Cold-Inducible RNA Binding Protein as a Vaccination Platform to Enhance Immunotherapeutic Responses against Hepatocellular Carcinoma


 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Immunogenicity of a CIRP-Based Vaccine Harboring a Polyepitopic Antigen Is Enhanced by Combination with ICPI
2.2. Therapeutic Vaccination with a CIRP-Containing Immunogen Increases the Efficacy of ICPI
2.3. Vaccination Induces Peripheral Antitumor Immunity but Weakly Rescues the Exhausted Phenotype Displayed by Hepatic Tumor-Infiltrating Lymphocytes
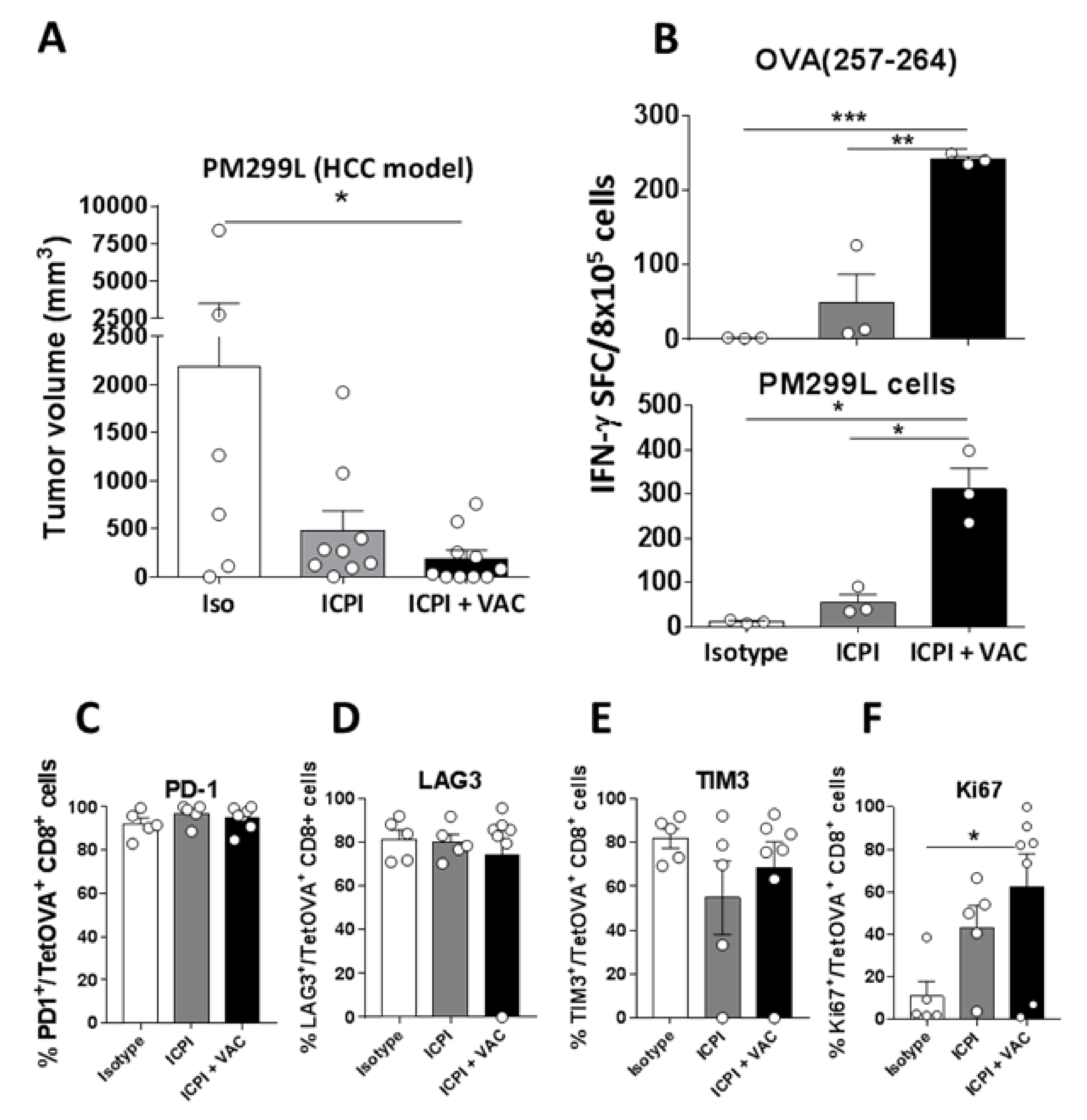
2.4. The CIRP-Based Vaccine Enhances Therapeutic Responses of ICPI-Based Protocols in an HCC Model
2.5. Immunization with Human HCC Antigen Glypican 3 Conjugated to CIRP (GPC3-CIRP) Induces Anti GPC3 Immunity, Enhanced by Combination with ICPI
2.6. Combination of Vaccine with ICPI Does not Enhance Liver Toxicity in Mice
3. Discussion
4. Materials and Methods
4.1. Immunogens
4.2. Mice
4.3. Tumor Cell Lines
4.4. Vaccination to Evaluate Protein Immunogenicity
4.5. ELISPOT
4.6. Tumor Treatment Experiments
4.7. Flow Cytometry
4.8. Serum Markers
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Flecken, T.; Schmidt, N.; Hild, S.; Gostick, E.; Drognitz, O.; Zeiser, R.; Schemmer, P.; Bruns, H.; Eiermann, T.; Price, D.A.; et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8(+) T-cell responses in hepatocellular carcinoma. Hepatology 2014, 59, 1415–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, Y.; Nakashima, O.; Kutami, R.; Yamamoto, O.; Kojiro, M. Clinicopathological study on hepatocellular carcinoma with lymphocytic infiltration. Hepatology 1998, 27, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Unitt, E.; Marshall, A.; Gelson, W.; Rushbrook, S.M.; Davies, S.; Vowler, S.L.; Morris, L.S.; Coleman, N.; Alexander, G.J. Tumour lymphocytic infiltrate and recurrence of hepatocellular carcinoma following liver transplantation. J. Hepatol. 2006, 45, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Sangro, B.; Gomez-Martin, C.; de la Mata, M.; Inarrairaegui, M.; Garralda, E.; Barrera, P.; Riezu-Boj, J.I.; Larrea, E.; Alfaro, C.; Sarobe, P.; et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 2013, 59, 81–88. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): A non-randomised, open-label phase 2 trial. Lancet Oncol. 2018, 19, 940–952. [Google Scholar] [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Yau, T.; Kang, Y.K.; Kim, T.Y.; El-Khoueiry, A.B.; Santoro, A.; Sangro, B.; Melero, I.; Kudo, M.; Hou, M.M.; Matilla, A.; et al. Nivolumab (NIVO) + ipilimumab (IPI) combination therapy in patients (pts) with advanced hepatocellular carcinoma (aHCC): Results from CheckMate 040. J. Clin. Oncol. 2019, 37, 4012. [Google Scholar] [CrossRef]
