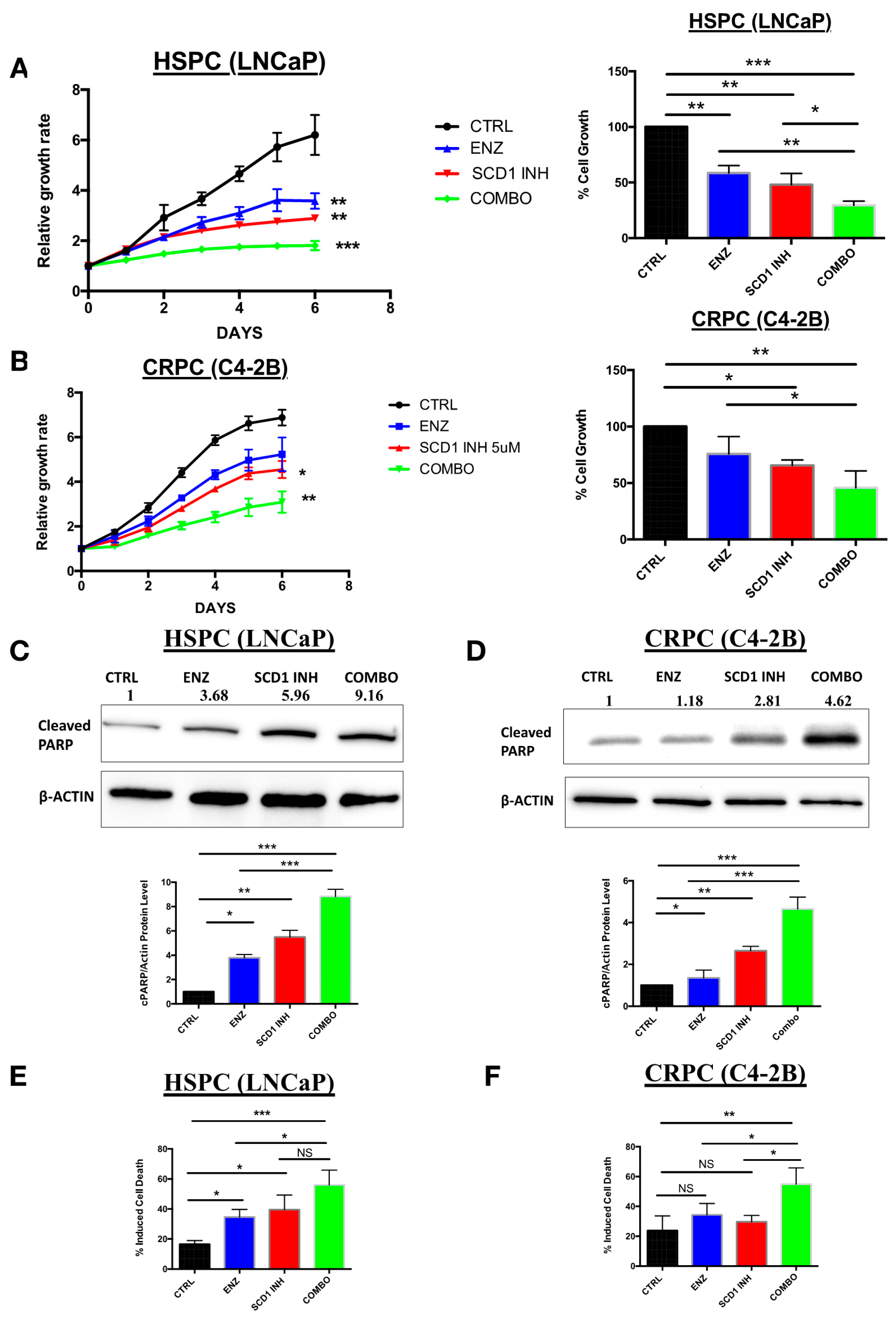
2.3. In Vitro Inhibition of SCD1 Combined with Enzalutamide Significantly Attenuates Prostate Cancer Cell Proliferation
2.4. Combination Therapy Induces Significant ER Stress and Produces High Levels of Reactive Oxygen Species Associated with PC Cell Death
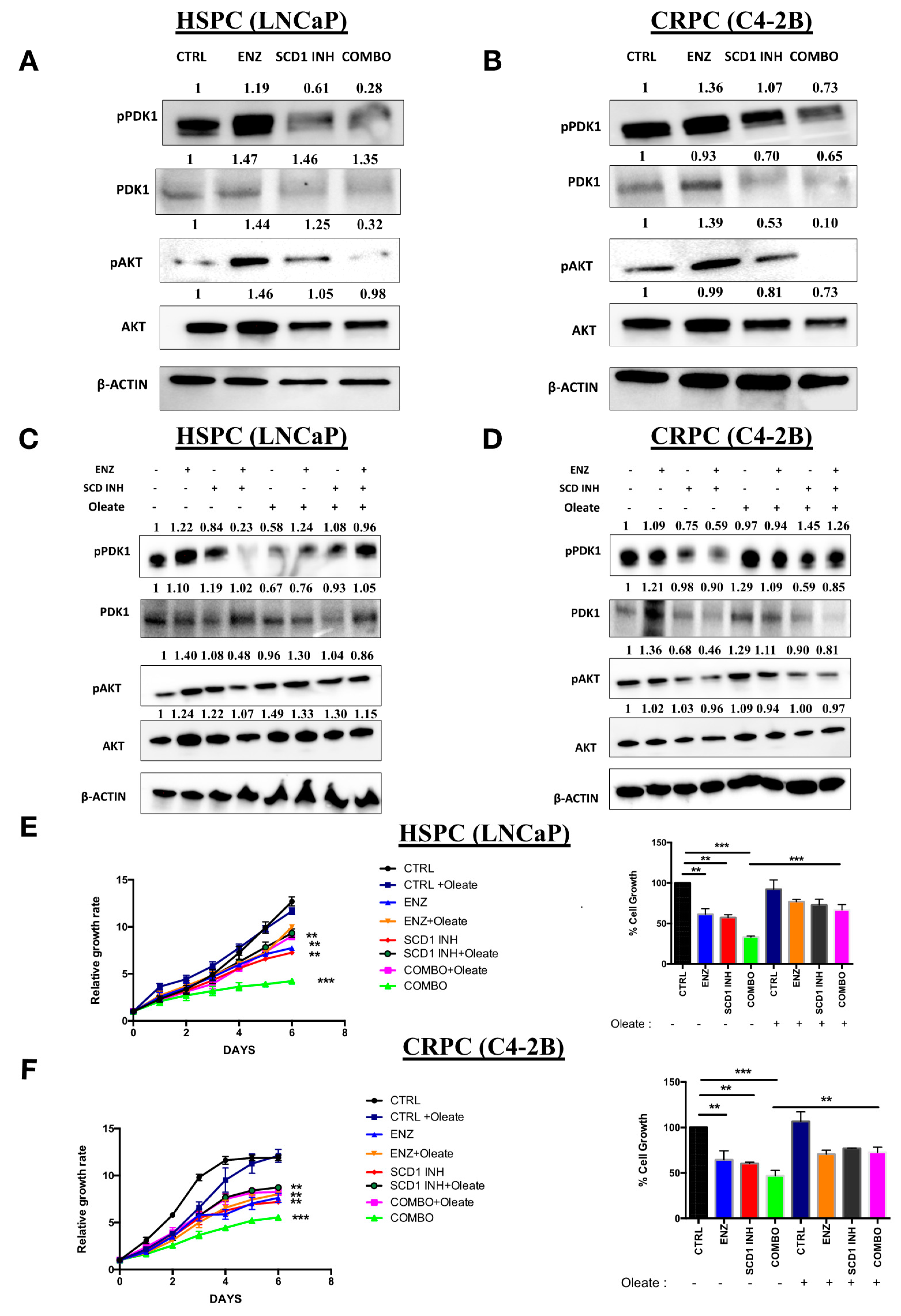
2.5. Combination Therapy Induces Inhibition of the Oncogenic P13K/AKT Pathway via a Decrease in Cellular Oleate
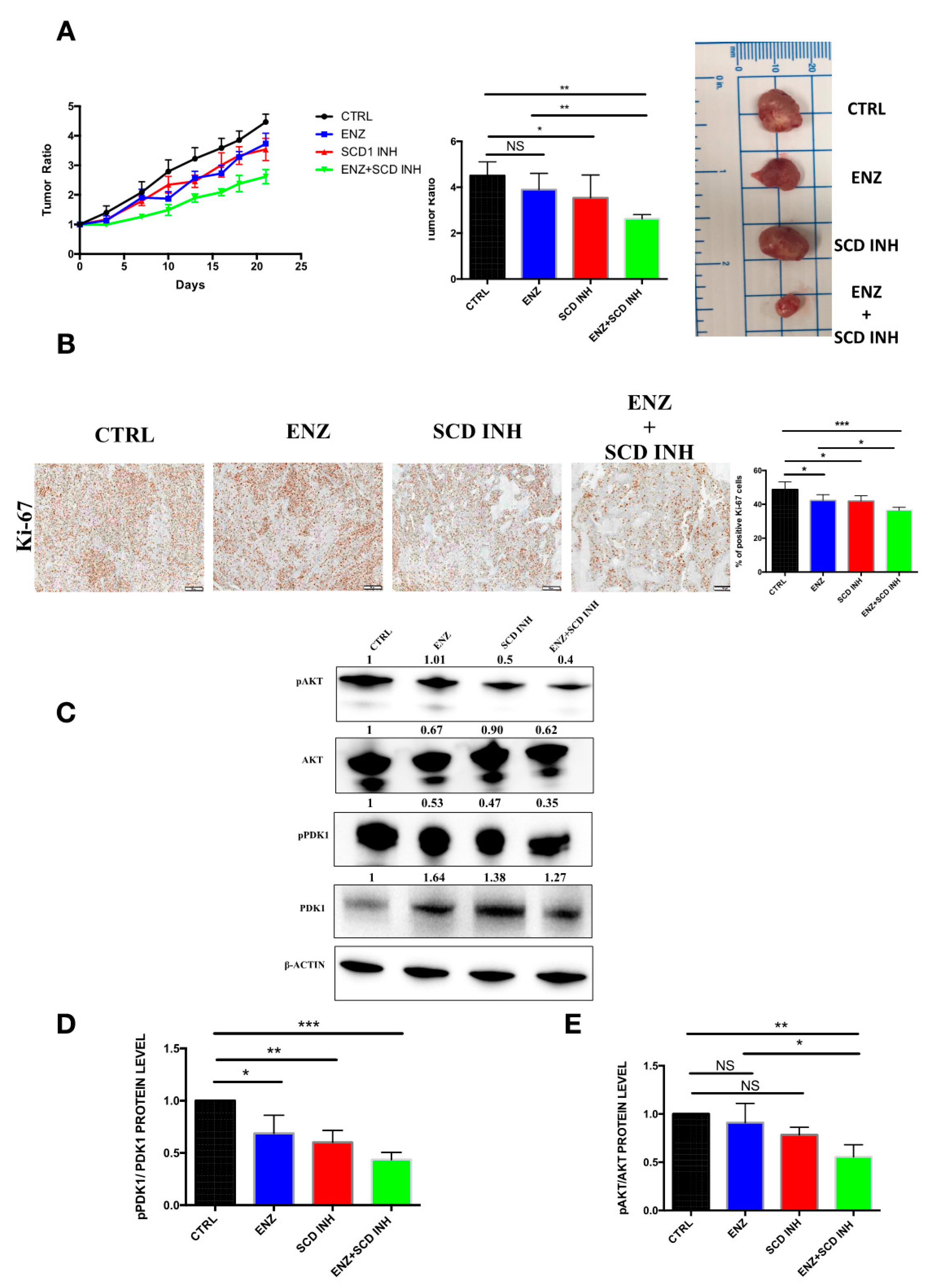
2.6. Pharmacological Combination of Enzalutamide and SCD1 Inhibitor Targets Tumor Growth More Efficiently than Monotherapy in Prostate Cancer Xenograft Models
2.7. Combination Therapy Induces Changes in the Monounsaturated and Saturated Fatty Acid Content in Prostate Cancer Tumors
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Transfection
4.3. RNAseq
4.4. Gene Set Enrichment Analysis (GSEA)
4.5. IncuCyte Cell Proliferation Phase Contrast Imaging Assay
4.6. Protein Extraction and Western Blot Analysis
4.7. Oxidative Stress Detection (ROS)
4.8. Real-Time PCR Analysis
4.9. Analysis of Cell Death by Flow Cytometry
4.10. Murine Xenograft Model
4.11. Tumor Lipid Content
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Canadian Cancer Statistics Advisory Committee. Canadian Cancer Statistics 2018; Canadian Cancer Society: Toronto, ON, Canada, 2018. [Google Scholar]
- Huggins, C. Prostatic cancer treated by orchiectomy; the five year results. J. Am. Med. Assoc. 1946, 131, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Centenera, M.M.; Swinnen, J.V. Androgen control of lipid metabolism in prostate cancer: Novel insights and future applications. Endocr. Relat. Cancer 2016, 23, R219–R227. [Google Scholar] [CrossRef] [PubMed]
