Protein Expression in Metastatic Melanoma and the Link to Disease Presentation in a Range of Tumor Phenotypes

 , , , , , ,
, , , , , , 
Abstract
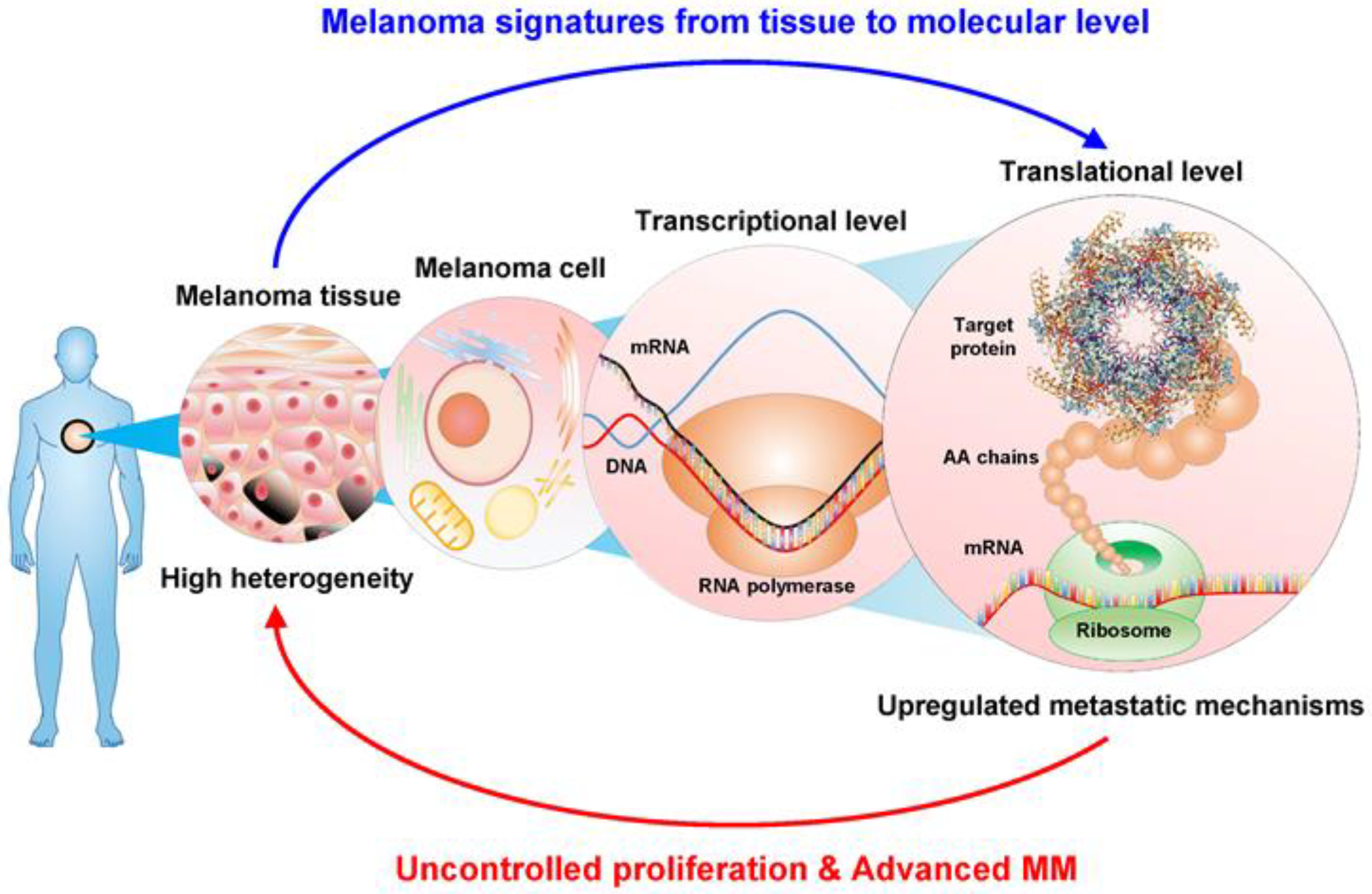
:1. Introduction
2. Disease Presentation of MM Incorporating Histopathology
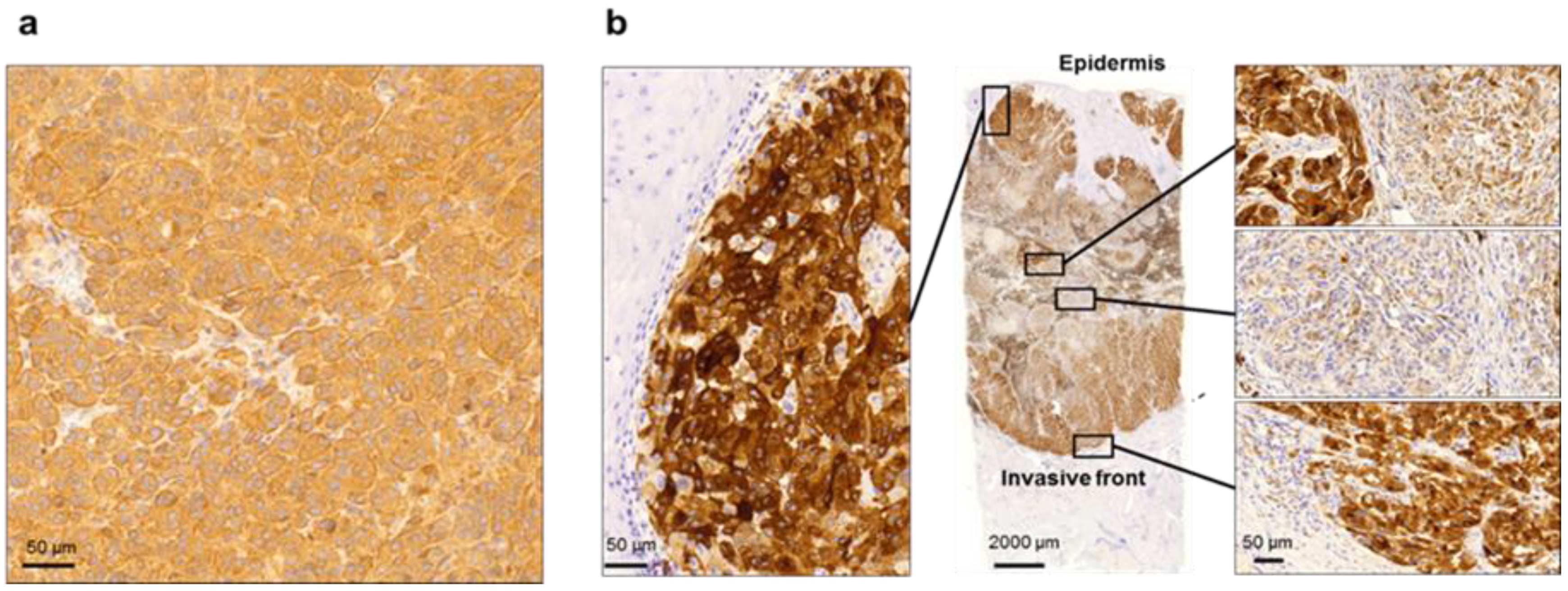
2.1. Disease Presentation with BRAFV600E in MM Tissue
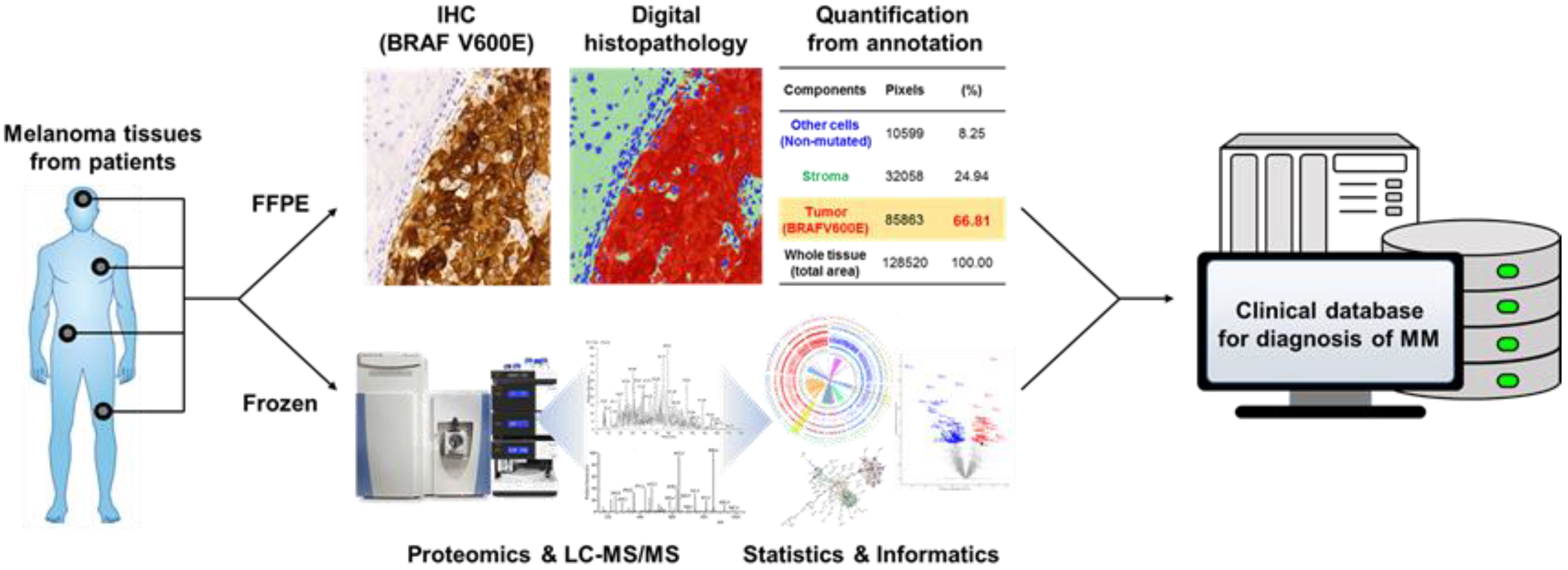
2.2. Comparison of Fresh Frozen and FFPE Metastatic Tumor Tissue Images Indicating the Associated Challenges of Histopathological Evaluation of MM
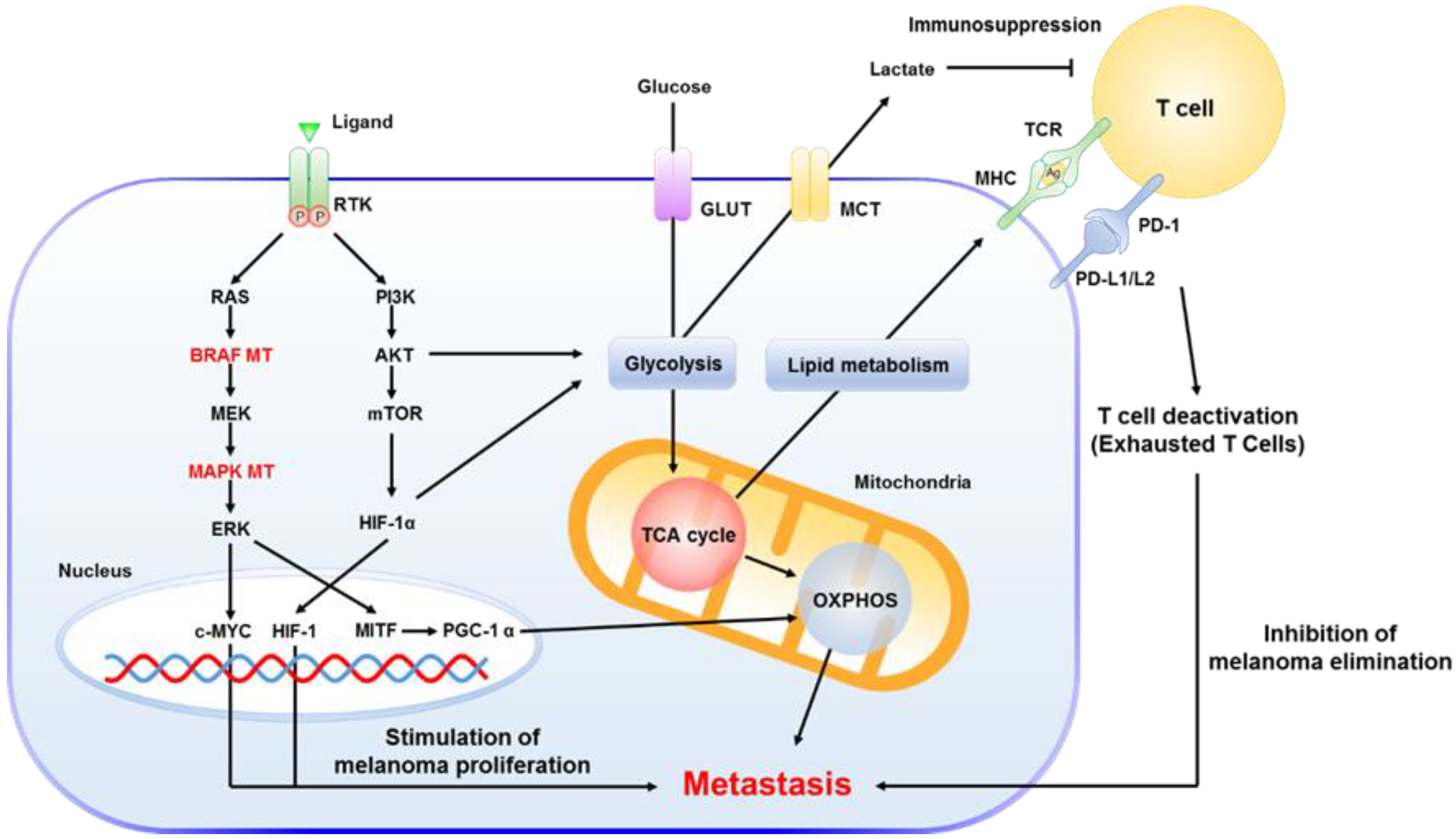
3. Metastatic Mechanisms in Melanoma
3.1. Targeted Therapies to Inhibit the Mutated BRAF Signaling Pathway in MM
3.2. The Key Role of Mitochondria in MM
3.3. Drug-Treatment Impacts from Protein Expression in Melanoma Patients
3.4. Immunotherapy with Checkpoint Inhibitors
4. Therapeutic Opportunities from New Approaches for MM
4.1. Drug Resistance in MM
4.2. Combinatorial Treatment and Promising Therapies in MM
4.2.1. Triple Combination of Checkpoint Inhibitor with Therapies Targeting MEK and BRAF
4.2.2. Combination of the HDAC Inhibitor on MM
4.2.3. New Effective Approaches to Evaluate MM
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Erdmann, F.; Lortet-Tieulent, J.; Schüz, J.; Zeeb, H.; Greinert, R.; Breitbart, E.W.; Bray, F. International trends in the incidence of malignant melanoma 1953–2008—Are recent generations at higher or lower risk? Int. J. Cancer 2013, 132, 385–400. [Google Scholar] [CrossRef]
- Grzywa, T.M.; Paskal, W.; Włodarski, P.K. Intratumor and intertumor heterogeneity in melanoma. Transl. Oncol. 2017, 10, 956–975. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghì, A.; Spandidos, D.A.; Mccubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statistical Databases—Socialstyrelsen. Available online: https://www.socialstyrelsen.se/en/statistics-and-data/statistics/statistical-databases/ (accessed on 14 February 2020).
- Mcgranahan, N.; Swanton, C. Cancer cell perspective biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef]
- Finn, L.; Markovic, S.N.; Joseph, R.W. Therapy for metastatic melanoma: The past, present, and future. BMC Med. 2012, 10, 23. [Google Scholar] [CrossRef] [Green Version]
- Tarhini, A.A.; Garwala, S.S.A. Cutaneous melanoma: Available therapy for metastatic disease. Dermatol. Ther. 2006, 19, 19–25. [Google Scholar] [CrossRef]
- Ko, J.M.; Fisher, D.E. A new era: Melanoma genetics and therapeutics. J. Pathol. 2011, 223, 241–250. [Google Scholar] [CrossRef]
- Pasquali, S.; Hadjinicolaou, A.V.; Chiarion Sileni, V.; Rossi, C.R.; Mocellin, S. Systemic treatments for metastatic cutaneous melanoma. Cochrane Database Syst. Rev. 2018. [Google Scholar] [CrossRef]
- Domingues, B.; Lopes, J.; Soares, P.; Populo, H. Melanoma treatment in review. ImmunoTargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Das, M.; Liu, Y.; Huang, L. Targeted drug delivery to melanoma. Adv. Drug Deliv. Rev. 2018, 127, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, S.; Seidel, D.; Jahnke, H.G.; Eichler, M.; Simon, J.C.; Robitzki, A.A. Induced cross-resistance of BRAF V600E melanoma cells to standard chemotherapeutic dacarbazine after chronic PLX4032 treatment. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.; Wellbrock, C.; Marais, R. Melanoma biology and new targeted therapy. Nature 2007, 445, 851–857. [Google Scholar] [CrossRef] [PubMed]
- Dimitriou, F.; Krattinger, R.; Ramelyte, E.; Barysch, M.J.; Micaletto, S.; Dummer, R.; Goldinger, S.M. The world of melanoma: Epidemiologic, genetic, and anatomic differences of melanoma across the globe. Curr. Oncol. Rep. 2018, 20, 87. [Google Scholar] [CrossRef] [PubMed]