- Finn, R.S.; Ikeda, M.; Zhu, A.X.; Sung, M.W.; Baron, A.D.; Kudo, M.; Okusaka, T.; Kobayashi, M.; Kumada, H.; Kaneko, S.; et al. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients With Unresectable Hepatocellular Carcinoma. J. Clin. Oncol. 2020, 38, 2960–2970. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangro, B.; Melero, I.; Wadhawan, S.; Finn, R.S.; Abou-Alfa, G.K.; Cheng, A.L.; Yau, T.; Furuse, J.; Park, J.W.; Boyd, Z.; et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2020. In Press. [Google Scholar] [CrossRef]
- Melero, I.; Gaudernack, G.; Gerritsen, W.; Huber, C.; Parmiani, G.; Scholl, S.; Thatcher, N.; Wagstaff, J.; Zielinski, C.; Faulkner, I.; et al. Therapeutic vaccines for cancer: An overview of clinical trials. Nat. Rev. Clin. Oncol. 2014, 11, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment-targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef] [Green Version]
- Hui, E.; Cheung, J.; Zhu, J.; Su, X.; Taylor, M.J.; Wallweber, H.A.; Sasmal, D.K.; Huang, J.; Kim, J.M.; Mellman, I.; et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science 2017, 355, 1428–1433. [Google Scholar] [CrossRef]
- Villanueva, L.; Silva, L.; Llopiz, D.; Ruiz, M.; Iglesias, T.; Lozano, T.; Casares, N.; Hervas-Stubbs, S.; Rodriguez, M.J.; Carrascosa, J.L.; et al. The Toll like receptor 4 ligand cold-inducible RNA-binding protein as vaccination platform against cancer. Oncoimmunology 2018, 7, e1409321. [Google Scholar] [CrossRef] [Green Version]
- Qiang, X.; Yang, W.L.; Wu, R.; Zhou, M.; Jacob, A.; Dong, W.; Kuncewitch, M.; Ji, Y.; Yang, H.; Wang, H.; et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 2013, 19, 1489–1495. [Google Scholar] [CrossRef] [Green Version]
- Kudo-Saito, C.; Schlom, J.; Hodge, J.W. Induction of an antigen cascade by diversified subcutaneous/intratumoral vaccination is associated with antitumor responses. Clin. Cancer Res. 2005, 11, 2416–2426. [Google Scholar] [CrossRef] [Green Version]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Lozano, T.; Chocarro, S.; Martin, C.; Lasarte-Cia, A.; Del Valle, C.; Gorraiz, M.; Sarrion, P.; Ruiz de Galarreta, M.; Lujambio, A.; Hervas-Stubbs, S.; et al. Genetic Modification of CD8(+) T Cells to Express EGFR: Potential Application for Adoptive T Cell Therapies. Front. Immunol. 2019, 10, 2990. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sanchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. beta-Catenin Activation Promotes Immune Escape and Resistance to Anti-PD-1 Therapy in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef] [PubMed]
