Tribbles Pseudokinase 3 Contributes to Cancer Stemness of Endometrial Cancer Cells by Regulating β-Catenin Expression

 , and
, and 
Abstract
:Simple Summary
Abstract
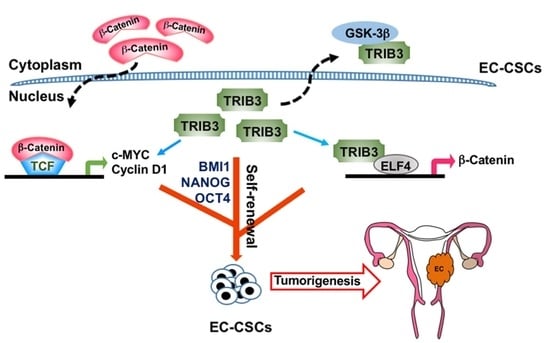
1. Introduction
2. Results
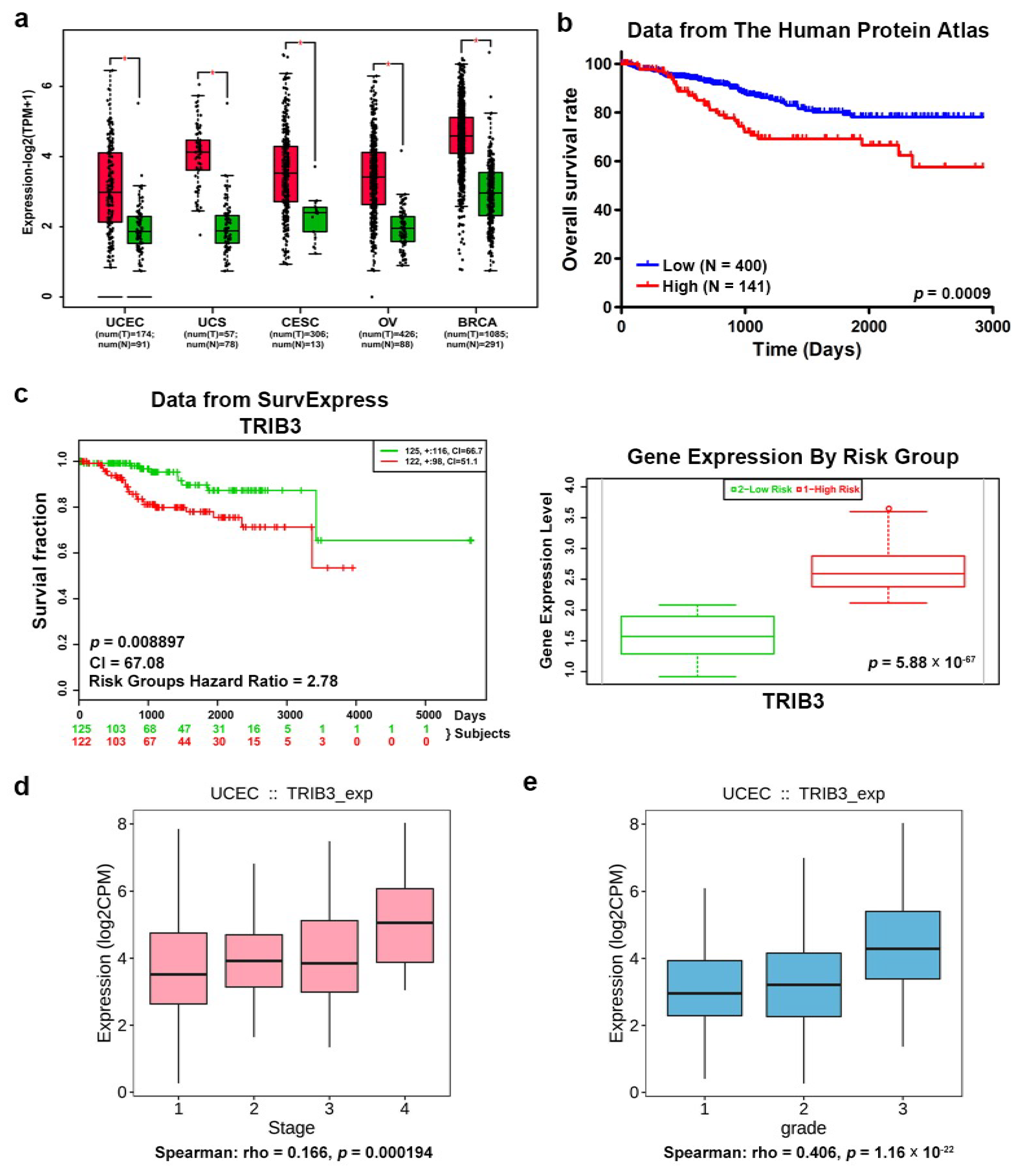
2.1. The TRIB3 Expression Level Is Positively Correlated with Shorter Overall Survival in EC Patients
2.2. TRIB3 Knockdown Suppresses the Growth of EC Cells
2.3. Silencing of TRIB3 Attenuates EC Cell Migration and Invasion and Decreases the Expression Levels of EMT-Associated Factors
2.4. Knockdown of TRIB3 Decreases the Self-Renewal Capability and Tumorigenicity of EC-CSCs
2.5. TRIB3 Participates in the Expression and Activity of β-Catenin in EC Cells
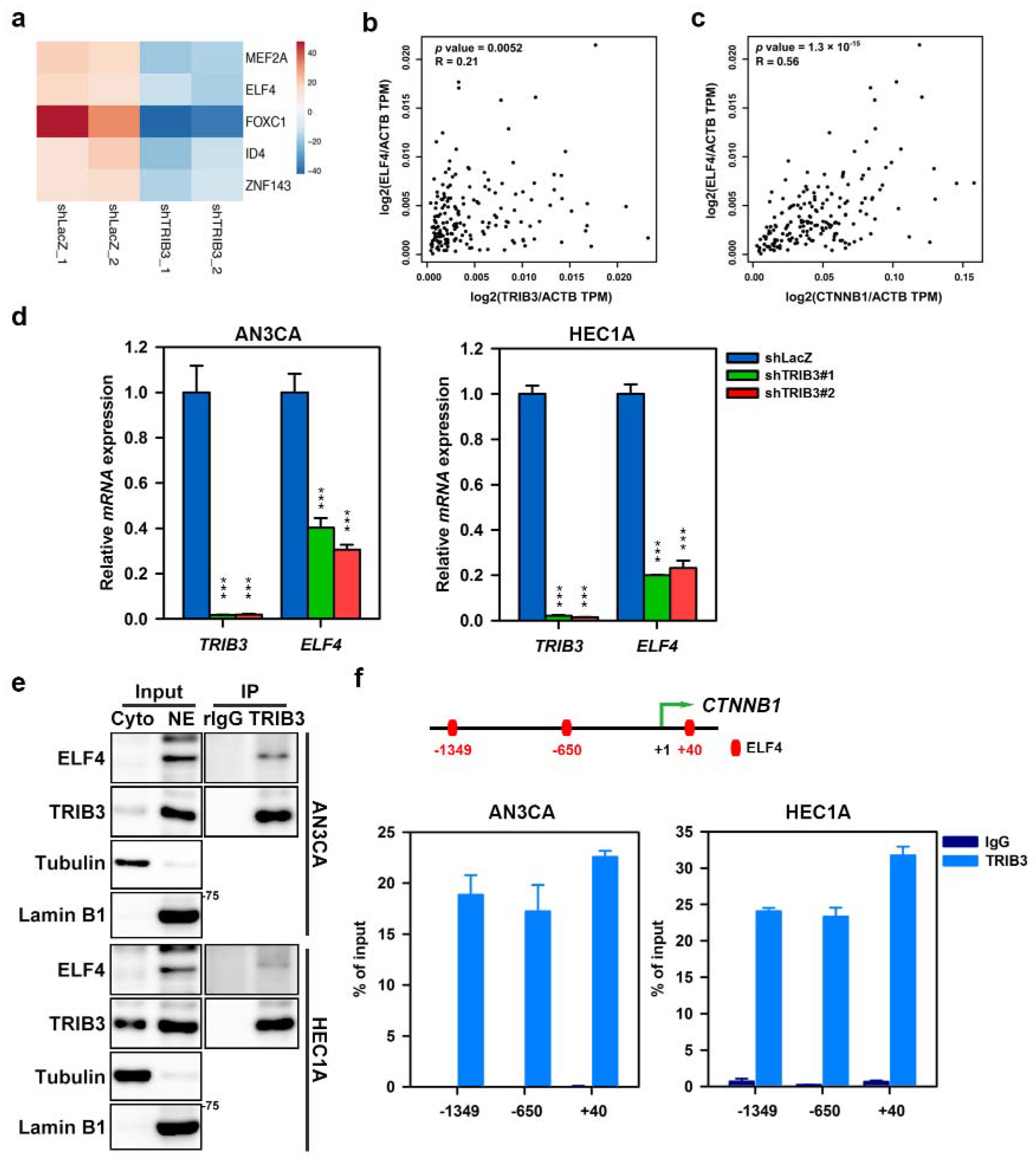
2.6. TRIB3 Interacts with ELF4 to Transcriptionally Regulate the Expression of β-Catenin
2.7. The Regulation Role of TRIB3 in β-Catenin and Cancer Stemness Genes Is Observed in Patient-Derived Primary EC Cells
3. Discussion
4. Materials and Methods
4.1. Endometrial Cancer Cell Lines
4.2. Establishment of Primary EC Cell Lines from Patients’ Endometrial Cancer Tissue
4.3. TRIB3 Knockdown with Lentiviral shRNA
4.4. Tumorsphere Cultivation
4.5. Determination of Cell Growth and Colony Formation Assay
4.6. Western Blot and Coimmunoprecipitation (Co-IP)
4.7. RNA Isolation and Real-Time PCR
4.8. RNA Sequencing Analysis
4.9. Migration and Invasion
4.10. Isolation of Cytosolic and Nuclear Fractions
4.11. Chromatin Immunoprecipitation (ChIP) qPCR
4.12. NOD/SCID Xenograft Mouse Model
4.13. Immunohistochemistry
4.14. Statistical Analysis
4.15. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.C.; Weng, C.S.; Huang, S.M.; Huang, N.; Chou, Y.J.; Wang, C.C.; Wang, K.L. Incidence and lifetime risk of uterine corpus cancer in Taiwanese women from 1991 to 2010. Taiwan J. Obstet. Gynecol. 2017, 56, 68–72. [Google Scholar] [CrossRef]
- Yang, H.P.; Wentzensen, N.; Trabert, B.; Gierach, G.L.; Felix, A.S.; Gunter, M.J.; Hollenbeck, A.; Park, Y.; Sherman, M.E.; Brinton, L.A. Endometrial cancer risk factors by 2 main histologic subtypes: The NIH-AARP Diet and Health Study. Am. J. Epidemiol. 2013, 177, 142–151. [Google Scholar] [CrossRef] [Green Version]
