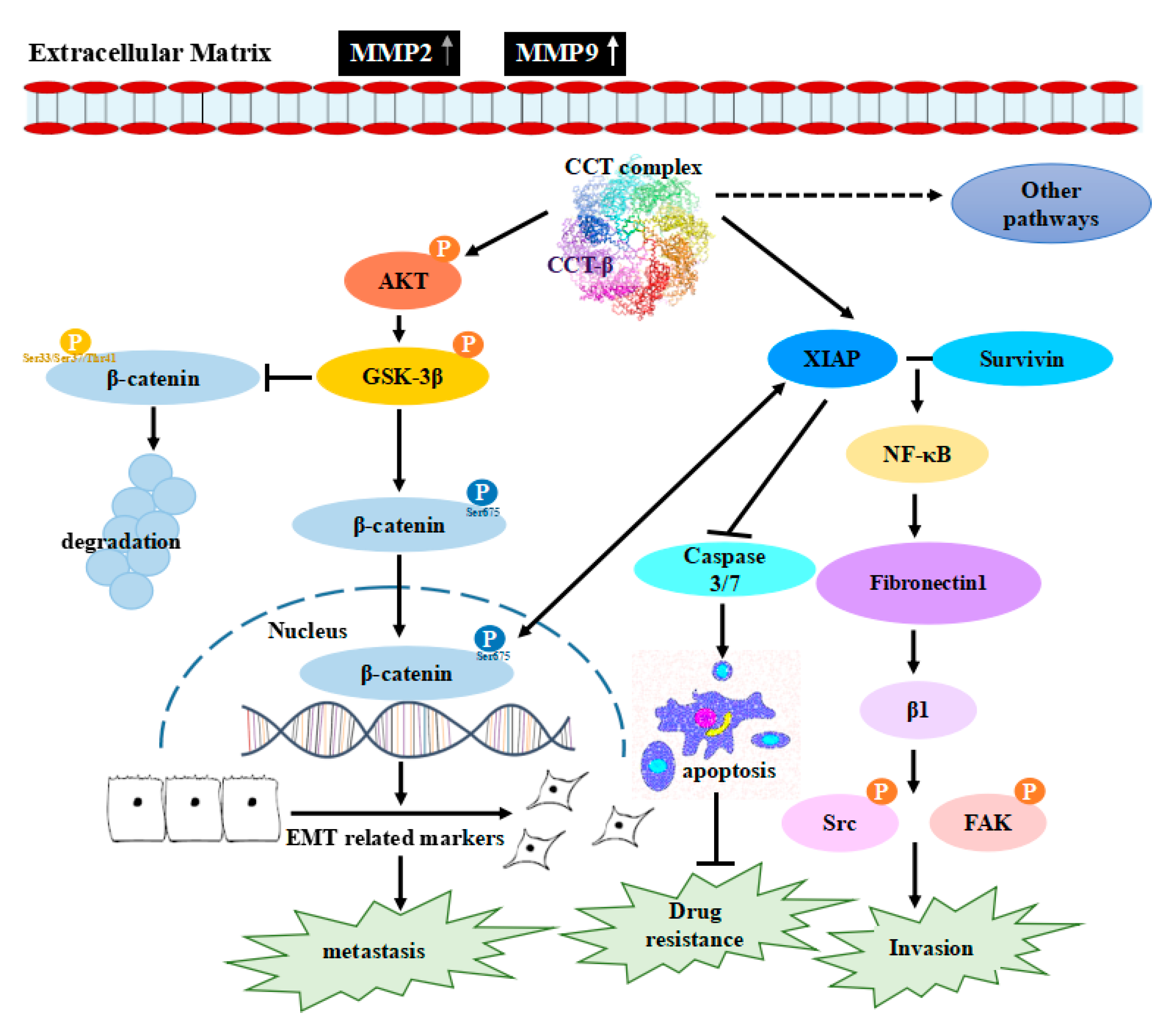
Chaperonin-Containing TCP-1 Promotes Cancer Chemoresistance and Metastasis through the AKT-GSK3β-β-Catenin and XIAP-Survivin Pathways

 ,
,  ,
,  and
and
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
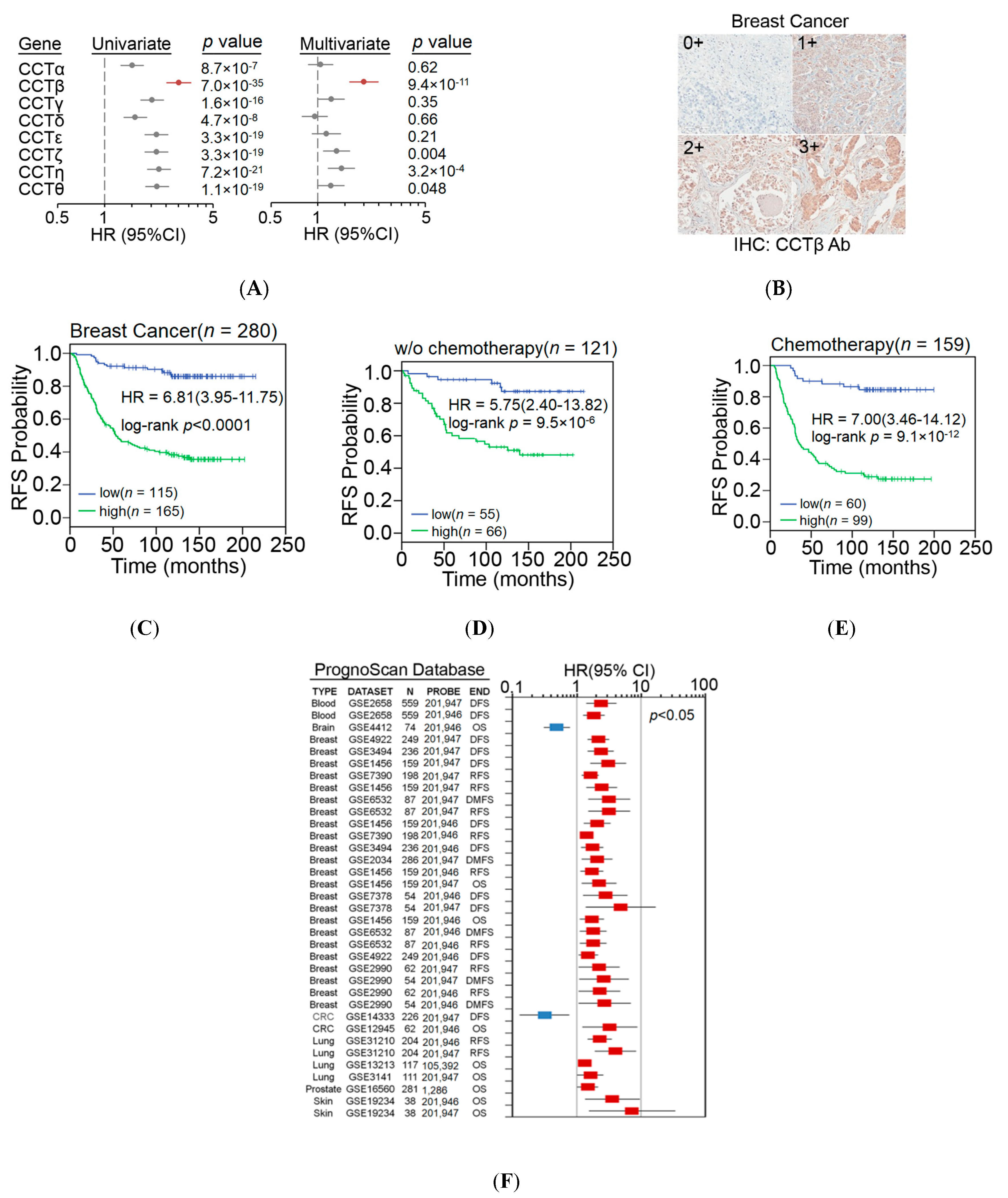
2.1. Endogenously High Expression of CCT-β Is Associated with a Poorer Chemotherapy Response in Clinical Cancer Patients
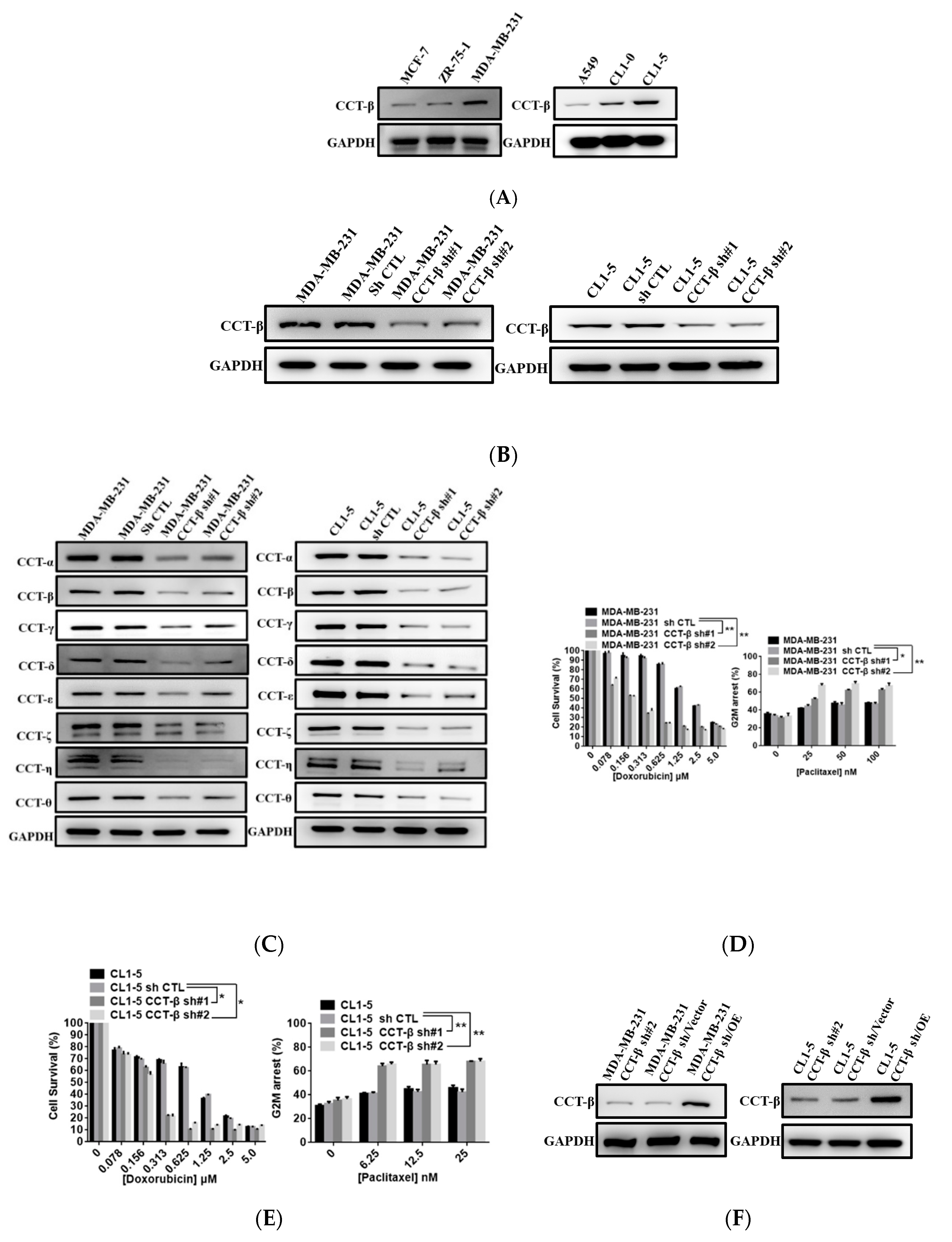
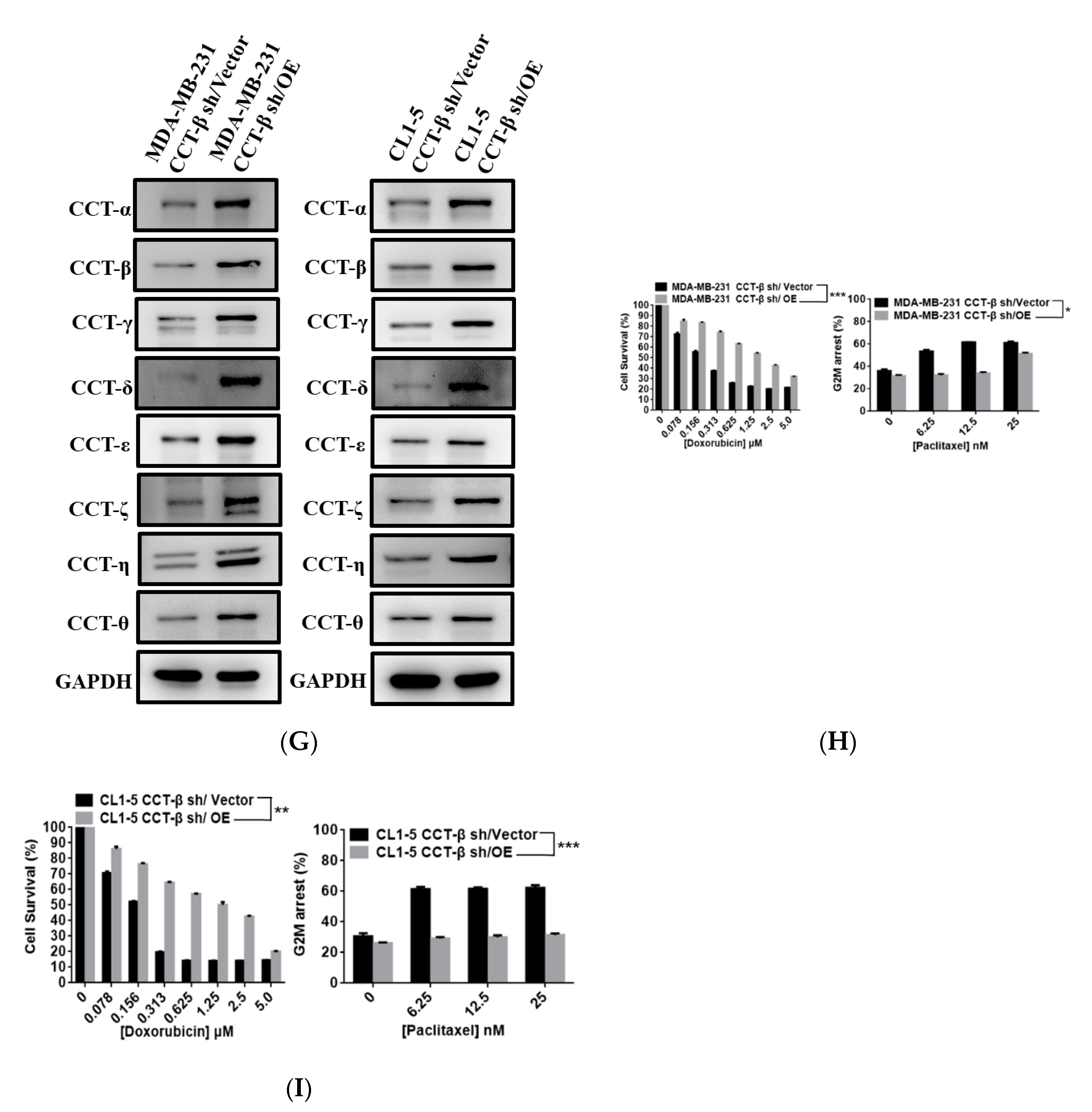
2.2. Upregulation of CCT-β Expression Correlated with Chemoresistance in MDA-MB-231 and CL1-5 Cells
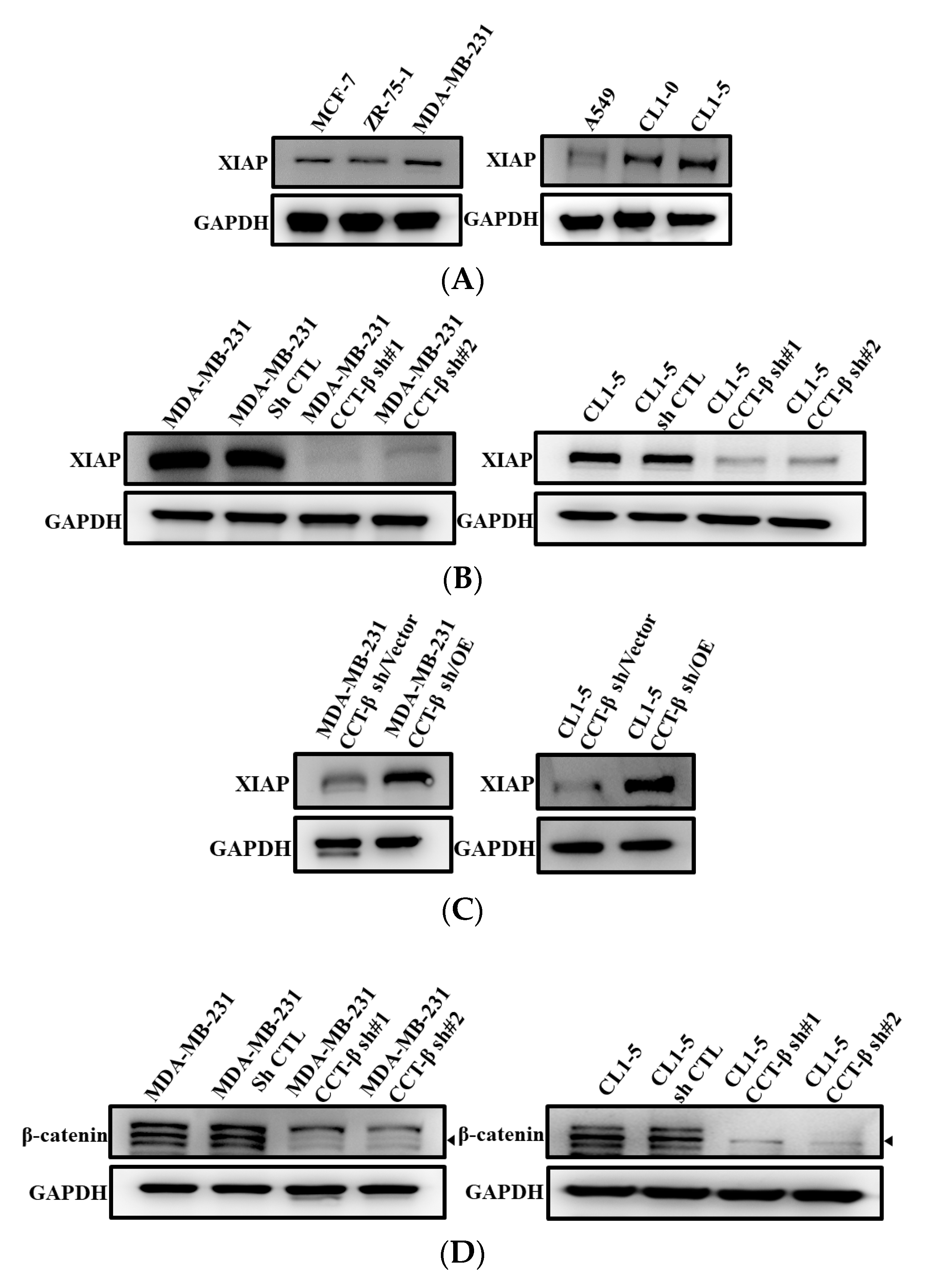
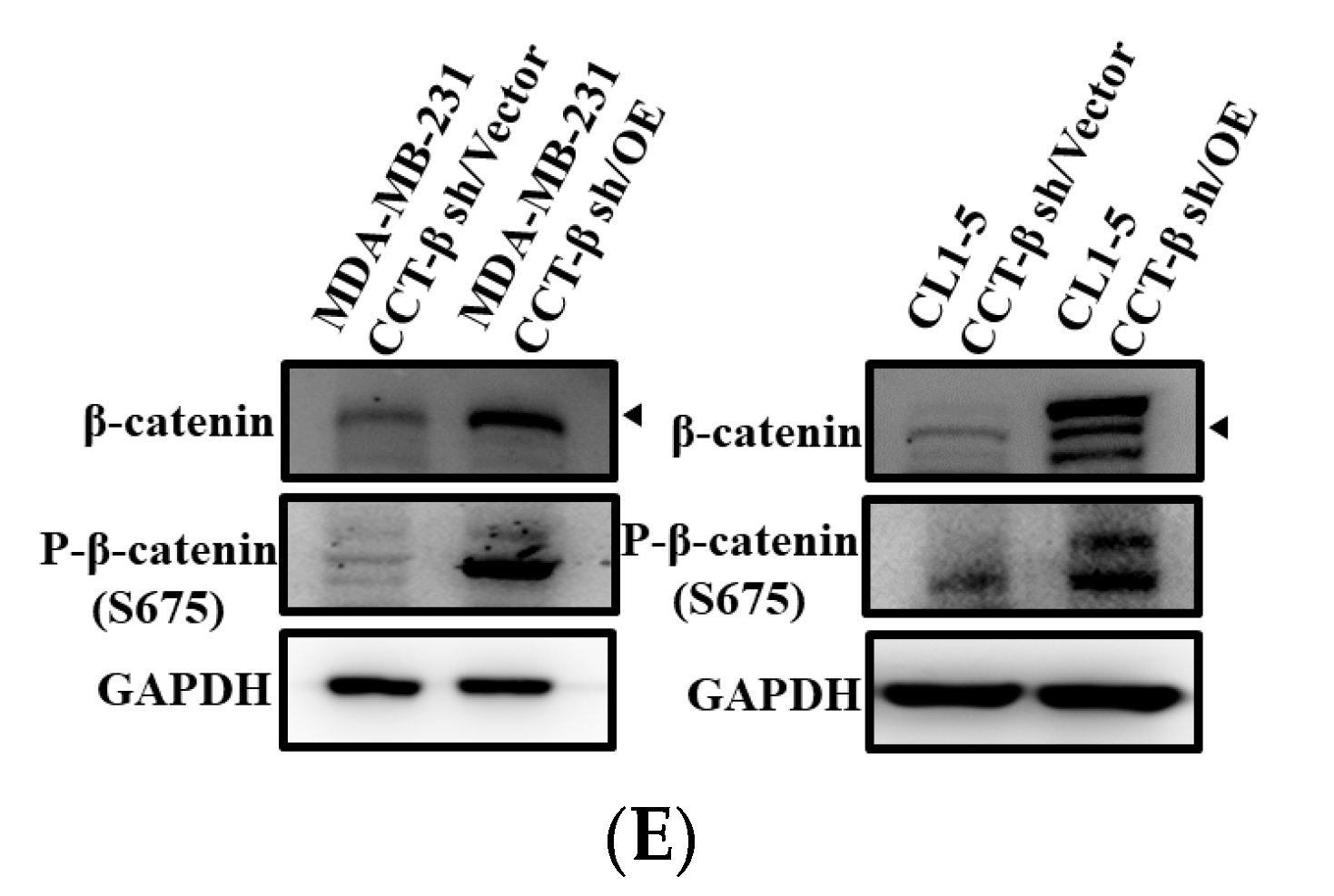
2.3. Correlation of X-Linked Inhibitor of Apoptosis (XIAP) and β-Catenin Expression with CCT-β Expression
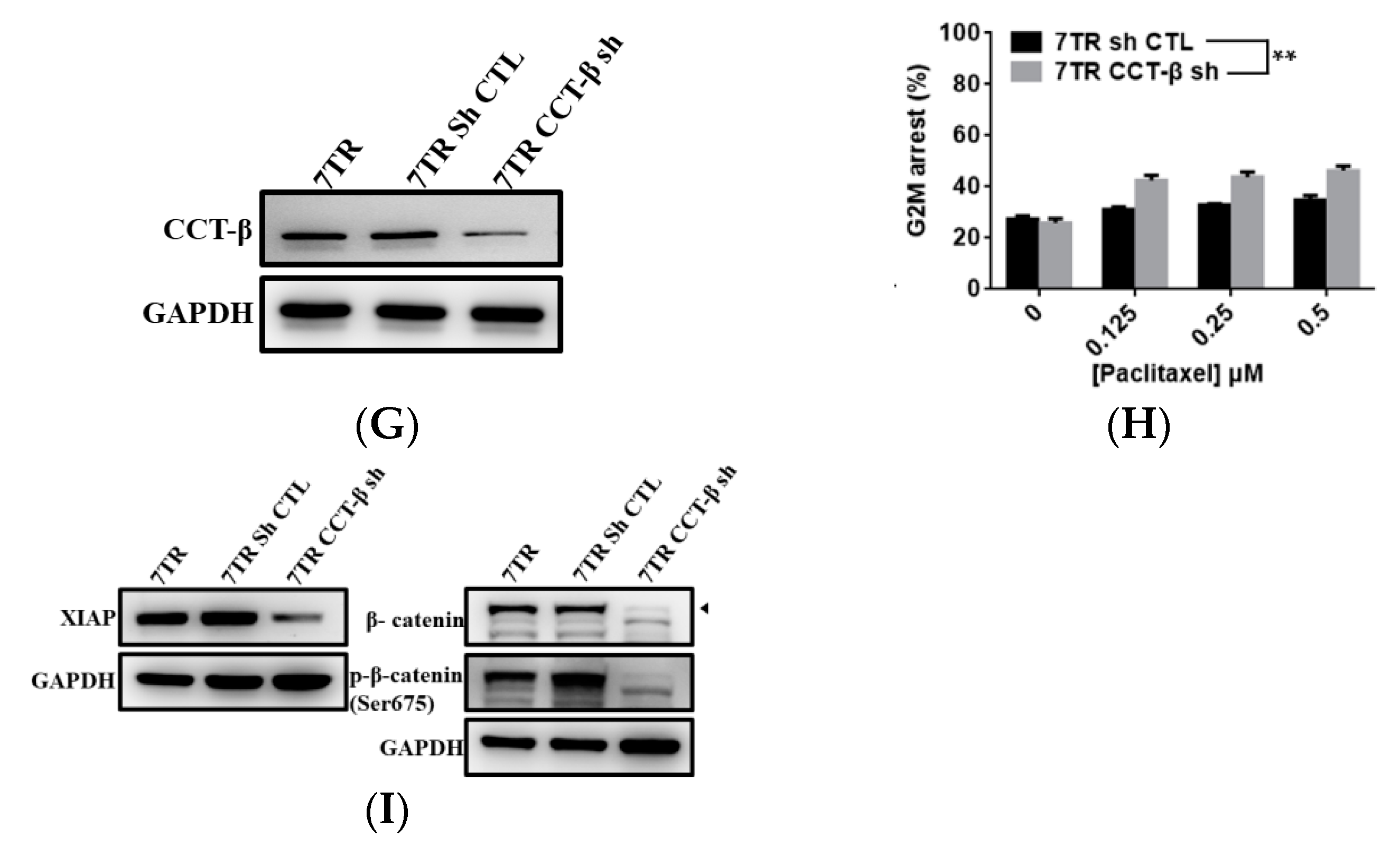
2.4. Correlation of CCT-β Expression with Chemoresistance in MCF-7 and 7TR
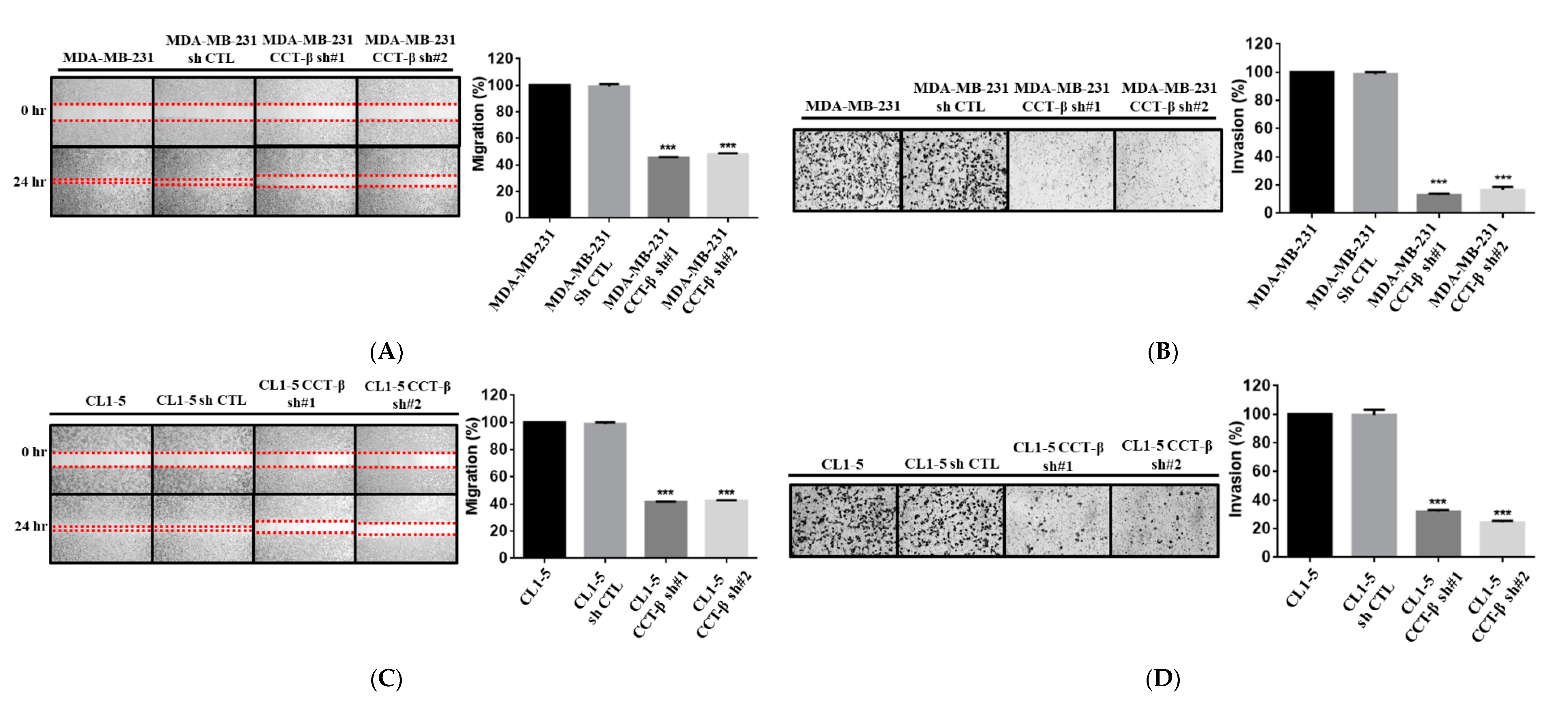
2.5. Knocking down CCT-β Inhibited the Invasion/Migration of MDA-MB-231 and CL1-5 Cells
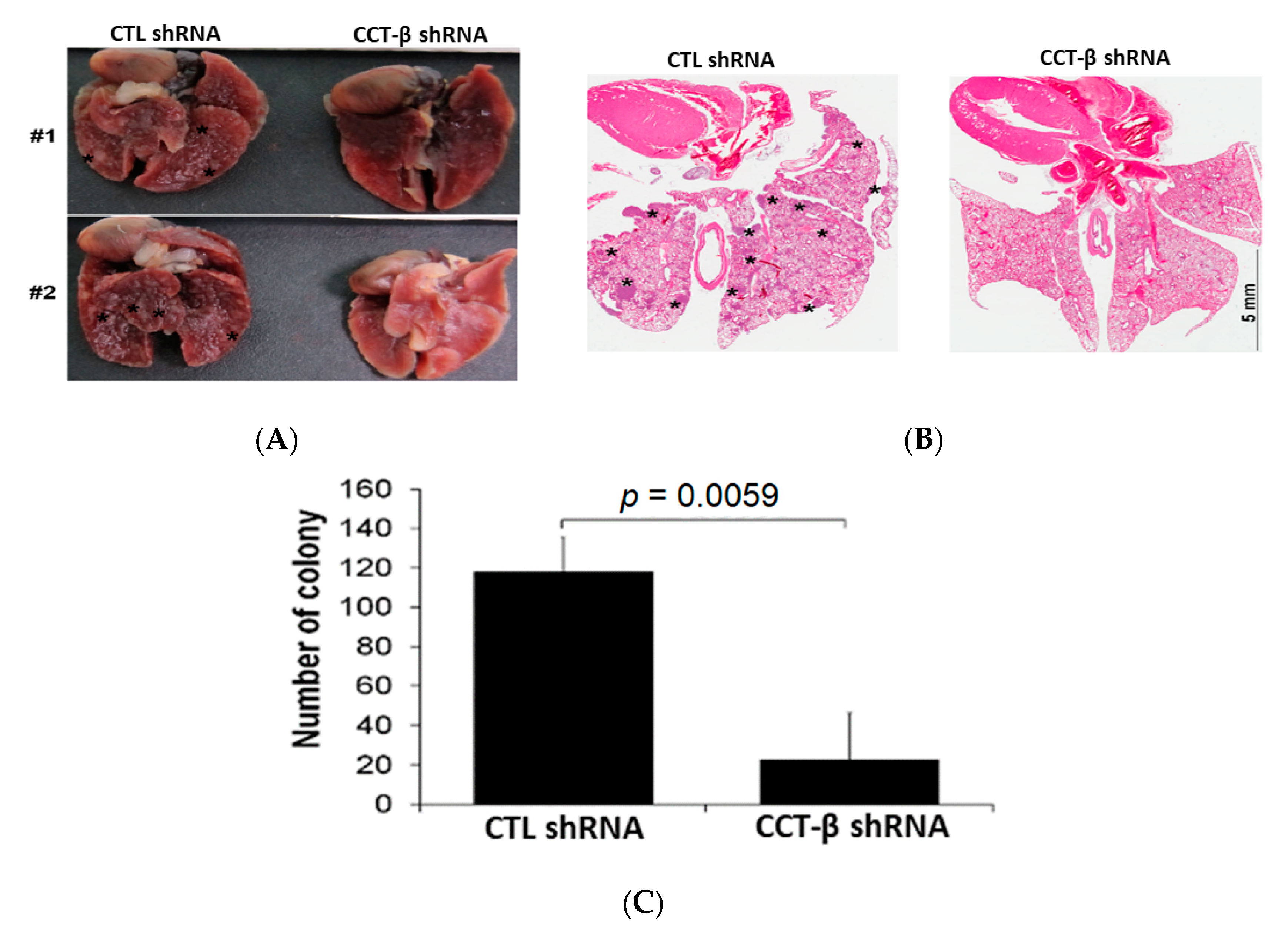
2.6. Knocking down CCT-β Inhibited MDA-MB-231 Cell Metastasis In Vivo
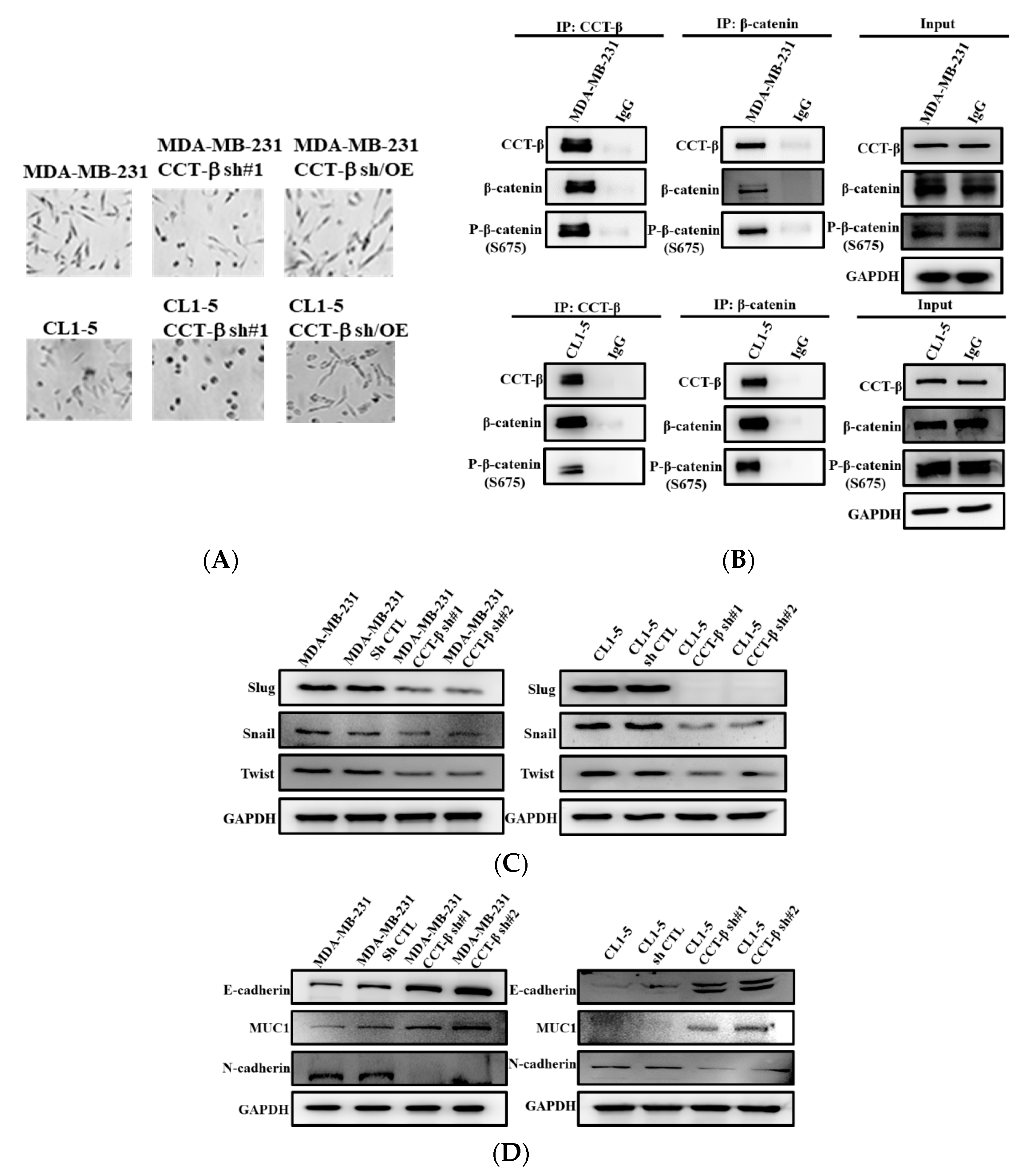
2.7. Knocking down CCT-β Altered Cell Morphology, Decreased the Levels of EMT Markers, and Inhibited the Activity and Expression of MMP2/9
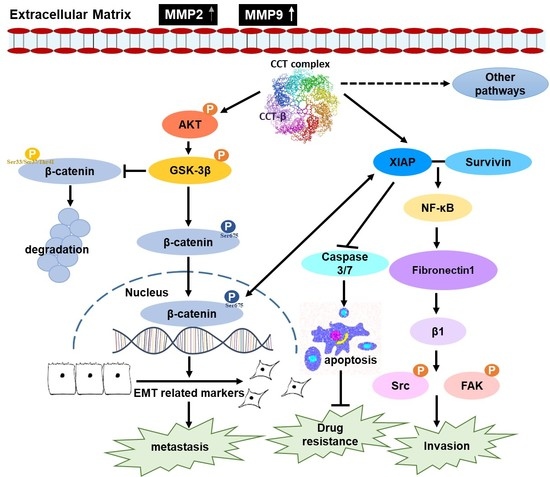
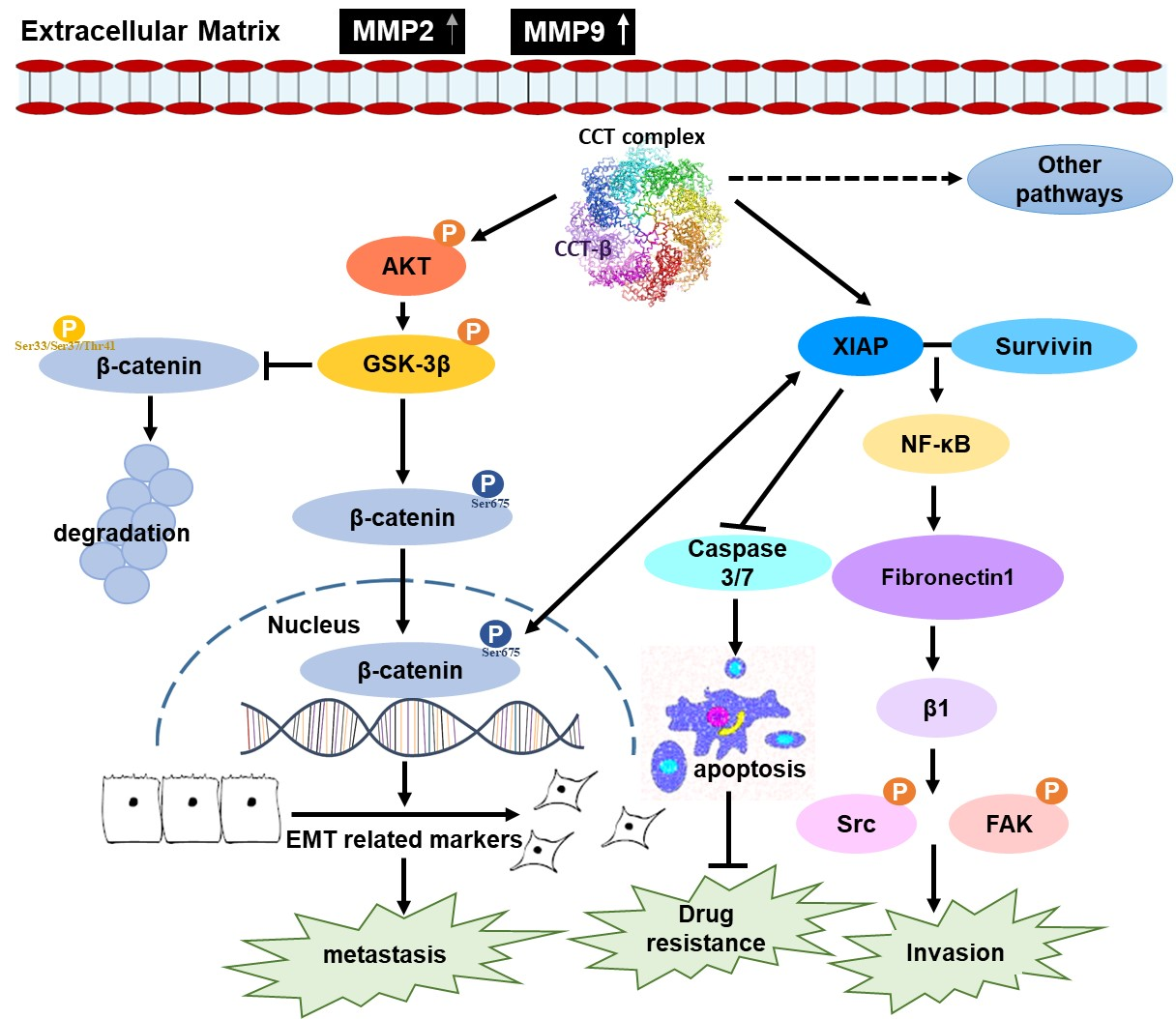
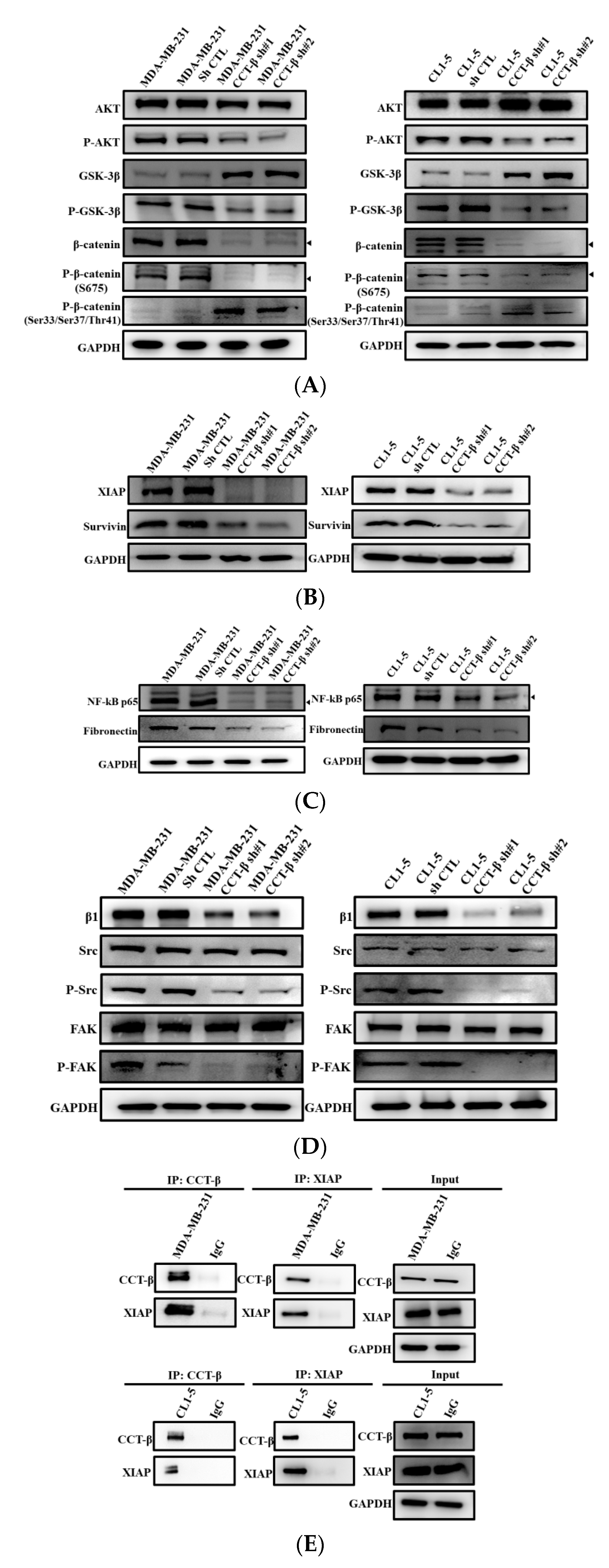
2.8. Knocking down CCT-β Inhibited Metastasis by Downregulating the AKT-GSK3β-β-catenin and XIAP-Survivin Pathways
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Collection of Clinical Samples and Immunohistochemistry
4.3. Cell Culture
4.4. Plasmids and Cell Transfection
4.5. Western Blot Analysis
4.6. Coimmunoprecipitation Assay
4.7. Flow Cytometry Analysis
4.8. MTT Assay
4.9. Wound-Healing Assay
4.10. Matrigel Invasion Assay
4.11. Animal Studies
4.12. Gelatin Zymography