- Filmus, J.; Capurro, M. Glypican-3: A marker and a therapeutic target in hepatocellular carcinoma. FEBS J. 2013, 280, 2471–2476. [Google Scholar] [CrossRef] [PubMed]
- O’Beirne, J.; Farzaneh, F.; Harrison, P.M. Generation of functional CD8+ T cells by human dendritic cells expressing glypican-3 epitopes. J. Exp. Clin. Cancer Res. 2010, 29, 48. [Google Scholar] [CrossRef] [Green Version]
- Sangro, B.; Chan, S.L.; Meyer, T.; Reig, M.; El-Khoueiry, A.; Galle, P.R. Diagnosis and management of toxicities of immune checkpoint inhibitors in hepatocellular carcinoma. J. Hepatol. 2020, 72, 320–341. [Google Scholar] [CrossRef] [Green Version]
- Inarrairaegui, M.; Melero, I.; Sangro, B. Immunotherapy of Hepatocellular Carcinoma: Facts and Hopes. Clin. Cancer Res. 2018, 24, 1518–1524. [Google Scholar] [CrossRef] [Green Version]
- Feng, G.S.; Hanley, K.L.; Liang, Y.; Lin, X. Improving the Efficacy of Liver Cancer Immunotherapy: The Power of Combined Preclinical and Clinical Studies. Hepatology 2020. [Google Scholar] [CrossRef]
- Cheng, A.L.; Hsu, C.; Chan, S.L.; Choo, S.P.; Kudo, M. Challenges of combination therapy with immune checkpoint inhibitors for hepatocellular carcinoma. J. Hepatol. 2020, 72, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Rizvi, N.A.; Hellmann, M.D.; Brahmer, J.R.; Juergens, R.A.; Borghaei, H.; Gettinger, S.; Chow, L.Q.; Gerber, D.E.; Laurie, S.A.; Goldman, J.W.; et al. Nivolumab in Combination With Platinum-Based Doublet Chemotherapy for First-Line Treatment of Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 2969–2979. [Google Scholar] [CrossRef]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, S.; Castven, D.; Galle, P.R.; Marquardt, D.T. Translational Considerations to Improve Response and Overcome Therapy Resistance in Immunotherapy for Hepatocellular Carcinoma. Cancers 2020, 12, 2495. [Google Scholar] [CrossRef]
- Tagliamonte, M.; Petrizzo, A.; Mauriello, A.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Potentiating cancer vaccine efficacy in liver cancer. Oncoimmunology 2018, 7, e1488564. [Google Scholar] [CrossRef] [PubMed]
- Llopiz, D.; Ruiz, M.; Infante, S.; Villanueva, L.; Silva, L.; Hervas-Stubbs, S.; Alignani, D.; Guruceaga, E.; Lasarte, J.J.; Sarobe, P. IL-10 expression defines an immunosuppressive dendritic cell population induced by antitumor therapeutic vaccination. Oncotarget 2016, 8, 2659–2671. [Google Scholar] [CrossRef]
- Albershardt, T.C.; Leleux, J.; Parsons, A.J.; Krull, J.E.; Berglund, P.; Ter Meulen, J. Intratumoral immune activation with TLR4 agonist synergizes with effector T cells to eradicate established murine tumors. NPJ Vaccines 2020, 5, 50. [Google Scholar] [CrossRef]