- Asmane, I.; Ceraline, J.; Duclos, B.; Rob, L.; Litique, V.; Barthelemy, P.; Bergerat, J.-P.; Dufour, P.; Kurtz, J.-E. New strategies for medical management of castration-resistant prostate cancer. Oncology 2011, 80, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mills, I.G. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat. Rev. Cancer 2014, 14, 187–198. [Google Scholar] [CrossRef]
- Knudsen, K.E.; Scher, H.I. Starving the addiction: New opportunities for durable suppression of AR signaling in prostate cancer. Clin. Cancer Res. 2009, 15, 4792–4798. [Google Scholar] [CrossRef] [Green Version]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Bilancio, A.; Perillo, B.; Sinisi, A.A.; Migliaccio, A.; Castoria, G. Estrogen Receptors in Epithelial-Mesenchymal Transition of Prostate Cancer. Cancers 2019, 11, 1418. [Google Scholar] [CrossRef] [Green Version]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Di Santi, A.; Cernera, G.; Rossi, V.; Abbondanza, C.; Moncharmont, B.; Sinisi, A.A.; et al. Prostate cancer stem cells: The role of androgen and estrogen receptors. Oncotarget 2016, 7, 193–208. [Google Scholar] [CrossRef] [Green Version]
- Cai, C.; Balk, S.P. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr. Relat. Cancer 2011, 18, R175–R182. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, D.N.; Butler, L.M.; Estelles, D.L.; de Bono, J.S. Molecular pathology and prostate cancer therapeutics: From biology to bedside. J. Pathol. 2014, 232, 178–184. [Google Scholar] [CrossRef]
- Culig, Z. Molecular Mechanisms of Enzalutamide Resistance in Prostate Cancer. Curr. Mol. Biol. Rep. 2017, 3, 230–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoykova, G.E.; Schlaepfer, I.R. Lipid Metabolism and Endocrine Resistance in Prostate Cancer, and New Opportunities for Therapy. Int. J. Mol. Sci. 2019, 20, 2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mounier, C.; Bouraoui, L.; Rassart, E. Lipogenesis in cancer progression (review). Int. J. Oncol. 