- Tronnier, M.; Mitteldorf, C. Treating advanced melanoma: Current insights and opportunities. Cancer Manag. Res. 2014, 6, 349–356. [Google Scholar] [CrossRef] [Green Version]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [Green Version]
- Poulikakos, P.I.; Zhang, C.; Bollag, G.; Shokat, K.M.; Rosen, N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 2010, 464, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Prasad, C.P.; Mohapatra, P.; Andersson, T. Therapy for BRAFI-resistant melanomas: Is WNT5A the answer? Cancers 2015, 7, 1900–1924. [Google Scholar] [CrossRef] [Green Version]
- Gray-Schopfer, V.C.; Karasarides, M.; Hayward, R.; Marais, R. Tumor necrosis factor-α blocks apoptosis in melanoma cells when BRAF signaling is inhibited. Cancer Res. 2007, 67, 122–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Wilmott, J.S.; Menzies, A.M.; Haydu, L.E.; Capper, D.; Preusser, M.; Zhang, Y.E.; Thompson, J.F.; Kefford, R.F.; Von Deimling, A.; Scolyer, R.A.; et al. BRAF V600E protein expression and outcome from BRAF inhibitor treatment in BRAF V600E metastatic melanoma. Br. J. Cancer 2013, 108, 924–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearlstein, M.V.; Zedek, D.C.; Ollila, D.W.; Treece, A.; Gulley, M.L.; Groben, P.A.; Thomas, N.E. Validation of the VE1 immunostain for the BRAF V600E mutation in melanoma. J. Cutan. Pathol. 2014, 41, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Schirosi, L.; Strippoli, S.; Gaudio, F.; Graziano, G.; Popescu, O.; Guida, M.; Simone, G.; Mangia, A. Is immunohistochemistry of BRAF V600E useful as a screening tool and during progression disease of melanoma patients? BMC Cancer 2016, 16, 905. [Google Scholar] [CrossRef] [Green Version]
- Betancourt, L.H.; Szasz, A.M.; Kuras, M.; Murillo, J.R.; Sugihara, Y.; Pla, I.; Horvath, Z.; Pawłowski, K.; Rezeli, M.; Miharada, K.; et al. The hidden story of heterogeneous B-raf V600E mutation quantitative protein expression in metastatic melanoma—Association with clinical outcome and tumor phenotypes. Cancers 2019, 11, 1981. [Google Scholar] [CrossRef]
- Gustafsson, O.J.R.; Arentz, G.; Hoffmann, P. Proteomic developments in the analysis of formalin-fixed tissue. Biochim. Biophys. Acta Proteins Proteom. 2015, 1854, 559–580. [Google Scholar] [CrossRef]
- O’Rourke, M.B.; Padula, M.P. Analysis of formalin-fixed, paraffin-embedded (FFPE) tissue via proteomic techniques and misconceptions of antigen retrieval. Biotechniques 2016, 60, 229–238. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.S.; Mcconechy, M.K.; Cochrane, D.R.; Nazeran, T.; Karnezis, A.N.; Huntsman, D.G.; Morin, G.B. Quantitative profiling of single formalin fixed tumour sections: Proteomics for translational research. Sci. Rep. 2016, 6, 34949. [Google Scholar] [CrossRef]
- Föll, M.C.; Fahrner, M.; Oria, V.O.; Kühs, M.; Biniossek, M.L.; Werner, M.; Bronsert, P.; Schilling, O. Reproducible proteomics sample preparation for single FFPE tissue slices using acid-labile surfactant and direct trypsinization. Clin. Proteom. 2018, 15, 11. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Weiss, T.; Zhang, Q.; Sun, R.; Wang, B.; Yi, X.; Wu, Z.; Gao, H.; Cai, X.; Ruan, G.; et al. High-throughput proteomic analysis of FFPE tissue samples facilitates tumor stratification. Mol. Oncol. 2019, 13, 2305–2328. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Kuras, M.; Betancourt, L.H.; Rezeli, M.; Rodriguez, J.; Szasz, M.; Zhou, Q.; Miliotis, T.; Andersson, R.; Marko-Varga, G. Assessing automated sample preparation technologies for high-throughput proteomics of frozen well characterized tissues from Swedish biobanks. J. Proteome Res. 2019, 18, 548–556. [Google Scholar] [CrossRef]