- Kelley, R.K.; Sangro, B.; Harris, W.P.; Ikeda, M.; Okusaka, T.; Kang, Y.K.; Qin, S.; Taj, W.M.D.; Lim, H.Y.; Yau, T.; et al. Efficacy, tolerability, and biologic activity of a novel regimen of tremelimumab (T) in combination with durvalumab (D) for patients (pts) with advanced hepatocellular carcinoma (aHCC). J. Clin. Oncol. 2020, 38, 4508. [Google Scholar] [CrossRef]
- Zhou, G.; Sprengers, D.; Boor, P.P.C.; Doukas, M.; Schutz, H.; Mancham, S.; Pedroza-Gonzalez, A.; Polak, W.G.; de Jonge, J.; Gaspersz, M.; et al. Antibodies Against Immune Checkpoint Molecules Restore Functions of Tumor-Infiltrating T Cells in Hepatocellular Carcinomas. Gastroenterology 2017, 153, 1107–1119. [Google Scholar] [CrossRef]
- Ma, J.; Zheng, B.; Goswami, S.; Meng, L.; Zhang, D.; Cao, C.; Li, T.; Zhu, F.; Ma, L.; Zhang, Z.; et al. PD1(Hi) CD8(+) T cells correlate with exhausted signature and poor clinical outcome in hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 331. [Google Scholar] [CrossRef]
- Kudo, M. Scientific Rationale for Combined Immunotherapy with PD-1/PD-L1 Antibodies and VEGF Inhibitors in Advanced Hepatocellular Carcinoma. Cancers 2020, 12, 1089. [Google Scholar] [CrossRef]
- Shigeta, K.; Datta, M.; Hato, T.; Kitahara, S.; Chen, I.X.; Matsui, A.; Kikuchi, H.; Mamessier, E.; Aoki, S.; Ramjiawan, R.R.; et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology 2020, 71, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Silva, L.; Egea, J.; Villanueva, L.; Ruiz, M.; Llopiz, D.; Repáraz, D.; Aparicio, B.; Lasarte-Cia, A.; Lasarte, J.J.; Ruiz de Galarreta, M.; et al. Cold-Inducible RNA Binding Protein as a Vaccination Platform to Enhance Immunotherapeutic Responses against Hepatocellular Carcinoma. Cancers 2020, 12, 3397. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113397
Silva L, Egea J, Villanueva L, Ruiz M, Llopiz D, Repáraz D, Aparicio B, Lasarte-Cia A, Lasarte JJ, Ruiz de Galarreta M, et al. Cold-Inducible RNA Binding Protein as a Vaccination Platform to Enhance Immunotherapeutic Responses against Hepatocellular Carcinoma. Cancers. 2020; 12(11):3397. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113397
Chicago/Turabian StyleSilva, Leyre, Josune Egea, Lorea Villanueva, Marta Ruiz, Diana Llopiz, David Repáraz, Belén Aparicio, Aritz Lasarte-Cia, Juan José Lasarte, Marina Ruiz de Galarreta, and et al. 2020. "Cold-Inducible RNA Binding Protein as a Vaccination Platform to Enhance Immunotherapeutic Responses against Hepatocellular Carcinoma" Cancers 12, no. 11: 3397. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12113397