Accessing a New Dimension in TP53 Biology: Multiplex Long Amplicon Digital PCR to Specifically Detect and Quantitate Individual TP53 Transcripts

 ,
,
Abstract
:1. Introduction
2. Results
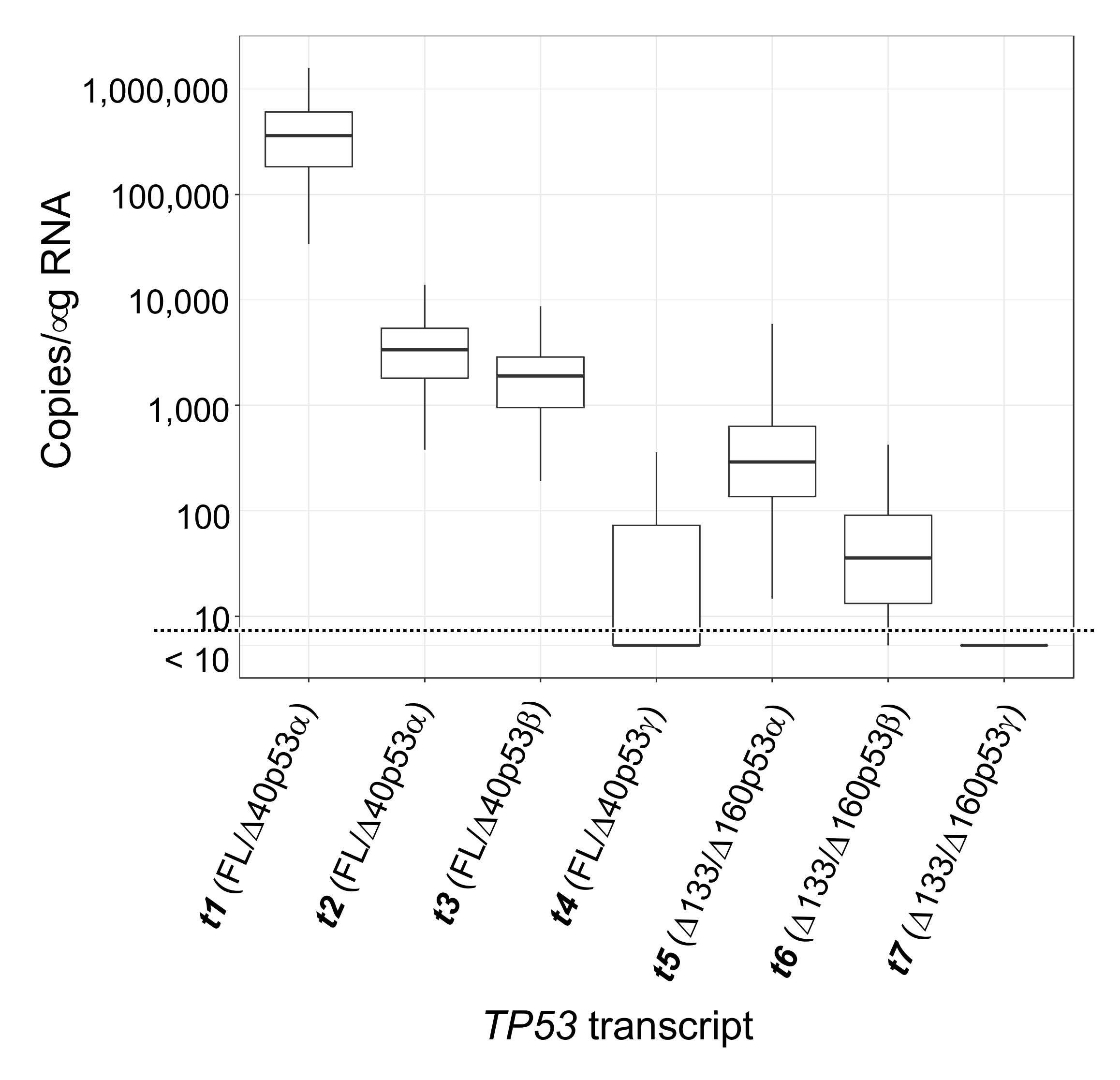
2.1. Quantitation of TP53 t1/t3/t4 (Encoding FL/Δ40p53α/β/γ) and t5/t6/t7 (Encoding Δ133/Δ160p53α/β/γ) Transcripts
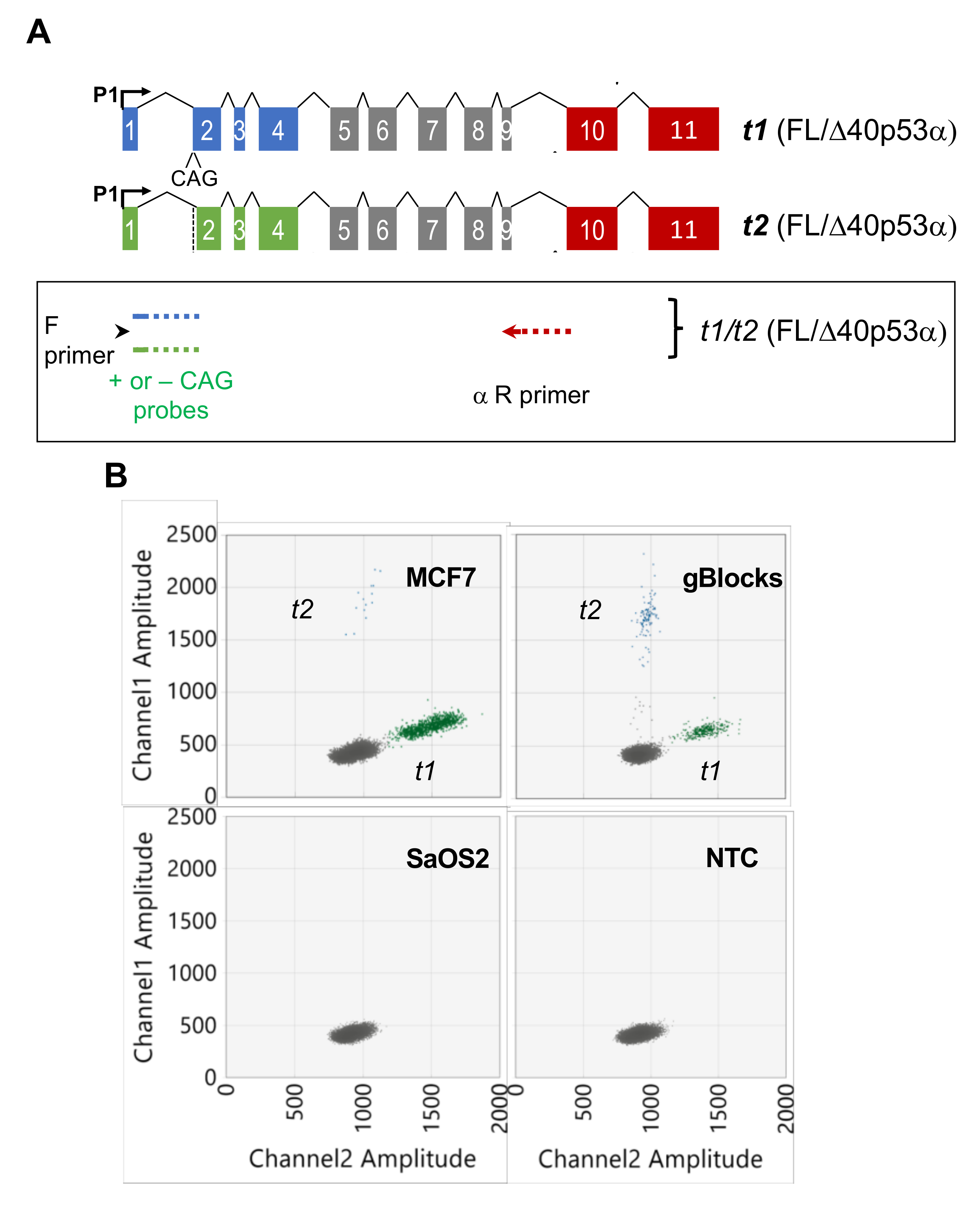
2.2. Quantitation of TP53 t1 and t2 Transcripts
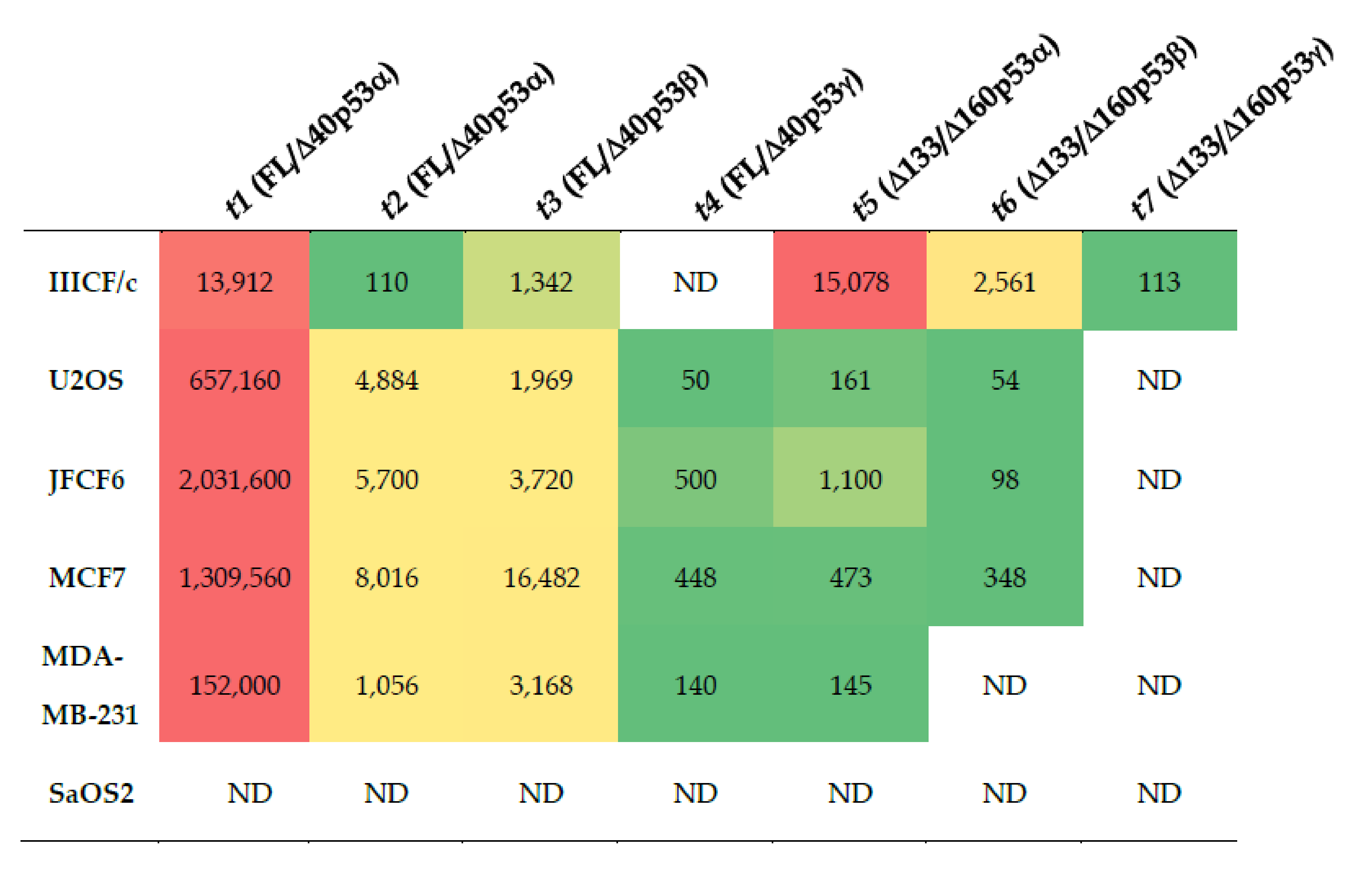
2.3. Demonstration of TP53 Isoform Analysis in Cancer Cell Lines
2.4. Demonstration of TP53 Isoform Analysis in Breast Cancer Patient Samples
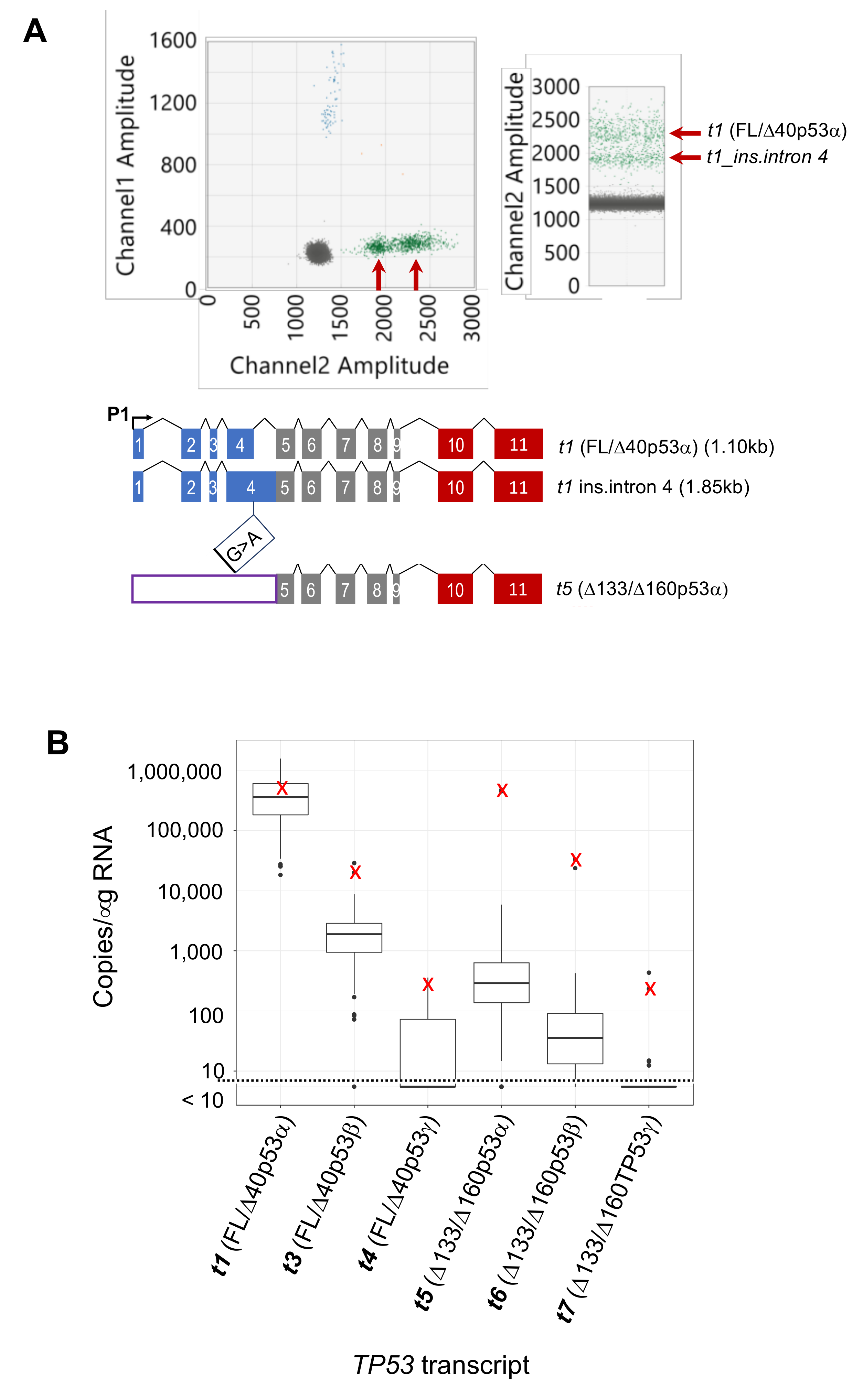
2.5. Identification of Additional TP53 Transcript Complexity
3. Discussion
4. Materials and Methods
4.1. Multiplex Long Amplicon ddPCR Assays
4.1.1. ddPCR Primer and Probe Design
4.1.2. Multiplex Long Amplicon ddPCR Reactions
Optimisation of Cycling Conditions for the Development of New Assays
4.2. Cell Line Information
4.3. Breast Tumour Sample Information
4.4. RNA Isolation and cDNA Synthesis
4.5. Cloning of Synthetic DNA Fragments (gBlocks)
4.6. RNA-Sequence Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tazi, J.; Bakkour, N.; Stamm, S. Alternative splicing and disease. Biochim. Biophys. Acta. 2009, 1792, 14–26. [Google Scholar] [CrossRef]
- Climente-Gonzalez, H.; Porta-Pardo, E.; Godzik, A.; Eyras, E. The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 2017, 20, 2215–2226. [Google Scholar] [CrossRef] [Green Version]
- El Marabti, E.; Younis, I. The Cancer Spliceome: Reprograming of Alternative Splicing in Cancer. Front. Mol. Biosci. 2018, 5, 80. [Google Scholar] [CrossRef]
- Oltean, S.; Bates, D.O. Hallmarks of alternative splicing in cancer. Oncogene 2014, 33, 5311–5318. [Google Scholar] [CrossRef] [Green Version]