- Murillo, J.R.; Kuras, M.; Rezeli, M.; Milliotis, T.; Betancourt, L.; Marko-Varga, G. Automated phosphopeptide enrichment from minute quantities of frozen malignant melanoma tissue. PLoS ONE 2018, 13, e0208562. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, A.; Kuras, M.; Murillo, J.R.; Pla, I.; Pawlowski, K.; Szasz, A.M.; Gil, J.; Nogueira, F.C.S.; Perez-Riverol, Y.; Eriksson, J.; et al. Novel functional proteins coded by the human genome discovered in metastases of melanoma patients. Cell Biol. Toxicol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Sharma, A.; Peng, H.H.; Robertson, G.; Dong, C. Targeting mutant (V600E) B-Raf in melanoma interrupts immunoediting of leukocyte functions and melanoma extravasation. Cancer Res. 2007, 67, 5814–5820. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.S.; Madhunapantula, S.V.; Robertson, G.P.; Drabick, J.J. Current and future trials of targeted therapies in cutaneous melanoma. In Impact of Genetic Targets on Cancer Therapy; Springer: New York, NY, USA, 2013; pp. 223–255. [Google Scholar]
- Boyle, G.M.; Pedley, J.; Martyn, A.C.; Banducci, K.J.; Strutton, G.M.; Brown, D.A.; Breit, S.N.; Parsons, P.G. Macrophage inhibitory cytokine-1 is overexpressed in malignant melanoma and is associated with tumorigenicity. J. Investig. Dermatol. 2009, 129, 383–391. [Google Scholar] [CrossRef]
- Eisen, T.; Ahmad, T.; Flaherty, K.T.; Gore, M.; Kaye, S.; Marais, R.; Gibbens, I.; Hackett, S.; James, M.; Schuchter, L.M.; et al. Sorafenib in advanced melanoma: A Phase II randomised discontinuation trial analysis. Br. J. Cancer 2006, 95, 581–586. [Google Scholar] [CrossRef] [Green Version]
- Jilaveanu, L.; Zito, C.; Lee, S.J.; Nathanson, K.L.; Camp, R.L.; Rimm, D.L.; Flaherty, K.T.; Kluger, H.M. Expression of sorafenib targets in melanoma patients treated with carboplatin, paclitaxel and sorafenib. Clin. Cancer Res. 2009, 15, 1076–1085. [Google Scholar] [CrossRef] [Green Version]
- Mattia, G.; Puglisi, R.; Ascione, B.; Malorni, W.; Carè, A.; Matarrese, P. Cell death-based treatments of melanoma:conventional treatments and new therapeutic strategies review-Article. Cell Death Dis. 2018, 9, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Bröcker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Karaszewska, B.; Schachter, J.; Rutkowski, P.; Mackiewicz, A.; Stroiakovski, D.; Lichinitser, M.; Dummer, R.; Grange, F.; Mortier, L.; et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N. Engl. J. Med. 2015, 372, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-year outcomes with dabrafenib plus trametinib in metastatic melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Pimiento, J.M.; Larkin, E.M.; Smalley, K.S.; Wiersma, G.L.; Monks, N.R.; Fedorenko, I.V.; Peterson, C.A.; Nickoloff, B.J. Melanoma genotypes and phenotypes get personal. Lab. Investig. 2013, 93, 858–867. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef]
- Renner, K.; Singer, K.; Koehl, G.E.; Geissler, E.K.; Peter, K.; Siska, P.J.; Kreutz, M. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front. Immunol. 2017, 8, 248. [Google Scholar] [CrossRef] [Green Version]