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roh, S.H.; Kasembeli, M.; Bakthavatsalam, D.; Chiu, W.; Tweardy, D.J. Contribution of the type II Chaperonin, TRiC/CCT, to oncogenesis. Int. J. Mol. Sci. 2015, 16, 26706–26720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valli, J.; Grantham, J. The role of the molecular chaperone CCT in protein folding and mediation of cytoskeleton-associated processes: Implications for cancer cell biology. Cell Stress Chaperones 2019, 24, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llorca, O.; Martin-Benito, J.; Gomez-Puertas, P.; Ritco-Vonsovici, M.; Willison, K.R.; Carrascosa, J.L.; Valpuesta, J.M. Analysis of the interaction between the eukaryotic chaperonin CCT and its substrates actin and tubulin. J. Struct. Biol. 2001, 135, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brackley, K.I.; Grantham, J. Activities of the chaperonin containing TCP-1 (CCT): Implications for cell cycle progression and cytoskeletal organisation. Cell Stress Chaperones 2009, 14, 23–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melki, R.; Batelier, G.; Soulié, S.; Williams, R.C., Jr. Cytoplasmic chaperonin containing TCP-1: Structural and functional characterization. Biochemistry 1997, 36, 5817–5826. [Google Scholar] [CrossRef] [PubMed]
- Won, K.A.; Schumacher, R.J.; Farr, G.W.; Horwich, A.L.; Reed, S.I. Maturation of human cyclin E requires the function of eukaryotic chaperonin CCT. Mol. Cell Biol. 1998, 18, 7584–7589. [Google Scholar] [CrossRef] [Green Version]
- Hansen, W.J.; Ohh, M.; Moslehi, J.; Kondo, K.; Kaelin, W.G.; Welch, W.J. Diverse effects of mutations in exon II of the von Hippel-Lindau (VHL) tumor suppressor gene on the interaction of pVHL with the cytosolic chaperonin and pVHL-dependent ubiquitin ligase activity. Mol. Cell Biol. 2002, 22, 1947–1960. [Google Scholar] [CrossRef] [Green Version]
- Boudiaf-Benmammar, C.; Cresteil, T.; Melki, R. The cytosolic chaperonin CCT/TRiC and cancer cell proliferation. PLoS ONE 2013, 8, e60895. [Google Scholar] [CrossRef]
- Zhao, M.; Spiess, M.; Johansson, H.J.; Olofsson, H.; Hu, J.; Lehtiö, J.; Strömblad, S. Identification of the PAK4 interactome reveals PAK4 phosphorylation of N-WASP and promotion of Arp2/3-dependent actin polymerization. Oncotarget 2017, 8, 77061–77074. [Google Scholar] [CrossRef] [Green Version]
