BAX Redistribution Induces Apoptosis Resistance and Selective Stress Sensitivity in Human HCC

 , and
, and
Abstract
:1. Introduction
2. Results
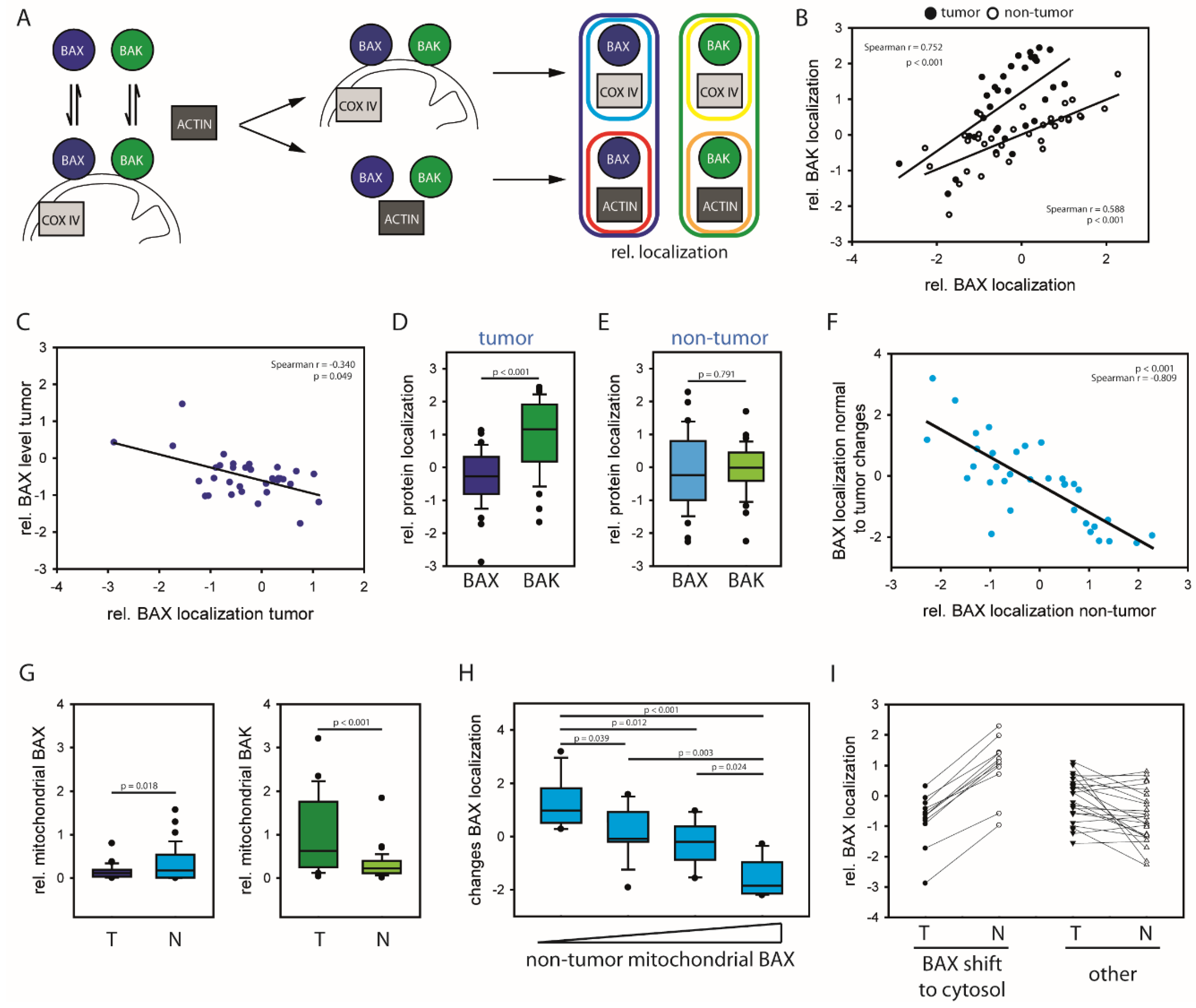
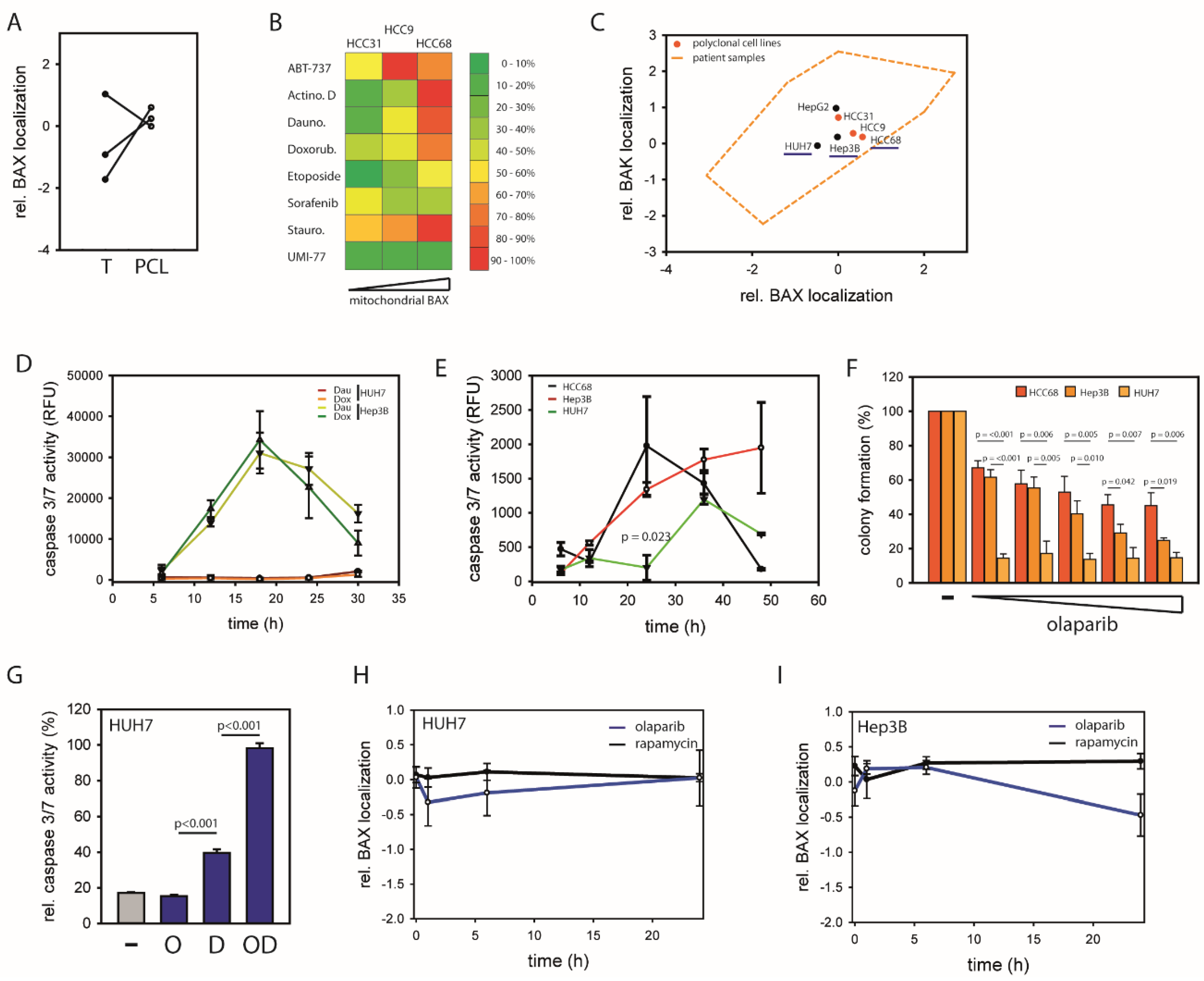
2.1. HCCs Show Selection towards Low Mitochondrial BAX Levels
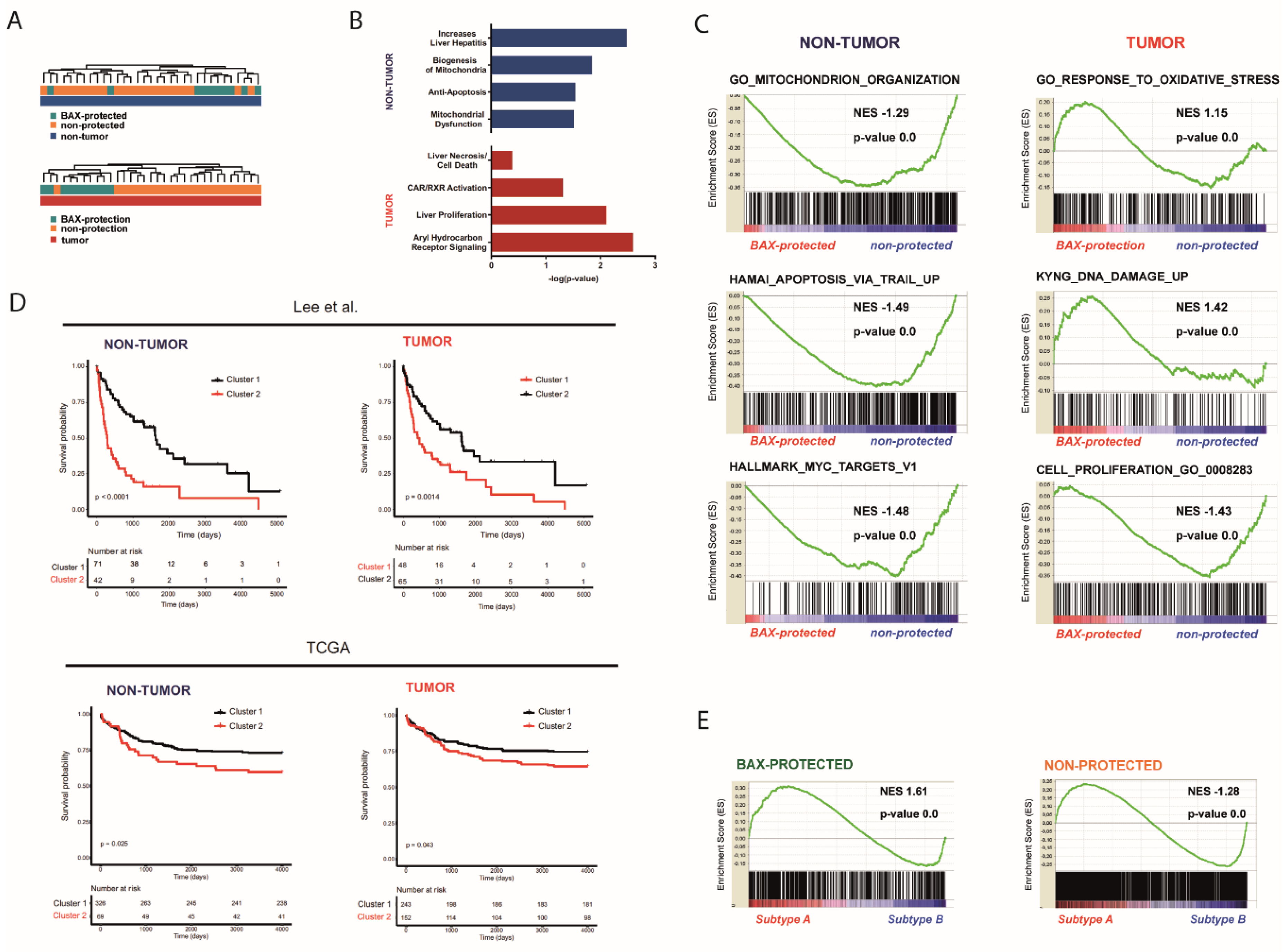
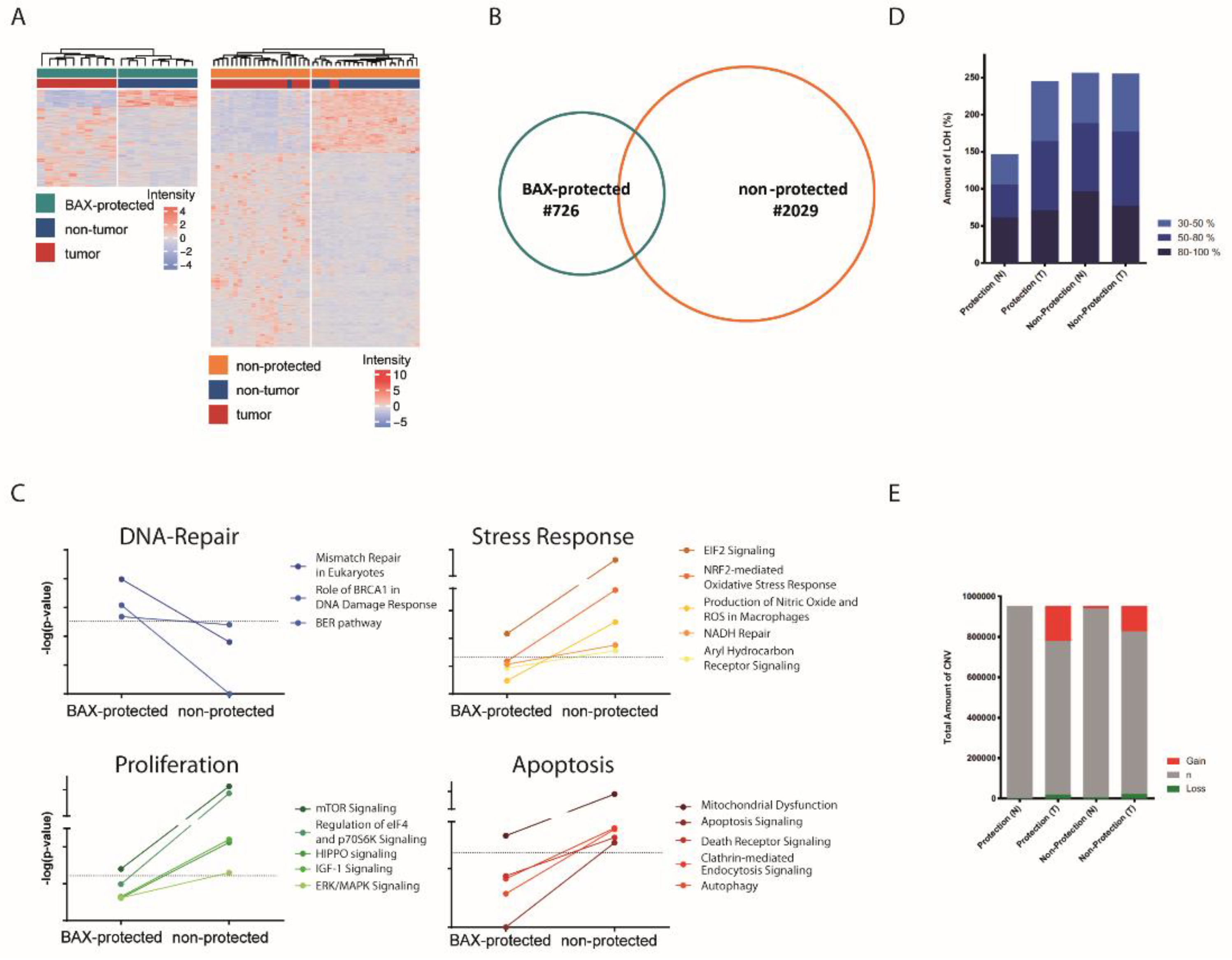
2.2. Distinct Molecular Alterations Drive Cancer Progression in BAX-Protected HCCs
2.3. BAX-Protected HCC Upregulate DNA Damage Repair
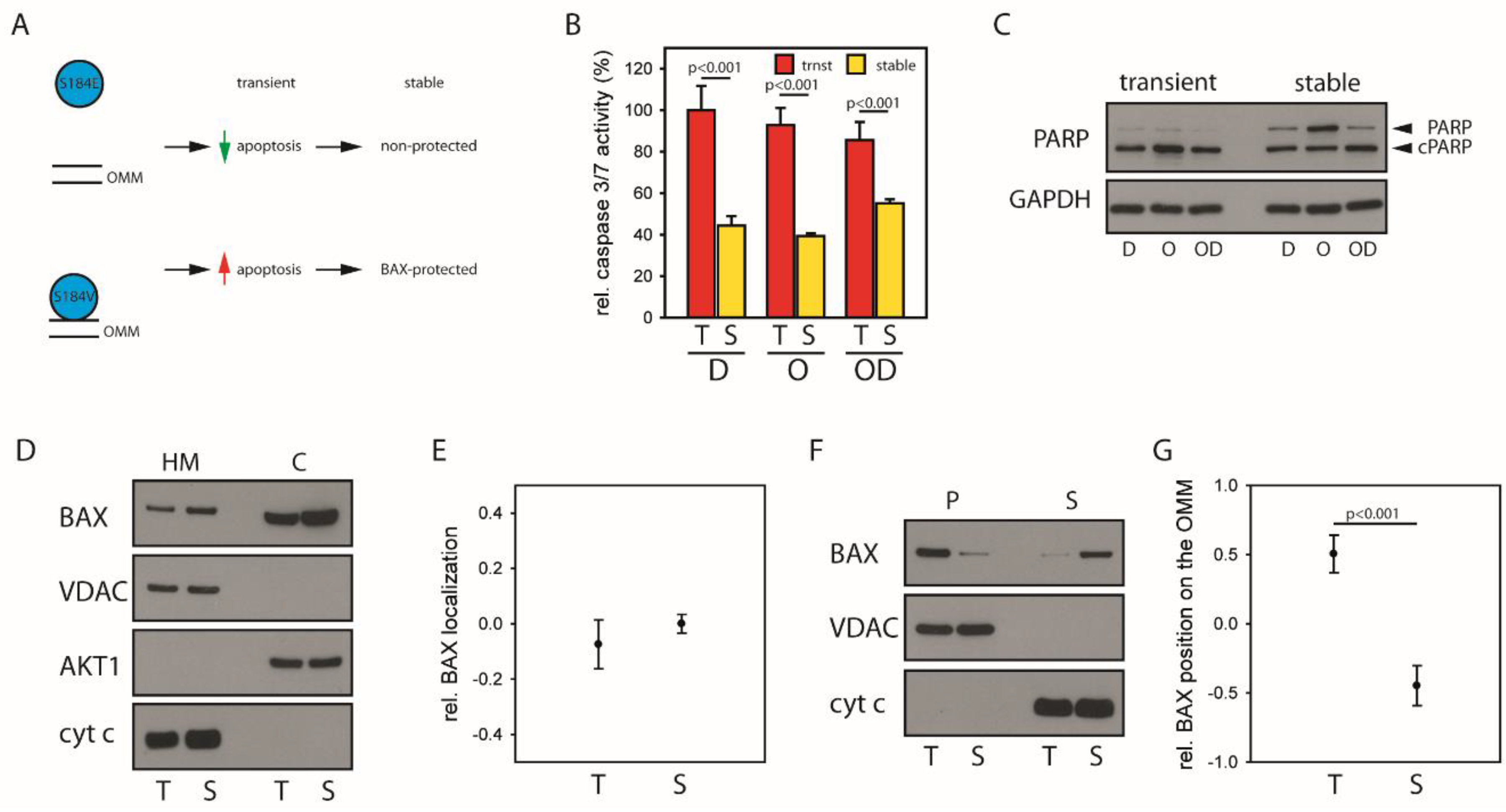
2.4. Mitochondrial BAX Induces Effects Mimicking Selection of BAX-Protected HCC
2.5. BAX-Specific Inhibition Increases Sensitivity to Inhibition of DNA Damage Repair
3. Discussion
4. Materials and Methods
4.1. Quantification of Western Blot Data
4.2. Patient Data
4.3. Subcellular Fractionation
4.4. Carbonate Extraction
4.5. Gene Expression Analysis
4.6. Genomic Analyses
4.7. Caspase 3/7 Assay and PARP Cleavage
4.8. Clonogenic Survival Assay
4.9. Cell Culture and BAX Expression
4.10. Preparation of Whole-Cell Lysates
4.11. Statistics, Databases and Patient Integration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Anderson, M.A.; Deng, J.; Seymour, J.F.; Tam, C.; Kim, S.Y.; Fein, J.; Yu, L.; Brown, J.R.; Westerman, D.; Si, E.G.; et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 2016, 127, 3215–3224. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, J.U.; Andersen, J.B.; Thorgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Luedde, T. Apoptosis and necroptosis in the liver: A matter of life and death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, T.; Ross, A.J.; King, A.; Zong, W.X.; Rathmell, J.C.; Shiels, H.A.; Ulrich, E.; Waymire, K.G.; Mahar, P.; Frauwirth, K.; et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 2000, 6, 1389–1399. [Google Scholar] [CrossRef] [Green Version]
- Martinou, J.-C.; Youle, R.J. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Edlich, F. The great migration of Bax and Bak. Mol. Cell. Oncol. 2015, 2, e995029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-x(L) retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Emschermann, F.; Lauterwasser, J.; Kaiser, A.; Ichim, G.; Tait, S.W.G.; Frank, S.; Langer, H.F.; et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015, 34, 67–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Youle, R.J.; Edlich, F. The C-terminal helix of Bcl-x(L) mediates Bax retrotranslocation from the mitochondria. Cell Death Differ. 2013, 20, 333–342. [Google Scholar] [CrossRef]
- Kim, H.; Rafiuddin-Shah, M.; Tu, H.-C.; Jeffers, J.R.; Zambetti, G.P.; Hsieh, J.J.-D.; Cheng, E.H.-Y. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat. Cell Biol. 2006, 8, 1348–1358. [Google Scholar] [CrossRef]