- Walenta, S.; Wetterling, M.; Lehrke, M.; Schwickert, G.; Sundfør, K.; Rofstad, E.K.; Mueller-Klieser, W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000, 60, 916–921. [Google Scholar]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018, 27, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Betancourt, L.H.; Pla, I.; Sanchez, A.; Appelqvist, R.; Miliotis, T.; Kuras, M.; Oskolas, H.; Kim, Y.; Horvath, Z.; et al. Clinical protein science in translational medicine targeting malignant melanoma. Cell Biol. Toxicol. 2019, 35, 293–332. [Google Scholar] [CrossRef] [Green Version]
- Yuan, P.; Ito, K.; Perez-Lorenzo, R.; Del Guzzo, C.; Lee, J.H.; Shen, C.H.; Bosenberg, M.W.; McMahon, M.; Cantley, L.C.; Zheng, B. Phenformin enhances the therapeutic benefit of BRAFV600E inhibition in melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18226–18231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, G.M.; Vashisht Gopal, Y.N.; McQuade, J.L.; Peng, W.; DeBerardinis, R.J.; Davies, M.A. Metabolic strategies of melanoma cells: Mechanisms, interactions with the tumor microenvironment, and therapeutic implications. Pigment Cell Melanoma Res. 2018, 31, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Theodosakis, N.; Micevic, G.; Kelly, D.P.; Bosenberg, M. Mitochondrial function in melanoma. Arch. Biochem. Biophys. 2014, 563, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Ortenberg, R.; Varanasi, S.K.; Mangalhara, K.C.; Mardamshina, M.; Markovits, E.; Baruch, E.N.; Tripple, V.; Arama-Chayoth, M.; Greenberg, E.; et al. Proteomics of melanoma response to immunotherapy reveals mitochondrial dependence. Cell 2019, 179, 236–250. [Google Scholar] [CrossRef]
- Eton, O.; Legha, S.S.; Bedikian, A.Y.; Lee, J.J.; Buzaid, A.C.; Hodges, C.; Ring, S.E.; Papadopoulos, N.E.; Plager, C.; East, M.J.; et al. Sequential biochemotherapy versus chemotherapy for metastatic melanoma: Results from a phase III randomized trial. J. Clin. Oncol. 2002, 20, 2045–2052. [Google Scholar] [CrossRef]
- Azimi, A.; Pernemalm, M.; Frostvik Stolt, M.; Hansson, J.; Lehtiö, J.; Egyházi Brage, S.; Hertzman Johansson, C. Proteomics analysis of melanoma metastases: Association between S100A13 expression and chemotherapy resistance. Br. J. Cancer 2014, 110, 2489–2495. [Google Scholar] [CrossRef] [Green Version]
- Marko-Varga, G.; Végvári, Á.; Welinder, C.; Lindberg, H.; Rezeli, M.; Edula, G.; Svensson, K.J.; Belting, M.; Laurell, T.; Fehniger, T.E. Standardization and utilization of biobank resources in clinical protein science with examples of emerging applications. J. Proteome Res. 2012, 11, 5124–5134. [Google Scholar] [CrossRef] [PubMed]
- Abdi, H.; Williams, L.J.; Valentin, D. Multiple factor analysis: Principal component analysis for multitable and multiblock data sets. Wiley Interdiscip. Rev. Comput. Stat. 2013, 5, 149–179. [Google Scholar] [CrossRef]
- Betancourt, L.H.; Pawłowski, K.; Eriksson, J.; Szasz, A.M.; Mitra, S.; Pla, I.; Welinder, C.; Ekedahl, H.; Broberg, P.; Appelqvist, R.; et al. Improved survival prognostication of node-positive malignant melanoma patients utilizing shotgun proteomics guided by histopathological characterization and genomic data. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Coit, D.G.; Andtbacka, R.; Anker, C.J.; Bichakjian, C.K.; Carson, W.E.; Daud, A.; Dilawari, R.A.; DiMaio, D.; Guild, V.; Halpern, A.C.; et al. Melanoma. JNCCN J. Natl. Compr. Cancer Netw. 2012, 10, 366–400. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Dunn, G.P.; Okada, H. Principles of immunology and its nuances in the central nervous system. Neuro-Oncology 2015, 17, vii3–vii8. [Google Scholar] [CrossRef] [Green Version]