- Robinson, T.J.; Freedman, J.A.; Al Abo, M.; Deveaux, A.E.; LaCroix, B.; Patierno, B.M.; George, D.J.; Patierno, S.R. Alternative RNA Splicing as a Potential Major Source of Untapped Molecular Targets in Precision Oncology and Cancer Disparities. Clin. Cancer. Res. 2019, 25, 2963–2968. [Google Scholar] [CrossRef] [Green Version]
- Joruiz, S.M.; Bourdon, J.C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Giron, C.G.; et al. Ensembl 2018. Nucleic. Acids. Res. 2018, 46, D754–D761. [Google Scholar] [CrossRef]
- Mehta, S.; Tsai, P.; Lasham, A.; Campbell, H.; Reddel, R.; Braithwaite, A.; Print, C. A Study of TP53 RNA Splicing Illustrates Pitfalls of RNA-seq Methodology. Cancer Res. 2016, 76, 7151–7159. [Google Scholar] [CrossRef] [Green Version]
- Avery-Kiejda, K.A.; Morten, B.; Wong-Brown, M.W.; Mathe, A.; Scott, R.J. The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 2014, 35, 586–596. [Google Scholar] [CrossRef]
- Bourdon, J.C.; Khoury, M.P.; Diot, A.; Baker, L.; Fernandes, K.; Aoubala, M.; Quinlan, P.; Purdie, C.A.; Jordan, L.B.; Prats, A.C.; et al. p53 mutant breast cancer patients expressing p53gamma have as good a prognosis as wild-type p53 breast cancer patients. Breast Cancer Res. BCR 2011, 13, R7. [Google Scholar] [CrossRef]
- Kazantseva, M.; Eiholzer, R.A.; Mehta, S.; Taha, A.; Bowie, S.; Roth, I.; Zhou, J.; Joruiz, S.M.; Royds, J.A.; Hung, N.A.; et al. Elevation of the TP53 isoform Delta133p53beta in glioblastomas: an alternative to mutant p53 in promoting tumor development. J. Pathol. 2018, 246, 77–88. [Google Scholar] [CrossRef] [Green Version]