- Kuwana, T.; Bouchier-Hayes, L.; Chipuk, J.E.; Bonzon, C.; Sullivan, B.A.; Green, D.R.; Newmeyer, D.D. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell 2005, 17, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Llambi, F.; Moldoveanu, T.; Tait, S.W.G.; Bouchier-Hayes, L.; Temirov, J.; McCormick, L.L.; Dillon, C.P.; Green, D.R. A Unified Model of Mammalian BCL-2 Protein Family Interactions at the Mitochondria. Mol. Cell 2011, 44, 517–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, S.N.; Chen, L.; Dewson, G.; Wei, A.; Naik, E.; Fletcher, J.I.; Adams, J.M.; Huang, D.C.S. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005, 19, 1294–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef] [Green Version]
- Lopez, J.; Bessou, M.; Riley, J.S.; Giampazolias, E.; Todt, F.; Rochegüe, T.; Oberst, A.; Green, D.R.; Edlich, F.; Ichim, G.; et al. Mito-priming as a method to engineer Bcl-2 addiction. Nat. Commun. 2016, 7, 10538. [Google Scholar] [CrossRef] [Green Version]
- Touzeau, C.; Ryan, J.; Guerriero, J.; Moreau, P.; Chonghaile, T.N.; Le Gouill, S.; Richardson, P.; Anderson, K.; Amiot, M.; Letai, A. BH3 profiling identifies heterogeneous dependency on Bcl-2 family members in multiple myeloma and predicts sensitivity to BH3 mimetics. Leukemia 2016, 30, 761–764. [Google Scholar] [CrossRef]