- Luke, J.J.; Flaherty, K.T.; Ribas, A.; Long, G.V. Targeted agents and immunotherapies: Optimizing outcomes in melanoma. Nat. Rev. Clin. Oncol. 2017, 14, 463–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, G.J.; Bi, W.L.; Wu, W.W.; Johanns, T.M.; Dunn, G.P.; Dunn, I.F. Management of intracranial melanomas in the era of precision medicine. Oncotarget 2017, 8, 89326–89347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khair, D.O.; Bax, H.J.; Mele, S.; Crescioli, S.; Pellizzari, G.; Khiabany, A.; Nakamura, M.; Harris, R.J.; French, E.; Hoffmann, R.M.; et al. Combining immune checkpoint inhibitors: Established and emerging targets and strategies to improve outcomes in melanoma. Front. Immunol. 2019, 10, 453. [Google Scholar] [CrossRef] [Green Version]
- Ascierto, P.A.; Ferrucci, P.F.; Fisher, R.; Del Vecchio, M.; Atkinson, V.; Schmidt, H.; Schachter, J.; Queirolo, P.; Long, G.V.; Di Giacomo, A.M.; et al. Dabrafenib, trametinib and pembrolizumab or placebo in BRAF-mutant melanoma. Nat. Med. 2019, 25, 941–946. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; MacConaill, L.E.; Hahn, W.C.; et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [Green Version]
- Trunzer, K.; Pavlick, A.C.; Schuchter, L.; Gonzalez, R.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; Kim, K.B.; Weber, J.S.; et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J. Clin. Oncol. 2013, 31, 1767–1774. [Google Scholar] [CrossRef]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Pelle, P.D.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, R.W.; Barbie, D.A. Refining targeted therapy opportunities for BRAF-mutant melanoma. Cancer Discov. 2017, 7, 799–801. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S.R.; Theurillat, J.P.; Van Allen, E.; Wagle, N.; Hsiao, J.; Cowley, G.S.; Schadendorf, D.; Root, D.E.; Garraway, L.A. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013, 3, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Moriceau, G.; Kong, X.; Lee, M.K.; Lee, H.; Koya, R.C.; Ng, C.; Chodon, T.; Scolyer, R.A.; Dahlman, K.B.; et al. Melanoma whole-exome sequencing identifies V600E B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat. Commun. 2012, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannessen, C.M.; Boehm, J.S.; Kim, S.Y.; Thomas, S.R.; Wardwell, L.; Johnson, L.A.; Emery, C.M.; Stransky, N.; Cogdill, A.P.; Barretina, J.; et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010, 468, 968–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef] [Green Version]
- Poulikakos, P.I.; Persaud, Y.; Janakiraman, M.; Kong, X.; Ng, C.; Moriceau, G.; Shi, H.; Atefi, M.; Titz, B.; Gabay, M.T.; et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 2011, 480, 387–390. [Google Scholar] [CrossRef] [Green Version]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef] [Green Version]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 2016, 167, 1540–1554. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, T.S.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of resistance to PD-1 and PD-L1 blockade. Cancer J. (United States) 2018, 24, 47–53. [Google Scholar] [CrossRef]
- Gao, J.; Shi, L.Z.; Zhao, H.; Chen, J.; Xiong, L.; He, Q.; Chen, T.; Roszik, J.; Bernatchez, C.; Woodman, S.E.; et al. Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016, 167, 397–404. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Little, A.S.; Smith, P.D.; Cook, S.J. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene 2013, 32, 1207–1215. [Google Scholar] [CrossRef]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Larkin, J.; Ascierto, P.A.; Dréno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandalà, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [Green Version]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hu-Lieskovan, S.; Mok, S.; Homet Moreno, B.; Tsoi, J.; Robert, L.; Goedert, L.; Pinheiro, E.M.; Koya, R.C.; Graeber, T.G.; Comin-Anduix, B.; et al. Improved antitumor activity of immunotherapy with BRAF and MEK inhibitors in BRAFV600E melanoma. Sci. Transl. Med. 2015, 7, 279ra41. [Google Scholar] [CrossRef] [Green Version]