- Fortner, R.T.; Husing, A.; Kuhn, T.; Konar, M.; Overvad, K.; Tjonneland, A.; Hansen, L.; Boutron-Ruault, M.C.; Severi, G.; Fournier, A.; et al. Endometrial cancer risk prediction including serum-based biomarkers: Results from the EPIC cohort. Int. J. Cancer 2017, 140, 1317–1323. [Google Scholar] [CrossRef]
- Huijgens, A.N.; Mertens, H.J. Factors predicting recurrent endometrial cancer. Facts Views Vis. Obgyn 2013, 5, 179–186. [Google Scholar]
- Moreno-Bueno, G.; Hardisson, D.; Sanchez, C.; Sarrio, D.; Cassia, R.; Garcia-Rostan, G.; Prat, J.; Guo, M.; Herman, J.G.; Matias-Guiu, X.; et al. Abnormalities of the APC/beta-catenin pathway in endometrial cancer. Oncogene 2002, 21, 7981–7990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Patel, L.; Mills, G.B.; Lu, K.H.; Sood, A.K.; Ding, L.; Kucherlapati, R.; Mardis, E.R.; Levine, D.A.; Shmulevich, I.; et al. Clinical significance of CTNNB1 mutation and Wnt pathway activation in endometrioid endometrial carcinoma. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ord, T.; Ord, T. Mammalian Pseudokinase TRIB3 in Normal Physiology and Disease: Charting the Progress in Old and New Avenues. Curr. Protein Pept. Sci. 2017, 18, 819–842. [Google Scholar] [CrossRef] [PubMed]
- Mondal, D.; Mathur, A.; Chandra, P.K. Tripping on TRIB3 at the junction of health, metabolic dysfunction and cancer. Biochimie 2016, 124, 34–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, Y.; Ohoka, N.; Hayashi, H.; Sato, R. TRB3 suppresses adipocyte differentiation by negatively regulating PPARgamma transcriptional activity. J. Lipid Res. 2008, 49, 880–892. [Google Scholar] [CrossRef] [Green Version]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef] [Green Version]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.M.; Sun, W.; Wang, Z.H.; Liang, X.; Hua, F.; Li, K.; Lv, X.X.; Zhang, X.W.; Liu, Y.Y.; Yu, J.J.; et al. TRIB3 supports breast cancer stemness by suppressing FOXO1 degradation and enhancing SOX2 transcription. Nat. Commun. 