- Reichenbach, F.; Wiedenmann, C.; Schalk, E.; Becker, D.; Funk, K.; Scholz-Kreisel, P.; Todt, F.; Wolleschak, D.; Döhner, K.; Marquardt, J.U.; et al. Mitochondrial BAX Determines the Predisposition to Apoptosis in Human AML. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4805–4816. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, I.; Roncero, C.; Fernández, M. Survival and apoptosis: A dysregulated balance in liver cancer. Liver Int. Off. J. Int. Assoc. Study Liver 2007, 27, 155–162. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address: [email protected]; Cancer Genome Atlas Research Network Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar] [CrossRef]
- Lee, J.-S.; Chu, I.-S.; Heo, J.; Calvisi, D.F.; Sun, Z.; Roskams, T.; Durnez, A.; Demetris, A.J.; Thorgeirsson, S.S. Classification and prediction of survival in hepatocellular carcinoma by gene expression profiling. Hepatol. Baltim. 2004, 40, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1′s inhibitory effect on Bak. Oncogene 2012, 31, 3177–3189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castven, D.; Becker, D.; Czauderna, C.; Wilhelm, D.; Andersen, J.B.; Strand, S.; Hartmann, M.; Heilmann-Heimbach, S.; Roth, W.; Hartmann, N.; et al. Application of patient-derived liver cancer cells for phenotypic characterization and therapeutic target identification. Int. J. Cancer 2019, 144, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.T.; Youle, R.J. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J. Biol. Chem. 1998, 273, 10777–10783. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.T.; Wolter, K.G.; Youle, R.J. Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. USA 1997, 94, 3668–3672. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Youle, R.J.; Tjandra, N. Structure of Bax: Coregulation of dimer formation and intracellular localization. Cell 2000, 103, 645–654. [Google Scholar] [CrossRef] [Green Version]
- Wolter, K.G.; Hsu, Y.T.; Smith, C.L.; Nechushtan, A.; Xi, X.G.; Youle, R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997, 139, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.J.; Dubrez, L.; Morgan, C.P.; Jones, N.A.; Whitehouse, J.; Corfe, B.M.; Dive, C.; Hickman, J.A. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 1999, 144, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Sarosiek, K.A.; Chi, X.; Bachman, J.A.; Sims, J.J.; Montero, J.; Patel, L.; Flanagan, A.; Andrews, D.W.; Sorger, P.; Letai, A. BID Preferentially Activates BAK while BIM Preferentially Activates BAX, Affecting Chemotherapy Response. Mol. Cell 2013, 51, 751–765. [Google Scholar] [CrossRef] [Green Version]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerheide, W.; Tan, Y.J.; Teng, E.; Ting, A.E.; Jedpiyawongse, A.; Srivatanakul, P. Downregulation of proapoptotic proteins Bax and Bcl-X(S) in p53 overexpressing hepatocellular carcinomas. Biochem. Biophys. Res. Commun. 