- Homet Moreno, B.; Mok, S.; Comin-Anduix, B.; Hu-Lieskovan, S.; Ribas, A. Combined treatment with dabrafenib and trametinib with immune-stimulating antibodies for BRAF mutant melanoma. Oncoimmunology 2016, 5, e105212. [Google Scholar] [CrossRef] [Green Version]
- Deken, M.A.; Gadiot, J.; Jordanova, E.S.; Lacroix, R.; Van Gool, M.; Kroon, P.; Pineda, C.; Geukes Foppen, M.H.; Scolyer, R.; Song, J.Y.; et al. Targeting the MAPK and PI3K pathways in combination with PD1 blockade in melanoma. Oncoimmunology 2016, 5, e1238557. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Lawrence, D.; Atkinson, V.; Agarwal, S.; Miller, W.H.; Carlino, M.S.; Fisher, R.; Long, G.V.; Hodi, F.S.; Tsoi, J.; et al. Combined BRAF and MEK inhibition with PD-1 blockade immunotherapy in BRAF-mutant melanoma. Nat. Med. 2019, 25, 936–940. [Google Scholar] [CrossRef]
- Sullivan, R.J.; Hamid, O.; Gonzalez, R.; Infante, J.R.; Patel, M.R.; Hodi, F.S.; Lewis, K.D.; Tawbi, H.A.; Hernandez, G.; Wongchenko, M.J.; et al. Atezolizumab plus cobimetinib and vemurafenib in BRAF-mutated melanoma patients. Nat. Med. 2019, 25, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Rozeman, E.A.; Blank, C.U. Combining checkpoint inhibition and targeted therapy in melanoma. Nat. Med. 2019, 25, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Reale, A.; Vitiello, A.; Conciatori, V.; Parolin, C.; Calistri, A.; Palù, G. Perspectives on immunotherapy via oncolytic viruses. Infect. Agent. Cancer 2019, 14, 5. [Google Scholar] [CrossRef] [Green Version]
- Lugowska, I.; Teterycz, P.; Rutkowski, P. Immunotherapy of melanoma. Wspolczesna Onkol. 2017, 2, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Rothermel, L.D.; Zager, J.S. Engineered oncolytic viruses to treat melanoma: Where are we now and what comes next? Expert Opin. Biol. Ther. 2018, 18, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Bishayee, A.; Pandey, A.K. Targeting histone deacetylases with natural and synthetic agents: An emerging anticancer strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics combination therapies: An overview of the role of HDACs in cancer immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.M.; Sodré, A.L.; Villagra, A.; Sarnaik, A.; Sotomayor, E.M.; Weber, J. HDAC inhibition upregulates PD-1 ligands in melanoma and augments immunotherapy with PD-1 blockade. Cancer Immunol. Res. 2015, 3, 1375–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzone, R.; Zwergel, C.; Mai, A.; Valente, S. Epi-drugs in combination with immunotherapy: A new avenue to improve anticancer efficacy. Clin. Epigenet. 2017, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Orillion, A.; Pili, R. Histone deacetylase inhibitors as immunomodulators in cancer therapeutics. Epigenomics 2016, 8, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Kirkwood, J.; Dent, P. HDAC inhibitors enhance the immunotherapy response of melanoma cells. Oncotarget 2017, 8, 83155–83170. [Google Scholar] [CrossRef] [Green Version]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Agarwala, S.S.; Moschos, S.J.; Johnson, M.L.; Opyrchal, M.; Gabrilovich, D.; Danaher, P.; Wang, F.; Brouwer, S.; Ordentlich, P.; Sankoh, S.; et al. Efficacy and safety of entinostat (ENT) and pembrolizumab (PEMBRO) in patients with melanoma progressing on or after a PD-1/L1 blocking antibody. J. Clin. Oncol. 2018, 36, 9530. [Google Scholar] [CrossRef]