2019, 10, 5720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.C.; Wang, W.L.; Chang, W.C.; Huang, Y.H.; Hong, G.C.; Wang, H.L.; Chou, Y.H.; Tseng, H.C.; Lee, H.T.; Li, S.T.; et al. Tribbles Homolog 3 Involved in Radiation Response of Triple Negative Breast Cancer Cells by Regulating Notch1 Activation. Cancers 2019, 11, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, F.; Shang, S.; Yang, Y.W.; Zhang, H.Z.; Xu, T.L.; Yu, J.J.; Zhou, D.D.; Cui, B.; Li, K.; Lv, X.X.; et al. TRIB3 Interacts With beta-Catenin and TCF4 to Increase Stem Cell Features of Colorectal Cancer Stem Cells and Tumorigenesis. Gastroenterology 2019, 156, 708–721.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, N.; Ishii, H.; Mimori, K.; Takatsuno, Y.; Kim, H.; Hirose, H.; Sekimoto, M.; Doki, Y.; Mori, M. Abnormal expression of TRIB3 in colorectal cancer: A novel marker for prognosis. Br. J. Cancer 2009, 101, 1664–1670. [Google Scholar] [CrossRef] [Green Version]
- Hua, F.; Mu, R.; Liu, J.; Xue, J.; Wang, Z.; Lin, H.; Yang, H.; Chen, X.; Hu, Z. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J. Cell Sci. 2011, 124, 3235–3246. [Google Scholar] [CrossRef] [Green Version]
- Hua, F.; Li, K.; Yu, J.J.; Lv, X.X.; Yan, J.; Zhang, X.W.; Sun, W.; Lin, H.; Shang, S.; Wang, F.; et al. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat. Commun. 2015, 6, 7951. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Wang, F.; Cao, W.B.; Lv, X.X.; Hua, F.; Cui, B.; Yu, J.J.; Zhang, X.W.; Shang, S.; Liu, S.S.; et al. TRIB3 Promotes APL Progression through Stabilization of the Oncoprotein PML-RARalpha and Inhibition of p53-Mediated Senescence. Cancer Cell 2017, 31, 697–710.e7. [Google Scholar] [CrossRef] [Green Version]
- Abbaszadegan, M.R.; Bagheri, V.; Razavi, M.S.; Momtazi, A.A.; Sahebkar, A.; Gholamin, M. Isolation, identification, and characterization of cancer stem cells: A review. J. Cell. Physiol. 2017, 232, 2008–2018. [Google Scholar] [CrossRef]
- O’Connor, M.L.; Xiang, D.; Shigdar, S.; Macdonald, J.; Li, Y.; Wang, T.; Pu, C.; Wang, Z.; Qiao, L.; Duan, W. Cancer stem cells: A contentious hypothesis now moving forward. Cancer Lett. 2014, 344, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Yu, C.C.; Wang, B.Y.; Chang, W.W. Tumorsphere as an effective in vitro platform for screening anti-cancer stem cell drugs. Oncotarget 2016, 7, 1215–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, T.; Hamdan, D.; Leboeuf, C.; El