2014, 45, 485–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migita, T.; Ruiz, S.; Fornari, A.; Fiorentino, M.; Priolo, C.; Zadra, G.; Inazuka, F.; Grisanzio, C.; Palescandolo, E.; Shin, E. Fatty acid synthase: A metabolic enzyme and candidate oncogene in prostate cancer. J. Natl. Cancer Inst. 2009, 101, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Heemers, H.V.; Verhoeven, G.; Swinnen, J.V. Androgen activation of the sterol regulatory element-binding protein pathway: Current insights. Mol. Endocrinol. 2006, 20, 2265–2277. [Google Scholar] [CrossRef]
- Tennakoon, J.B.; Shi, Y.; Han, J.J.; Tsouko, E.; White, M.A.; Burns, A.R.; Zhang, A.; Xia, X.; Ilkayeva, O.R.; Xin, L.; et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene 2014, 33, 5251–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef] [PubMed]
- Zadra, G.; Ribeiro, C.F.; Chetta, P.; Ho, Y.; Cacciatore, S.; Gao, X.; Syamala, S.; Bango, C.; Photopoulos, C.; Huang, Y.; et al. Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, V.; Benfodda, Z.; Rodier, G.; Henriquet, C.; Iborra, F.; Avances, C.; Allory, Y.; De La Taille, A.; Culine, S.; Blancou, H.; et al. Abrogation of de novo lipogenesis by stearoyl-CoA desaturase 1 inhibition interferes with oncogenic signaling and blocks prostate cancer progression in mice. Mol. Cancer Ther. 2010, 9, 1740–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; Van Veldhoven, P.P.; Waltregny, D.; Daniels, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [Green Version]
- Alasiri, G.; Fan, L.Y.; Zona, S.; Goldsbrough, I.G.; Ke, H.L.; Auner, H.W.; Lam, E.W.-F. ER stress and cancer: The FOXO forkhead transcription factor link. Mol. Cell Endocrinol. 2018, 462 Pt B, 67–81. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, S.L.; Sobel, R.; Whitmore, T.G.; Akbari, M.; Bradley, D.R.; Gleave, M.E.; Nelson, C.C. Dysregulation of sterol response element-binding proteins and downstream effectors in prostate cancer during progression to androgen independence. Cancer Res. 2004, 64, 