- Campbell, H.; Fleming, N.; Roth, I.; Mehta, S.; Wiles, A.; Williams, G.; Vennin, C.; Arsic, N.; Parkin, A.; Pajic, M.; et al. 133p53 isoform promotes tumour invasion and metastasis via interleukin-6 activation of JAK-STAT and RhoA-ROCK signalling. Nature commun. 2018, 9, 254. [Google Scholar] [CrossRef] [Green Version]
- Kazantseva, M.; Mehta, S.; Eiholzer, R.A.; Gimenez, G.; Bowie, S.; Campbell, H.; Reily-Bell, A.L.; Roth, I.; Ray, S.; Drummond, C.J.; et al. The Delta133p53beta isoform promotes an immunosuppressive environment leading to aggressive prostate cancer. Cell Death Dis 2019, 10, 631. [Google Scholar] [CrossRef]
- Bischof, K.; Knappskog, S.; Hjelle, S.M.; Stefansson, I.; Woie, K.; Salvesen, H.B.; Gjertsen, B.T.; Bjorge, L. Influence of p53 Isoform Expression on Survival in High-Grade Serous Ovarian Cancers. Sci. Rep. 2019, 9, 5244. [Google Scholar] [CrossRef] [Green Version]
- Bischof, K.; Knappskog, S.; Stefansson, I.; McCormack, E.M.; Trovik, J.; Werner, H.M.J.; Woie, K.; Gjertsen, B.T.; Bjorge, L. High expression of the p53 isoform gamma is associated with reduced progression-free survival in uterine serous carcinoma. BMC Cancer 2018, 18, 684. [Google Scholar] [CrossRef] [Green Version]
- McManus, C.J.; Graveley, B.R. RNA structure and the mechanisms of alternative splicing. Curr. Opin. Genet. Dev. 2011, 21, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Marcel, V.; Tran, P.L.; Sagne, C.; Martel-Planche, G.; Vaslin, L.; Teulade-Fichou, M.P.; Hall, J.; Mergny, J.L.; Hainaut, P.; Van Dyck, E. G-quadruplex structures in TP53 intron 3: role in alternative splicing and in production of p53 mRNA isoforms. Carcinogenesis 2011, 32, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Ray, P.S.; Grover, R.; Das, S. Two internal ribosome entry sites mediate the translation of p53 isoforms. EMBO