- Brackley, K.I.; Grantham, J. Interactions between the actin filament capping and severing protein gelsolin and the molecular chaperone CCT: Evidence for nonclassical substrate. Cell Stress Chaperones 2011, 16, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Trinidad, A.G.; Muller, P.A.; Cuellar, J.; Klejnot, M.; Nobis, M.; Valpuesta, J.M.; Vousden, K.H. Interaction of p53 with the CCT complex promotes protein folding and wild-type p53 activity. Mol. Cell 2013, 50, 805–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tracy, C.M.; Gray, A.J.; Cuéllar, J.; Shaw, T.S.; Howlett, A.C.; Taylor, R.M.; Prince, J.T.; Ahn, N.G.; Valpuesta, J.M.; Willardson, B.M. Programmed cell death protein 5 interacts with the cytosolic chaperonin containing tailless complex polypeptide 1 (CCT) to regulate β-tubulin folding. J. Biol. Chem. 2014, 289, 4490–4502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokota, S.; Yamamoto, Y.; Shimizu, K.; Momoi, H.; Kamikawa, T.; Yamaoka, Y.; Yanagi, H.; Yura, T.; Kubota, H. Increased expression of cytosolic chaperonin CCT in human hepatocellular and colonic carcinoma. Cell Stress Chaperones 2001, 6, 345–350. [Google Scholar] [CrossRef] [Green Version]
- Coghlin, C.; Carpenter, B.; Dundas, S.R.; Lawrie, L.C.; Telfer, C.; Murray, G.I. Characterization and over-expression of chaperonin t-complex proteins in colorectal cancer. J. Pathol. 2006, 210, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Yang, Z.L.; Yuan, Y.; Li, J.H.; Liang, L.F.; Zeng, G.X.; Chen, S.L. Clinicopathological features and CCT2 and PDIA2 expression in gallbladder squamous/adenosquamous carcinoma and gallbladder adenocarcinoma. World J. Surg. Oncol. 2013, 11, 143. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Yoon, S.O.; Kubota, K.; Mendoza, M.C.; Gygi, S.P.; Blenis, J. p90 ribosomal S6 kinase and p70 ribosomal S6 kinase link phosphorylation of the eukaryotic chaperonin containing TCP-1 to growth factor, insulin, and nutrient signaling. J. Biol. Chem. 2009, 284, 14939–14948. [Google Scholar] [CrossRef] [Green Version]