2212–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.; Gao, S.; Barrett, D.; Ahmed, M.; Han, D.; Macoska, J.A.; He, H.H.; Cai, C. Reactivation of androgen receptor-regulated lipid biosynthesis drives the progression of castration-resistant prostate cancer. Oncogene 2018, 37, 710–721. [Google Scholar] [CrossRef] [Green Version]
- Barelli, H.; Antonny, B. Lipid unsaturation and organelle dynamics. Curr. Opin. Cell Biol. 2016, 41, 25–32. [Google Scholar] [CrossRef]
- Huang, W.C.; Zhau, H.E.; Chung, L.W. Androgen receptor survival signaling is blocked by anti-beta2-microglobulin monoclonal antibody via a MAPK/lipogenic pathway in human prostate cancer cells. J. Biol. Chem. 2010, 285, 7947–7956. [Google Scholar] [CrossRef] [Green Version]
- Lounis, M.A.; Bergeron, K.F.; Burhans, M.S.; Ntambi, J.M.; Mounier, C. Oleate activates SREBP-1 signaling activity in SCD1-deficient hepatocytes. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E710–E720. [Google Scholar] [CrossRef]
- Solinas, G.; Boren, J.; Dulloo, A.G. De novo lipogenesis in metabolic homeostasis: More friend than foe? Mol. Metab. 2015, 4, 367–377. [Google Scholar] [CrossRef]
- Fu, S.; Yang, L.; Li, P.; Hofmann, O.; Dicker, L.; Hide, W.; Lin, X.; Watkins, S.M.; Ivanov, A.R.; Hotamisligil, G.S. Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 2011, 473, 528–531. [Google Scholar] [CrossRef] [Green Version]
- Volmer, R.; Ron, D. Lipid-dependent regulation of the unfolded protein response. Curr. Opin. Cell Biol. 2015, 33, 67–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunelle, J.K.; Bell, E.L.; Quesada, N.M.; Vercauteren, K.; Tiranti, V.; Zeviani, M.; Scarpulla, R.C.; Chandel, N.S. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005, 1, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzy, R.D.; Hoyos, B.; Robin, E.; Chen, H.; Liu, L.; Mansfield, K.D.; Simon, M.C.; Hammerling, U.; Schumacker, P.T. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005, 1, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Robbins, H.L.; Hague, A. The PI3K/Akt Pathway in Tumors of Endocrine Tissues. Front. Endocrinol. (Lausanne) 2015, 6, 188. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhou, L.; Wu, X.; Li, R.; Wen, J.; Sha, J.; Wen, X. The PI3K/AKT pathway in the pathogenesis of prostate cancer. Front. Biosci (Landmark Ed.) 2016, 21, 1084–1091. [Google Scholar]
- Bitting, R.L.; Armstrong, A.J. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr. Relat. Cancer 2013, 20, R83–R99. [Google Scholar] [CrossRef] [Green Version]
- Edlind, M.P.; Hsieh, A.C. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J. Androl. 2014, 16, 378–386. [Google Scholar] [CrossRef]
- Rini, B.I.; Atkins, M.B. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009, 10, 992–1000. [Google Scholar] [CrossRef]
- Kudo, Y.; Tanaka, Y.; Tateishi, K.; Yamamoto, K.; Yamamoto, S.; Mohri, D.; Isomura, Y.; Seto, M.; Nakagawa, H.; Asaoka, Y.; et al. Altered composition of fatty acids exacerbates hepatotumorigenesis during activation of the phosphatidylinositol 3-kinase pathway. J. Hepatol. 2011, 55, 1400–1408. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Tsai, J.P.; Shen, C.J.; Liao, Y.H.; Chen, B.K. Oleate-induced PTX3 promotes head and neck squamous cell carcinoma metastasis through the up-regulation of vimentin. Oncotarget 2017, 8, 41364–41378. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.R.; Lee, J.Y.; Park, H.S.; Heo, H.J.; Park, J.Y.; Bae, S.S.; Hong, K.W.; Sung, S.M.; Kim, C.D. Oleic acid enhances vascular smooth muscle cell proliferation via phosphatidylinositol 3-kinase/Akt signaling pathway. Pharmacol. Res. 2006, 54, 97–102. [Google Scholar] [CrossRef] [PubMed]
- She, K.; Fang, S.; Du, W.; Fan, X.; He, J.; Pan, H. SCD1 is required for EGFR-targeting cancer therapy of lung cancer via re-activation of EGFR/PI3K/AKT signals. Cancer Cell Int. 2019, 19, 103. [Google Scholar] [CrossRef] [PubMed]