Rep. 2006, 7, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.Q.; Halaby, M.J.; Zhang, Y. The identification of an internal ribosomal entry site in the 5’-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene 2006, 25, 4613–4619. [Google Scholar] [CrossRef] [Green Version]
- Fernando, M.R.; Jiang, C.; Krzyzanowski, G.D.; Ryan, W.L. Analysis of human blood plasma cell-free DNA fragment size distribution using EvaGreen chemistry based droplet digital PCR assays. Clin. Chim. Acta. 2018, 483, 39–47. [Google Scholar] [CrossRef]
- Miotke, L.; Lau, B.T.; Rumma, R.T.; Ji, H.P. High sensitivity detection and quantitation of DNA copy number and single nucleotide variants with single color droplet digital PCR. Anal. Chem. 2014, 86, 2618–2624. [Google Scholar] [CrossRef]
- Bio-Rad Laboratories. ddPCR HDR Genome Edit Detection Reference Assays. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/10000065279.pdf (accessed on 23 March 2020).
- Rogan, E.M.; Bryan, T.M.; Hukku, B.; Maclean, K.; Chang, A.C.; Moy, E.L.; Englezou, A.; Warneford, S.G.; Dalla-Pozza, L.; Reddel, R.R. Alterations in p53 and p16INK4 expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol. Cell. Biol. 1995, 15, 4745–4753. [Google Scholar] [CrossRef] [Green Version]
- Aoubala, M.; Murray-Zmijewski, F.; Khoury, M.P.; Fernandes, K.; Perrier, S.; Bernard, H.; Prats, A.C.; Lane, D.P.; Bourdon, J.C. p53 directly transactivates Delta133p53alpha, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011, 18, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Muthukaruppan, A.; Lasham, A.; Blenkiron, C.; Woad, K.J.; Black, M.A.; Knowlton, N.; McCarthy, N.; Findlay, M.P.; Print, C.G.; Shelling, A.N. Genomic profiling of breast tumours from New Zealand patients. N.Z. Med. J. 2017, 130, 40–56. [Google Scholar]