- Murphy, L.; Henry, M.; Meleady, P.; Clynes, M.; Keenan, J. Proteomic investigation of taxol and taxotere resistance and invasiveness in a squamous lung carcinoma cell line. Biochim. Biophys. Acta 2008, 1784, 1184–1191. [Google Scholar] [CrossRef]
- Di, M.M.; Della, C.A.; Cicchillitti, L.; Del, B.P.; Urbani, A.; Ferlini, C.; Scambia, G.; Donati, M.B.; Rotilio, D. A proteomic approach to paclitaxel chemoresistance in ovarian cancer cell lines. Biochim. Biophys. Acta 2009, 1794, 225–236. [Google Scholar]
- Lin, Y.F.; Tsai, W.P.; Liu, H.G.; Liang, P.H. Intracellular beta-tubulin/chaperonin containing TCP1-beta complex serves as a novel chemotherapeutic target against drug-resistant tumors. Cancer Res. 2009, 69, 6879–6888. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.F.; Lee, Y.F.; Liang, P.H. Targeting β-tubulin: CCT-β complexes incurs Hsp90- and VCP-related protein degradation and induces ER stress-associated apoptosis by triggering capacitative Ca2+ entry, mitochondrial perturbation and caspase overactivation. Cell Death Dis. 2012, 3, e434. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J.; Kumar, V.; Lin, Y.F.; Liang, P.H. Disrupting CCT-β: β-tubulin selectively kills CCT-β overexpressed cancer cells through MAPKs activation. Cell Death Dis. 2017, 8, e3052. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Chang, Y.J.; Kuo, Y.T.; Liang, P.H. Targeting β-tubulin/CCT-β complex induces apoptosis and suppresses migration and invasion of highly metastatic lung adenocarcinoma. Carcinogenesis 2020, 41, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Y.; Liu, F.C.; Chou, P.Y.; Chien, Y.C.; Chang, W.S.; Huang, G.J.; Wu, C.H.; Sheu, M.J. Ethanol extracts of fruiting bodies of Antrodia cinnamomea suppress CL1-5 human lung adenocarcinoma cells migration by inhibiting matrix metalloproteinase-2/9 through ERK, JNK, p38, and PI3K/Akt signaling pathways. Evid. Based Complement. Alternat. Med. 2012, 2012, 378415. [Google Scholar] [PubMed] [Green Version]
- Toth, M.; Sohail, A.; Fridman, R. Assessment of gelatinases (MMP-2 and MMP-9) by gelatin zymography. Methods Mol. Biol. 2012, 878, 121–135. [Google Scholar]