- Noto, A.; De Vitis, C.; Pisanu, M.E.; Roscilli, G.; Ricci, G.; Catizone, A.; Sorrentino, G.; Chianese, G.; Taglialatela-Scafati, O.; Trisciuoglio, D.; et al. Stearoyl-CoA-desaturase 1 regulates lung cancer stemness via stabilization and nuclear localization of YAP/TAZ. Oncogene 2017, 36, 4573–4584. [Google Scholar] [CrossRef]
- Tracz-Gaszewska, Z.; Dobrzyn, P. Stearoyl-CoA Desaturase 1 as a Therapeutic Target for the Treatment of Cancer. Cancers 2019, 11, 948. [Google Scholar] [CrossRef] [Green Version]
- Mason, P.; Liang, B.; Li, L.; Fremgen, T.; Murphy, E.; Quinn, A.; Madden, S.L.; Biemann, H.-P.; Wang, B.; Cohen, A.; et al. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS ONE 2012, 7, e33823. [Google Scholar] [CrossRef] [Green Version]
- Karaskov, E.; Scott, C.; Zhang, L.; Teodoro, T.; Ravazzola, M.; Volchuk, A. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 2006, 147, 3398–3407. [Google Scholar] [CrossRef] [Green Version]
- Nemcova-Furstova, V.; James, R.F.; Kovar, J. Inhibitory effect of unsaturated fatty acids on saturated fatty acid-induced apoptosis in human pancreatic beta-cells: Activation of caspases and ER stress induction. Cell Physiol. Biochem. 2011, 27, 525–538. [Google Scholar] [CrossRef] [Green Version]
- Pineau, L.; Colas, J.; Dupont, S.; Beney, L.; Fleurat-Lessard, P.; Berjeaud, J.M.; Berges, T.; Ferreira, T. Lipid-induced ER stress: Synergistic effects of sterols and saturated fatty acids. Traffic 2009, 10, 673–690. [Google Scholar] [CrossRef]
- Matsui, H.; Yokoyama, T.; Sekiguchi, K.; Iijima, D.; Sunaga, H.; Maniwa, M.; Ueno, M.; Iso, T.; Arai, M.; Kurabayashi, M. Stearoyl-CoA desaturase-1 (SCD1) augments saturated fatty acid-induced lipid accumulation and inhibits apoptosis in cardiac myocytes. PLoS ONE 2012, 7, e33283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelinas, R.; Thompson-Legault, J.; Bouchard, B.; Daneault, C.; Mansour, A.; Gillis, M.A.; Charron, G.; Gavino, V.; Labarthe, F.; Des