- Warneford, S.G.; Witton, L.J.; Townsend, M.L.; Rowe, P.B.; Reddel, R.R.; Dalla-Pozza, L.; Symonds, G. Germ-line splicing mutation of the p53 gene in a cancer-prone family. Cell Growth Differ. 1992, 3, 839–846. [Google Scholar]
- Rogan, E.M. Mechanisms of cellular immortalisation: A study in Li-Fraumeni Syndrome Fibroblasts. Ph.D. Thesis, University of Sydney, Sydney, Australia, 1995. [Google Scholar]
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 2015, 16, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Hayman, L.; Chaudhry, W.R.; Revin, V.V.; Zhelev, N.; Bourdon, J.C. What is the potential of p53 isoforms as a predictive biomarker in the treatment of cancer? Expert Rev. Mol. Diagn. 2019, 19, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Gadea, G.; Arsic, N.; Fernandes, K.; Diot, A.; Joruiz, S.M.; Abdallah, S.; Meuray, V.; Vinot, S.; Anguille, C.; Remenyi, J.; et al. TP53 drives invasion through expression of its Delta133p53beta variant. eLife 2016, 5. [Google Scholar] [CrossRef]
- Huggett, J.F.; Cowen, S.; Foy, C.A. Considerations for digital PCR as an accurate molecular diagnostic tool. Clin. Chem. 2015, 61, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Soares, E.; Zhou, H. Master regulatory role of p63 in epidermal development and disease. Cell. Mol. Life Sci. 2018, 75, 1179–1190. [Google Scholar] [CrossRef] [Green Version]
- Cowen, L.E.; Tang, Y. Identification of nonsense-mediated mRNA decay pathway as a critical regulator of p53 isoform beta. Sci. Rep. 2017, 7, 17535. [Google Scholar] [CrossRef] [Green Version]