- Mehrotra, S.; Languino, L.R.; Raskett, C.M.; Mercurio, A.M.; Dohi, T.; Altieri, D.C. IAP regulation of metastasis. Cancer Cell 2010, 17, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Hehlgans, S.; Petraki, C.; Reichert, S.; Cordes, N.; Rödel, C.; Rödel, F. Double targeting of Survivin and XIAP radiosensitizes 3D grown human colorectal tumor cells and decreases migration. Radiother. Oncol. 2013, 108, 32–39. [Google Scholar] [CrossRef]
- Chai, J.; Shiozaki, E.; Srinivasula, S.M.; Wu, Q.; Datta, P.; Alnemri, E.S.; Shi, Y. Structural basis of caspase-7 inhibition by XIAP. Cell 2001, 104, 769–780. [Google Scholar] [CrossRef]
- Riedl, S.J.; Renatus, M.; Schwarzenbacher, R.; Zhou, Q.; Sun, C.; Fesik, S.W.; Liddington, R.C.; Salvesen, G.S. Structural basis for the inhibition of caspase-3 by XIAP. Cell 2001, 104, 791–800. [Google Scholar] [CrossRef]
- Shiozaki, E.N.; Chai, J.; Rigotti, D.J.; Riedl, S.J.; Li, P.; Srinivasula, S.M.; Alnemri, E.S.; Fairman, R.; Shi, Y. Mechanism of XIAP-mediated inhibition of caspase-9. Mol. Cell 2003, 11, 519–527. [Google Scholar] [CrossRef]
- Notarbartolo, M.; Cervello, M.; Poma, P.; Dusonchet, L.; Meli, M.; D’Alessandro, N. Expression of the IAPs in multidrug resistant tumor cells. Oncol. Rep. 2004, 11, 133–136. [Google Scholar] [CrossRef]
- Schimmer, A.D.; Welsh, K.; Pinilla, C.; Wang, Z.; Krajewska, M.; Bonneau, M.J.; Pedersen, I.M.; Kitada, S.; Scott, F.L.; Bailly-Maitre, B.; et al. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell 2004, 5, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, M.; Takano, M.; Iwaya, K.; Shinomiya, N.; Kato, M.; Aoyama, T.; Sasaki, N.; Goto, T.; Suzuki, A.; Hitrata, J.; et al. X-chromosome-linked inhibitor of apoptosis as a key factor for chemoresistance in clear cell carcinoma of the ovary. Br. J. Cancer 2014, 110, 2881–2886. [Google Scholar] [CrossRef] [PubMed]