- Azad, N.S.; Shirai, K.; McRee, A.J.; Opyrchal, M.; Johnson, D.B.; Ordentlich, P.; Brouwer, S.; Sankoh, S.; Schmidt, E.V.; Meyers, M.L.; et al. ENCORE 601: A phase 2 study of entinostat in combination with pembrolizumab in patients with microsatellite stable metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 3557. [Google Scholar] [CrossRef]
- Maertens, O.; Kuzmickas, R.; Manchester, H.E.; Emerson, C.E.; Gavin, A.G.; Guild, C.J.; Wong, T.C.; De Raedt, T.; Bowman-Colin, C.; Hatchi, E.; et al. MAPK pathway suppression unmasks latent dna repair defects and confers a chemical synthetic vulnerability in BRAF-, NRAS-, and NF1-mutant melanomas. Cancer Discov. 2019, 9, 526–545. [Google Scholar] [CrossRef] [Green Version]
- Price, J.C.; Guan, S.; Burlingame, A.; Prusiner, S.B.; Ghaemmaghami, S. Analysis of proteome dynamics in the mouse brain. Proc. Natl. Acad. Sci. USA 2010, 107, 14508–14513. [Google Scholar] [CrossRef] [Green Version]
- Hanash, S.; Taguchi, A. The grand challenge to decipher the cancer proteome. Nat. Rev. Cancer 2010, 10, 652–660. [Google Scholar] [CrossRef]
- Wulfkuhle, J.D.; Liotta, L.A.; Petricoin, E.F. Proteomic applications for the early detection of cancer. Nat. Rev. Cancer 2003, 3, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef]
- Moscow, J.A.; Fojo, T.; Schilsky, R.L. The evidence framework for precision cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, H.; Trocoli Drakensjö, I.R.; Girnita, A. Personalized medicine in malignant melanoma: Towards patient tailored treatment. Front. Oncol. 2018, 8, 202. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Categories | Clinical Information | Histopathological Information (Average) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Code | Age of Diagnosis | Gender | Disease Stage | Treatment | Connective Tissue Content | Lymphatic Score | Tumor Cells Content | Adjacent Lymph Node Area | Pigmentation Score | Predominant Cytoplasm | Lymphatic Distribution | Necrosis Content | Lymphocyte Density |
| MM16 | 55 | Female | 4 | Nontreated | 9.8 | 3 | 53.5 | 1.8 | 3 | 1 | 2 | 35 | 1 |
| MM23 | 71 | Female | 4 | Nontreated | 19 | 3.7 | 50.3 | 30.7 | 1 | 2 | 1.7 | 0 | 2 |
| MM59 | 38 | Female | 4 | Nontreated | 12.5 | 2 | 62 | 0 | 2 | 2 | 1 | 25.5 | 1 |
| MM65 | 59 | Female | 4 | Nontreated | 11.7 | 2 | 67.3 | 0 | 1 | 2 | 1 | 21 | 1 |
| MM84 | 52 | Female | 4 | Nontreated | 1.3 | 2 | 71.3 | 0 | 1.3 | 1 | 1 | 27.3 | 1 |
| MM93 | 70 | Male | 4 | Nontreated | 83 | 3 | 15 | 2 | 1 | 0 | 1 | 0 | 2 |
| MM87 | 29 | Female | 3 | BRAF | 7 | 3 | 62 | 30 | 0 | 1 | 1 | 1 | 2 |
| MM100 | 64 | Female | 3 | BRAF | 8 | 2 | 80 | 5 | 0 | 1 | 1 | 7 | 1 |
| MM103 | 61 | Male | 3 | BRAF | 2 | 2 | 98 | 0 | 0 | 2 | 1 | 0 | 1 |
| MM60 | 77 | Female | 3 | Chemotherapy | 2 | 0.7 | 98 | 0 | 0.7 | 2 | 0.3 | 0 | 0.3 |
| MM71 | 61 | Male | 4 | Chemotherapy | 7.7 | 3 | 92.3 | 0 | 3 | 2 | 1 | 0 | 2 |
| MM82 | 76 | Female | 3 | Chemotherapy | 13.3 | 2 | 65 | 16.7 | 0 | 1 | 1 | 5 | 1 |
| MM98 | 55 | Male | 4 | Chemotherapy | 17 | 4 | 75 | 0 | 2 | 0 | 2 | 8 | 2 |
| MM22 | 44 | Male | 3 | Interferon | 5 | 5 | 25.7 | 69 | 3 | 2 | 3 | 0.3 | 2 |
| MM52 | 68 | Female | 3 | Interferon | 7 | 2 | 93 | 0 | 1 | 1 | 1 | 0 | 1 |
| MM54 | 81 | Female | 4 | Interferon + Chemotherapy | 6.3 | 1.3 | 57.7 | 0 | 0 | 2 | 0.7 | 36 | 0.7 |