Bouchtaoui, M.; Gapihan, G.; Nguyen, T.T.; Meles, S.; Angeli, E.; Ratajczak, P.; Lu, H.; et al. Targeting Cancer Stem Cells to Overcome Chemoresistance. Int. J. Mol. Sci. 2018, 19, 4036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannone, G.; Attademo, L.; Scotto, G.; Genta, S.; Ghisoni, E.; Tuninetti, V.; Aglietta, M.; Pignata, S.; Valabrega, G. Endometrial Cancer Stem Cells: Role, Characterization and Therapeutic Implications. Cancers 2019, 11, 1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, Y.; Yamawaki, K.; Ishiguro, T.; Yoshihara, K.; Ueda, H.; Sato, A.; Ohata, H.; Yoshida, Y.; Minamino, T.; Okamoto, K.; et al. ALDH-Dependent Glycolytic Activation Mediates Stemness and Paclitaxel Resistance in Patient-Derived Spheroid Models of Uterine Endometrial Cancer. Stem Cell Rep. 2019, 13, 730–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.; Lee, S.; Park, W.H.; Suh, D.H.; Kim, K.; Kim, Y.B.; No, J.H. Silencing Bmi1 expression suppresses cancer stemness and enhances chemosensitivity in endometrial cancer cells. Biomed. Pharm. 2018, 108, 584–589. [Google Scholar] [CrossRef]
- Zhou, X.; Zhou, Y.P.; Huang, G.R.; Gong, B.L.; Yang, B.; Zhang, D.X.; Hu, P.; Xu, S.R. Expression of the stem cell marker, Nanog, in human endometrial adenocarcinoma. Int. J. Gynecol. Pathol. 2011, 30, 262–270. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. Gepia2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Navani, S. Manual evaluation of tissue microarrays in a high-throughput research project: The contribution of indian surgical pathology to the human protein atlas (hpa) project. Proteomics 2016, 16, 1266–1270. [Google Scholar] [CrossRef]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martinez-Ledesma, E.; Martinez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Pena, J.G.; Trevino, V. Survexpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [Green Version]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. Tisidb: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamos, J.L.; Weis, W.I. The beta-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 2013, 5, a007898. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.D.; Praz, V.; Delorenzi, M.; Perier, R.; Bucher, P. The eukaryotic promoter database epd: The impact of in silico primer extension. Nucleic Acids Res 2004, 32, D82–D85. [Google Scholar] [CrossRef] [Green Version]