Rosiers, C. Prolonged QT interval and lipid alterations beyond beta-oxidation in very long-chain acyl-CoA dehydrogenase null mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H813–H823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson Legault, J.; Strittmatter, L.; Tardif, J.; Sharma, R.; Tremblay-Vaillancourt, V.; Aubut, C.; Boucher, G.; Clish, C.B.; Cyr, D.; Daneault, C.; et al. A Metabolic Signature of Mitochondrial Dysfunction Revealed through a Monogenic Form of Leigh Syndrome. Cell Rep. 2015, 13, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcot, V.; Brunet, J.; Daneault, C.; Tardif, J.C.; Des Rosiers, C.; Lettre, G. Validation of fatty acid intakes estimated by a food frequency questionnaire using erythrocyte fatty acid profiling in the Montreal Heart Institute Biobank. J. Hum. Nutr. Diet. 2015, 28, 646–658. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lounis, M.A.; Péant, B.; Leclerc-Desaulniers, K.; Ganguli, D.; Daneault, C.; Ruiz, M.; Zoubeidi, A.; Mes-Masson, A.-M.; Saad, F. Modulation of de Novo Lipogenesis Improves Response to Enzalutamide Treatment in Prostate Cancer. Cancers 2020, 12, 3339. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113339
Lounis MA, Péant B, Leclerc-Desaulniers K, Ganguli D, Daneault C, Ruiz M, Zoubeidi A, Mes-Masson A-M, Saad F. Modulation of de Novo Lipogenesis Improves Response to Enzalutamide Treatment in Prostate Cancer. Cancers. 2020; 12(11):3339. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113339
Chicago/Turabian StyleLounis, Mohamed Amine, Benjamin Péant, Kim Leclerc-Desaulniers, Dwaipayan Ganguli, Caroline Daneault, Matthieu Ruiz, Amina Zoubeidi, Anne-Marie Mes-Masson, and Fred Saad. 2020. "Modulation of de Novo Lipogenesis Improves Response to Enzalutamide Treatment in Prostate Cancer" Cancers 12, no. 11: 3339. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113339