- Nestal de Moraes, G.; Delbue, D.; Silva, K.L.; Robaina, M.C.; Khongkow, P.; Gomes, A.R.; Zona, S.; Crocamo, S.; Mencalha, A.L.; Magalhães, L.M.; et al. FOXM1 targets XIAP and Survivin to modulate breast cancer survival and chemoresistance. Cell Signal. 2015, 27, 2496–2505. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Meares, G.; Song, L.; Jope, R.S. XIAP associates with GSK3 and inhibits the promotion of intrinsic apoptotic signaling by GSK3. Cell Signal. 2009, 21, 1857–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, A.J.; Wallace, H.A.; Freeman, T.J.; Beauchamp, R.D.; Lee, L.A.; Lee, E. XIAP monoubiquitylates Groucho/TLE to promote canonical Wnt signaling. Mol. Cell 2012, 45, 619–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, V.H.; Hang, B.I.; Sawyer, L.M.; Neitzel, L.R.; Crispi, E.E.; Rose, K.L.; Popay, T.M.; Zhong, A.; Lee, L.A.; Tansey, W.P.; et al. Phosphorylation of XIAP at threonine 180 controls its activity in Wnt signaling. J. Cell Sci. 2018, 131, jcs210575. [Google Scholar] [CrossRef] [Green Version]
- Silva, K.L.; de Souza, P.S.; Nestal de Moraes, G.; Moellmann-Coelho, A.; Vasconcelos, F.D.C.; Maia, R.C. XIAP and P-glycoprotein co-expression is related to imatinib resistance in chronic myeloid leukemia cells. Leuk. Res. 2013, 37, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Nagano, H.; Takemasa, I. Activation of Wnt/β-catenin signaling pathway induces chemoresistance to interferon-α/5-fluorouracil combination therapy for hepatocellular carcinoma. Br. J. Cancer 2009, 100, 1647–1658. [Google Scholar] [CrossRef]
- Chikazawa, N.; Tanaka, H.; Tasaka, T.; Nakamura, M.; Tanaka, M.; Onishi, H.; Katano, M. Inhibition of Wnt signaling pathway decreases chemotherapy-resistant side-population colon cancer cells. Anticancer Res. 2010, 30, 2041–2048. [Google Scholar]
- Cao, J.; Ma, J.; Sun, L.; Li, J.; Qin, T.; Zhou, C.; Cheng, L.; Chen, K.; Qian, W.; Duan, W.; et al. Targeting glypican-4 overcomes 5-FU resistance and attenuates stem cell-like properties via suppression of Wnt/β-catenin pathway in pancreatic cancer cells. J. Cell Biochem. 2018, 119, 9498–9512. [Google Scholar] [CrossRef]