- Ando, M.; Kawazu, M.; Ueno, T.; Koinuma, D.; Ando, K.; Koya, J.; Kataoka, K.; Yasuda, T.; Yamaguchi, H.; Fukumura, K.; et al. Mutational Landscape and Antiproliferative Functions of ELF Transcription Factors in Human Cancer. Cancer Res. 2016, 76, 1814–1824. [Google Scholar] [CrossRef] [Green Version]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [Green Version]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef]
- Bandapalli, O.R.; Dihlmann, S.; Helwa, R.; Macher-Goeppinger, S.; Weitz, J.; Schirmacher, P.; Brand, K. Transcriptional activation of the beta-catenin gene at the invasion front of colorectal liver metastases. J. Pathol. 2009, 218, 370–379. [Google Scholar] [CrossRef]
- Riazi, A.M.; Takeuchi, J.K.; Hornberger, L.K.; Zaidi, S.H.; Amini, F.; Coles, J.; Bruneau, B.G.; Van Arsdell, G.S. NKX2-5 regulates the expression of beta-catenin and GATA4 in ventricular myocytes. PLoS ONE 2009, 4, e5698. [Google Scholar] [CrossRef] [Green Version]
- Guigon, C.J.; Kim, D.W.; Zhu, X.; Zhao, L.; Cheng, S.Y. Tumor suppressor action of liganded thyroid hormone receptor beta by direct repression of beta-catenin gene expression. Endocrinology 2010, 151, 5528–5536. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Jiang, S.; Wang, C.; Jiang, W.; Liu, Z.; Liu, C.; Saiyin, H.; Yang, X.; Shen, S.; Jiang, D.; et al. Zinc finger transcription factor 191, directly binding to beta-catenin promoter, promotes cell proliferation of hepatocellular carcinoma. Hepatology 2012, 55, 1830–1839. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gao, B.; Ponnusamy, M.; Lin, Z.; Liu, J. MEF2 signaling and human diseases. Oncotarget 2017, 8, 112152–112165. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, E.; Hancock, W.W.; Brancolini, C. MEF2 and the tumorigenic process, hic sunt leones. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Sashida, G.; Liu, Y.; Elf, S.; Miyata, Y.; Ohyashiki, K.; Izumi, M.; Menendez, S.; Nimer, S.D. ELF4/MEF activates MDM2 expression and blocks oncogene-induced p16 activation to promote transformation. Mol. Cell. Biol. 2009, 29, 3687–3699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wang, W.; Huang, K.; Wang, Y.; Li, J.; Yang, X. MicroRNA-34a inhibits cells proliferation and invasion by downregulating Notch1 in endometrial cancer. Oncotarget 2017, 8, 111258–111270. [Google Scholar] [CrossRef]