- Clark, M.B.; Wrzesinski, T.; Garcia, A.B.; Hall, N.A.L.; Kleinman, J.E.; Hyde, T.; Weinberger, D.R.; Harrison, P.J.; Haerty, W.; Tunbridge, E.M. Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain. Mol. Psychiatry 2020, 25, 37–47. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Fair, B.J.; Dwyer, Z.W.; Gildea, M.; Pleiss, J.A. Detection of splice isoforms and rare intermediates using multiplexed primer extension sequencing. Nat. Methods 2019, 16, 55–58. [Google Scholar] [CrossRef]
- Zhang, Z.; Pan, Z.; Ying, Y.; Xie, Z.; Adhikari, S.; Phillips, J.; Carstens, R.P.; Black, D.L.; Wu, Y.; Xing, Y. Deep-learning augmented RNA-seq analysis of transcript splicing. Nat. Methods 2019, 16, 307–310. [Google Scholar] [CrossRef]
- Bio-Rad Laboratories. Droplet Digital PCR Applications Guide. Available online: https://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6407.pdf (accessed on 23 March 2020).
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The digital MIQE guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef]
- Lasham, A.; Samuel, W.; Cao, H.; Patel, R.; Mehta, R.; Stern, J.L.; Reid, G.; Woolley, A.G.; Miller, L.D.; Black, M.A.; et al. YB-1, the E2F pathway, and regulation of tumor cell growth. J. Natl. Cancer Inst. 2012, 104, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Lasham, A.; Mehta, S.Y.; Fitzgerald, S.J.; Woolley, A.G.; Hearn, J.I.; Hurley, D.G.; Ruza, I.; Algie, M.; Shelling, A.N.; Braithwaite, A.W.; et al. A novel EGR-1 dependent mechanism for YB-1 modulation of paclitaxel response in a triple negative breast cancer cell line. Int. J. Cancer 2016, 139, 1157–1170. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Key Assay Design Features | |
|---|---|
| 1. | Primers and probes designed with high melting temperatures (≥60 °C). |
| 2. | Probe design critical—with splice site location close to 5’ end of probe where possible. |