- Shen, D.Y.; Zhang, W.; Zeng, X.; Liu, C.Q. Inhibition of Wnt/β-catenin signaling downregulates P-glycoprotein and reverses multi-drug resistance of cholangiocarcinoma. Cancer Sci. 2013, 104, 1303–1308. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.J. Wnt signaling pathway in non-small cell lung cancer. J. Natl. Cancer Inst. 2014, 106, djt356. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Ashihara, E.; Maekawa, T. Targeting the Wnt/β-catenin signaling pathway in human cancers. Expert Opin. Ther. Targets 2011, 15, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.M.; O’Donovan, N.; Duffy, M.J. Survivin: A new target for anti-cancer therapy. Cancer Treat. Rev. 2009, 35, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Bian, F.; Zhang, X.; Qi, H.; Chuang, E.Y.; Pflugfelder, S.C.; Li, D.Q. The beta-catenin/Tcf4/survivin signaling maintains a less differentiated phenotype and high proliferative capacity of human corneal epithelial progenitor cells. Int. J. Biochem. Cell Biol. 2011, 43, 751–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenneth, N.S.; Duckett, C.S. IAP proteins: Regulators of cell migration and development. Curr. Opin. Cell Biol. 2012, 24, 871–875. [Google Scholar] [CrossRef]
- Huang, C.L.; Liu, D.; Ishikawa, S.; Nakashima, T.; Nakashima, N.; Yokomise, H.; Kadota, K.; Uenoc, M. Wnt1 overexpression promotes tumour progression in non-small cell lung cancer. Eur. J. Cancer 2008, 44, 2680–2688. [Google Scholar] [CrossRef]
- Teng, Y.; Wang, X.; Wang, Y.; Ma, D. Wnt/beta-catenin signaling regulates cancer stem cells in lung cancer A549 cells. Biochem. Biophys. Res. Commun. 2010, 392, 373–379. [Google Scholar] [CrossRef]
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/β-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef] [Green Version]
- Pacheco-Pinedo, E.C.; Durham, A.C.; Stewart, K.M.; Goss, A.M.; Lu, M.M.; Demayo, F.J.; Morrisey, E.E. Wnt/β-catenin signaling accelerates mouse lung tumorigenesis by imposing an embryonic distal progenitor phenotype on lung epithelium. J. Clin. Investig. 2011, 121, 1935–1945. [Google Scholar] [CrossRef] [Green Version]