- Ghahhari, N.M.; Babashah, S. Interplay between microRNAs and WNT/beta-catenin signalling pathway regulates epithelial-mesenchymal transition in cancer. Eur. J. Cancer 2015, 51, 1638–1649. [Google Scholar] [CrossRef]
- Chang, Y.C.; Tsai, C.H.; Lai, Y.L.; Yu, C.C.; Chi, W.Y.; Li, J.J.; Chang, W.W. Arecoline-induced myofibroblast transdifferentiation from human buccal mucosal fibroblasts is mediated by ZEB1. J. Cell. Mol. Med. 2014, 18, 698–708. [Google Scholar] [CrossRef]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′ to 3′) |
|---|---|
| TIRB3 | F: ACCGTATCCCTGAGCCTGA R: CTTGTCCCACAGGGAATCAT |
| c-Myc | F: AATGAAAAGGCCCCCAAGGTAGTTATCC R: GTCGTTTCCGCAACAAGTCCTCTTC |
| CTNNB1 | F: GAAACGGCTTTCAGTTGAGC R: CTGGCCATATCCACCAGAGT |
| MEF2A | F: CAAGGGCATGATGCCTCCACTA R: GCTGAGTACACAAGTCCTTGCG |
| ELF4 | F: AATTGGGACCGTCGCTAGACGA R: GTGGATGTTGCTGGGCACTGAA |
| GAPDH | F: CAATGACCCCTTCATTGACC R: TGGACTCCACGACGTACTCA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.-L.; Hong, G.-C.; Chien, P.-J.; Huang, Y.-H.; Lee, H.-T.; Wang, P.-H.; Lee, Y.-C.; Chang, W.-W. Tribbles Pseudokinase 3 Contributes to Cancer Stemness of Endometrial Cancer Cells by Regulating β-Catenin Expression. Cancers 2020, 12, 3785. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123785
Wang W-L, Hong G-C, Chien P-J, Huang Y-H, Lee H-T, Wang P-H, Lee Y-C, Chang W-W. Tribbles Pseudokinase 3 Contributes to Cancer Stemness of Endometrial Cancer Cells by Regulating β-Catenin Expression. Cancers. 2020; 12(12):3785. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123785
Chicago/Turabian StyleWang, Wen-Ling, Guan-Ci Hong, Peng-Ju Chien, Yu-Hao Huang, Hsueh-Te Lee, Po-Hui Wang, Yueh-Chun Lee, and Wen-Wei Chang. 2020. "Tribbles Pseudokinase 3 Contributes to Cancer Stemness of Endometrial Cancer Cells by Regulating β-Catenin Expression" Cancers 12, no. 12: 3785. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123785