| 3. | Long extension times (4–6 min) with high cycle numbers (50 cycles). |
| 4. | Two probes to one transcript can be used to allow detection of >2 transcripts in one reaction, if only two fluorophore channels available. |
| Design | Notes | |
|---|---|---|
| Primer 1 |
| Could be forward or reverse primer |
| Primer 2 |
| Could be forward or reverse primer |
| Probe/s |
| *If not possible to design with 5’ end within a few nucleotides of splice junction and also follow ddPCR probe guidelines, then design with probe 3’ end within a few nucleotides of splice junction. This not necessary if splice form has unique sequence. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lasham, A.; Tsai, P.; Fitzgerald, S.J.; Mehta, S.Y.; Knowlton, N.S.; Braithwaite, A.W.; Print, C.G. Accessing a New Dimension in TP53 Biology: Multiplex Long Amplicon Digital PCR to Specifically Detect and Quantitate Individual TP53 Transcripts. Cancers 2020, 12, 769. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030769
Lasham A, Tsai P, Fitzgerald SJ, Mehta SY, Knowlton NS, Braithwaite AW, Print CG. Accessing a New Dimension in TP53 Biology: Multiplex Long Amplicon Digital PCR to Specifically Detect and Quantitate Individual TP53 Transcripts. Cancers. 2020; 12(3):769. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030769
Chicago/Turabian StyleLasham, Annette, Peter Tsai, Sandra J. Fitzgerald, Sunali Y. Mehta, Nicholas S. Knowlton, Antony W. Braithwaite, and Cristin G. Print. 2020. "Accessing a New Dimension in TP53 Biology: Multiplex Long Amplicon Digital PCR to Specifically Detect and Quantitate Individual TP53 Transcripts" Cancers 12, no. 3: 769. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12030769