2000, 273, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Castven, D.; Fischer, M.; Becker, D.; Heinrich, S.; Andersen, J.B.; Strand, D.; Sprinzl, M.F.; Strand, S.; Czauderna, C.; Heilmann-Heimbach, S.; et al. Adverse genomic alterations and stemness features are induced by field cancerization in the microenvironment of hepatocellular carcinomas. Oncotarget 2017, 8, 48688–48700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Morganella, S.; Ceccarelli, M. VegaMC: A R/bioconductor package for fast downstream analysis of large array comparative genomic hybridization datasets. Bioinformatics 2012, 28, 2512–2514. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Jeong, S.-Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z. Missing data exploration: Highlighting graphical presentation of missing pattern. Ann. Transl. Med. 2015, 3, 356. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-Protected | ||
| Functions | p-Value Range | #Molecules |
| Protein Synthesis | −0.3 × 10−3–1.17 × 10−13 | 255 |
| Amino Acid Metabolism | −0.57 × 10−3–1.77 × 10−11 | 42 |
| Small Molecule Biochemistry | −0.57 × 10−3–1.77 × 10−11 | 285 |
| Lipid Metabolism | −0.57 × 10−3–7.14 × 10−8 | 214 |
| Molecular Transport | −0.57 × 10−3–7.14 × 10−8 | 262 |
| BAX-Protected | ||
| Functions | p-Value range | #Molecules |