- Prosperi, J.R.; Goss, K.H. A Wnt-ow of opportunity: Targeting the Wnt/β-catenin pathway in breast cancer. Curr. Drug Targets 2010, 11, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/β-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Geyer, F.C.; Lacroix-Triki, M.; Savage, K.; Arnedos, M.; Lambros, M.B.; MacKay, A.; Natrajan, R.; Reis-Filho, J.S. β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mol. Pathol. 2011, 24, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Bassiouni, R.; Nemec, K.N.; Iketani, A.; Flores, O.; Showalter, A.; Khaled, A.S.; Vishnubhotla, P.; Sprung, R.W., Jr.; Kaittanis, C.; Perez, J.M.; et al. Chaperonin containing TCP-1 protein level in breast cancer cells predicts therapeutic application of a cytotoxic peptide. Clin. Cancer Res. 2017, 22, 4366–4379. [Google Scholar] [CrossRef] [Green Version]
- Jan, Y.H.; Lai, T.C.; Yang, C.J.; Lin, Y.F.; Huang, M.S.; Hsiao, M. Adenylate kinase 4 modulates oxidative stress and stabilizes HIF-1α to drive lung adenocarcinoma metastasis. J. Hematol. Oncol. 2019, 12, 12. [Google Scholar] [CrossRef]
- Chu, Y.W.; Yang, P.C.; Yang, S.C.; Shyu, Y.C.; Hendrix, M.J.; Wu, R.; Wu, C.W. Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am. J. Respir. Cell Mol. Biol. 1997, 17, 353–360. [Google Scholar] [CrossRef] [Green Version]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, Y.-X.; Lin, Y.-F.; Chen, C.-L.; Huang, M.-S.; Hsiao, M.; Liang, P.-H. Chaperonin-Containing TCP-1 Promotes Cancer Chemoresistance and Metastasis through the AKT-GSK3β-β-Catenin and XIAP-Survivin Pathways. Cancers 2020, 12, 3865. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123865
Chang Y-X, Lin Y-F, Chen C-L, Huang M-S, Hsiao M, Liang P-H. Chaperonin-Containing TCP-1 Promotes Cancer Chemoresistance and Metastasis through the AKT-GSK3β-β-Catenin and XIAP-Survivin Pathways. Cancers. 2020; 12(12):3865. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123865
Chicago/Turabian StyleChang, Yun-Xun, Yuan-Feng Lin, Chi-Long Chen, Ming-Shyan Huang, Michael Hsiao, and Po-Huang Liang. 2020. "Chaperonin-Containing TCP-1 Promotes Cancer Chemoresistance and Metastasis through the AKT-GSK3β-β-Catenin and XIAP-Survivin Pathways" Cancers 12, no. 12: 3865. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12123865