| Accession | Gene | Log2.Fold Change | p-Value | |||
|---|---|---|---|---|---|---|
| Treatment | Age of Diagnosis | Gender | Disease Stage | |||
| P00747 | PLG * | −3.0721709 | 0.033233 | 0.083008 | 0.511929 | 0.576933 |
| P02679 | FGG * | −2.3908585 | 0.014045 | 0.269768 | 0.979767 | 0.189957 |
| P01024 | C3 * | −1.389545 | 0.048588 | 0.328285 | 0.96424 | 0.195198 |
| P01009 | SERPINA1 * | −1.2769647 | 0.027569 | 0.970118 | 0.84077 | 0.07742 |
| P84103 | SRSF3 * | 0.779147 | 0.049004 | 0.119298 | 0.649564 | 0.653696 |
| P69891 | HBG1 | −1.7589103 | 0.02118 | 0.352096 | 0.215719 | 0.071367 |
| P69892 | HBG2 | −1.7018783 | 0.031238 | 0.445556 | 0.295623 | 0.100417 |
| Q9P1F3 | ABRACL | 1.5688169 | 0.031732 | 0.631799 | 0.154456 | 0.077132 |
| P30085 | CMPK1 | 0.64424732 | 0.011524 | 0.131597 | 0.546014 | 0.062366 |
| Category | Applied Treatments for Melanoma Patients | Details (Clinical Trial ID) | References |
|---|---|---|---|
| Double combination | Trametinib (MEKi) Dabrafenib (BRAFi) | Improvement of PFS and OS than BRAFi as alone | [46,47,48,97] |
| Nivolumab (anti PD-1 Ab) Ipilimumab (anti CTLA-4 Ab) | Improvements of PFA and OS than ipilimumab (NCT01844505) | [74] | |
| Triple combination | Dabrafenib (BRAFi) Trametinib (MEKi) Pembrolizumab (anti PD-1 Ab) | BRAFV600-mutated metastatic melanoma patients (NCT02130466) | [102] |
| Dabrafenib (BRAFi) Trametinib (MEKi) Pembrolizumab (anti PD-1 Ab) | Phase 2, randomized double-blind placebo-control (NCT02130466) | [73] | |
| Cobimetinib (MEKi) Vemurafenib (BRAFi) Atezolizumab (anti-PD-L1 Ab) | Phase Ib, BRAFV600-mutated metastatic melanoma patients (NCT01656642) | [103] | |
| HDACi combination | Pembrolizumab (anti PD-1 Ab) Entinostat (HDACi) | Progression to metastatic melanoma on eye (NCT02697630) | [117] |
| Ipilimumab (anti CTLA-4 Ab) Panobinostat (HDACi) | Advanced and unresectable melanoma patients (NCT02032810) | [112] | |
| Pembrolizumab (anti PD-1 Ab) Entinostat (HDACi) | Melanoma patients, showing significant resistances on prior PD-1 blockade or prior PD-1/CTLA-4 blockade (NCT02437136) | [118,119] | |
| Dabrafenib (BRAFi) Trametinib (MEKi) Entinostat (HDACi) | On treating melanoma models of harboring mutations of BRAF, NRAS, and NF1 | [120] | |
| Anti PD-1 ab (from BioXCell) LBH589 (HDACi) | Slower tumor progression and increased survival compared with control and single treatments in B16F10 mice model | [113] | |
| Anti-PD-1 ab (from BioXCell) AR42 (HDACi) | Enhanced immunotherapy response with HDACi in B16 mice model | [116] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Gil, J.; Pla, I.; Sanchez, A.; Betancourt, L.H.; Lee, B.; Appelqvist, R.; Ingvar, C.; Lundgren, L.; Olsson, H.; et al. Protein Expression in Metastatic Melanoma and the Link to Disease Presentation in a Range of Tumor Phenotypes. Cancers 2020, 12, 767. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030767
Kim Y, Gil J, Pla I, Sanchez A, Betancourt LH, Lee B, Appelqvist R, Ingvar C, Lundgren L, Olsson H, et al. Protein Expression in Metastatic Melanoma and the Link to Disease Presentation in a Range of Tumor Phenotypes. Cancers. 2020; 12(3):767. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030767
Chicago/Turabian StyleKim, Yonghyo, Jeovanis Gil, Indira Pla, Aniel Sanchez, Lazaro Hiram Betancourt, Boram Lee, Roger Appelqvist, Christian Ingvar, Lotta Lundgren, Håkan Olsson, and et al. 2020. "Protein Expression in Metastatic Melanoma and the Link to Disease Presentation in a Range of Tumor Phenotypes" Cancers 12, no. 3: 767. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030767