| RNA Post-Transcriptional Modification | −0.28 × 10−2–6.00 × 10−5 | 10 |
| DNA Replication, Recombination and Repair | −0.17 × 10−2–1.46 × 10−4 | 12 |
| Cell Morphology | −0.17 × 10−2–4.75 × 10−4 | 23 |
| Cellular Function and Maintenance | −0.17 × 10−2–4.75 × 10−4 | 18 |
| Cell Cycle | −0.17 × 10−2–4.90 × 10−4 | 24 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funk, K.; Czauderna, C.; Klesse, R.; Becker, D.; Hajduk, J.; Oelgeklaus, A.; Reichenbach, F.; Fimm-Todt, F.; Lauterwasser, J.; Galle, P.R.; et al. BAX Redistribution Induces Apoptosis Resistance and Selective Stress Sensitivity in Human HCC. Cancers 2020, 12, 1437. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061437
Funk K, Czauderna C, Klesse R, Becker D, Hajduk J, Oelgeklaus A, Reichenbach F, Fimm-Todt F, Lauterwasser J, Galle PR, et al. BAX Redistribution Induces Apoptosis Resistance and Selective Stress Sensitivity in Human HCC. Cancers. 2020; 12(6):1437. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061437
Chicago/Turabian StyleFunk, Kathrin, Carolin Czauderna, Ramona Klesse, Diana Becker, Jovana Hajduk, Aline Oelgeklaus, Frank Reichenbach, Franziska Fimm-Todt, Joachim Lauterwasser, Peter R. Galle, and et al. 2020. "BAX Redistribution Induces Apoptosis Resistance and Selective Stress Sensitivity in Human HCC" Cancers 12, no. 6: 1437. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12061437