Ultra-Short Circulating Tumor DNA (usctDNA) in Plasma and Saliva of Non-Small Cell Lung Cancer (NSCLC) Patients

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
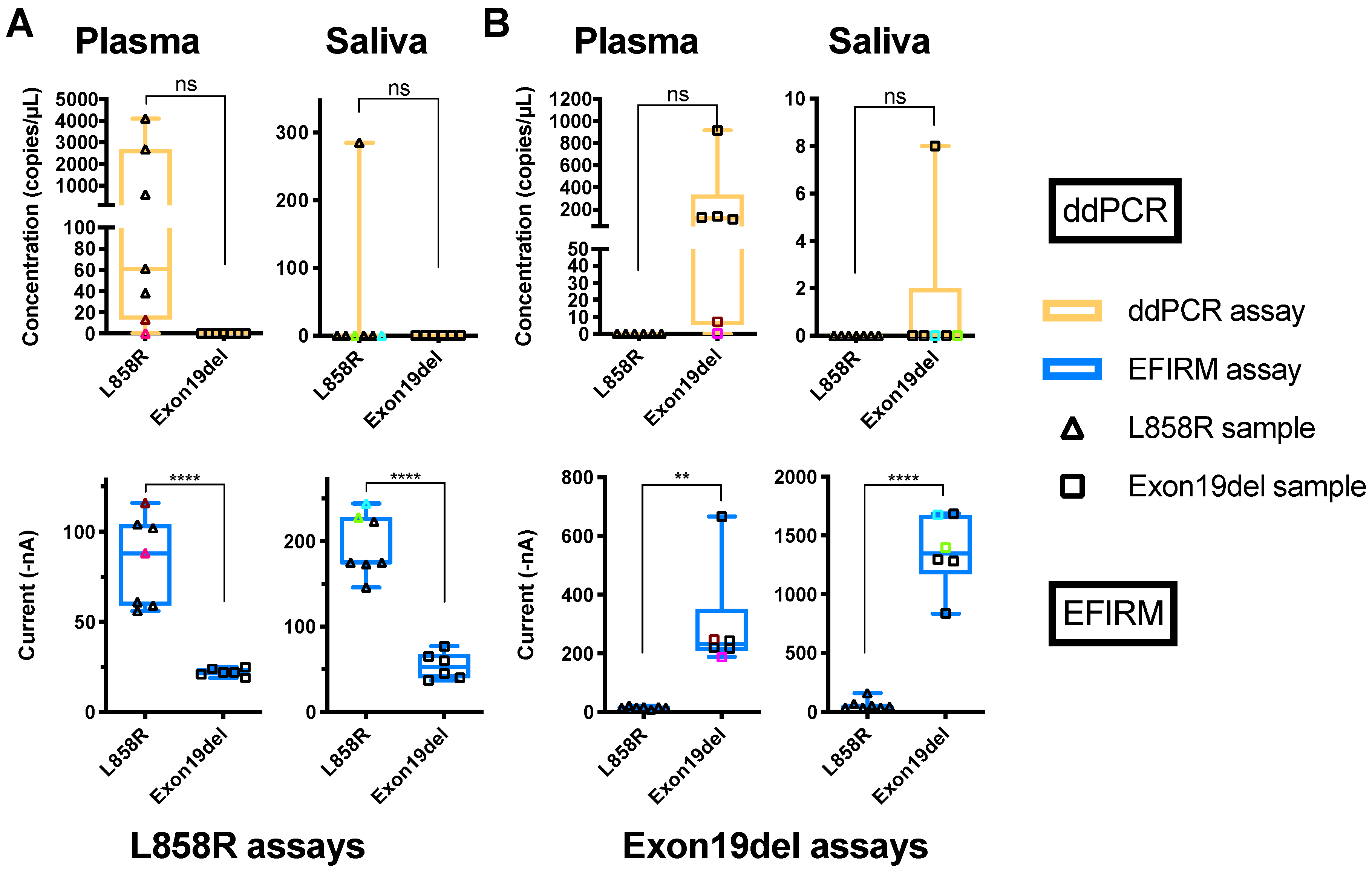
2.1. Comparisons of EFIRM and ddPCR Detection of EGFR ctDNA in Plasma and Saliva of NSCLC Patients
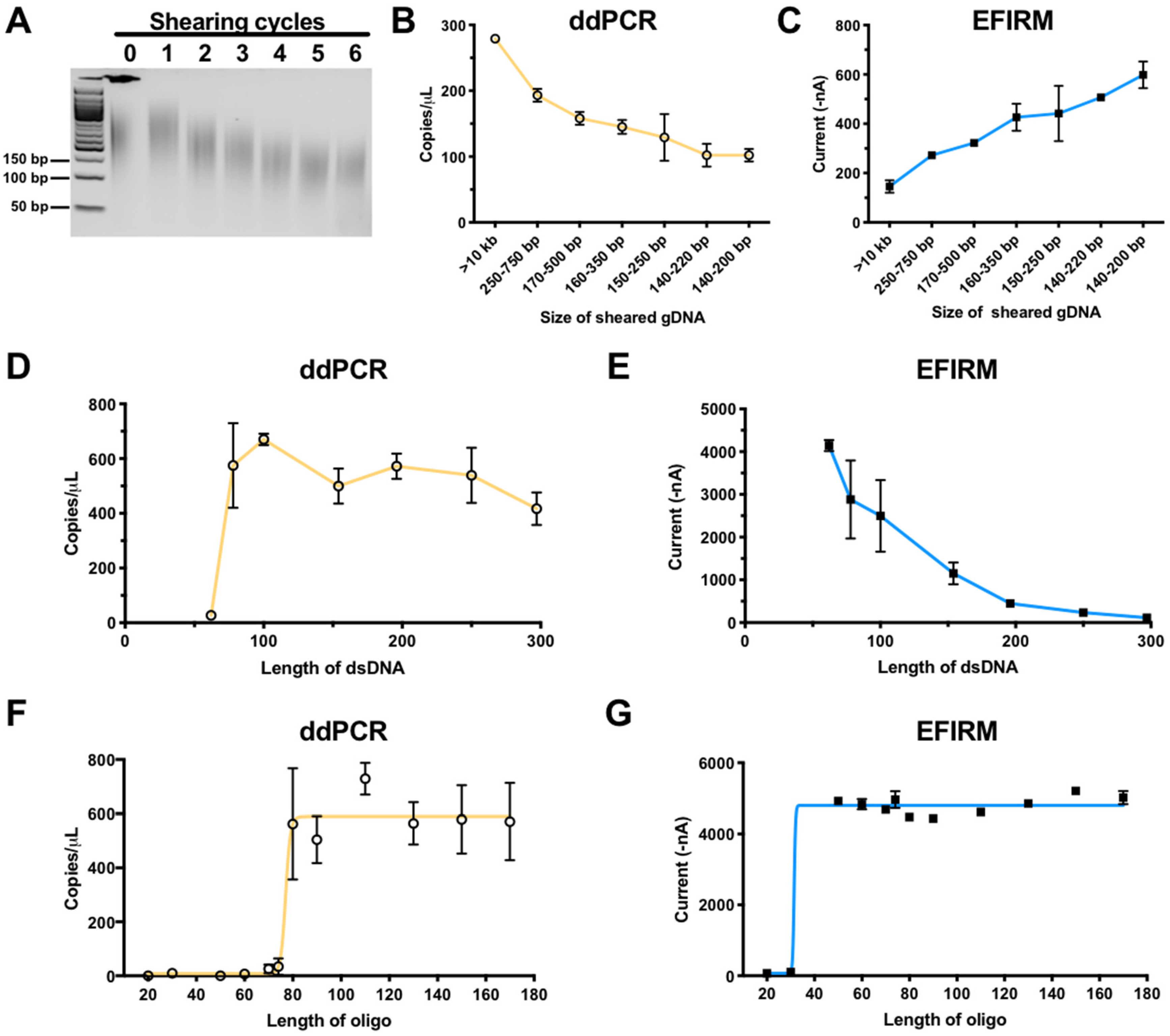
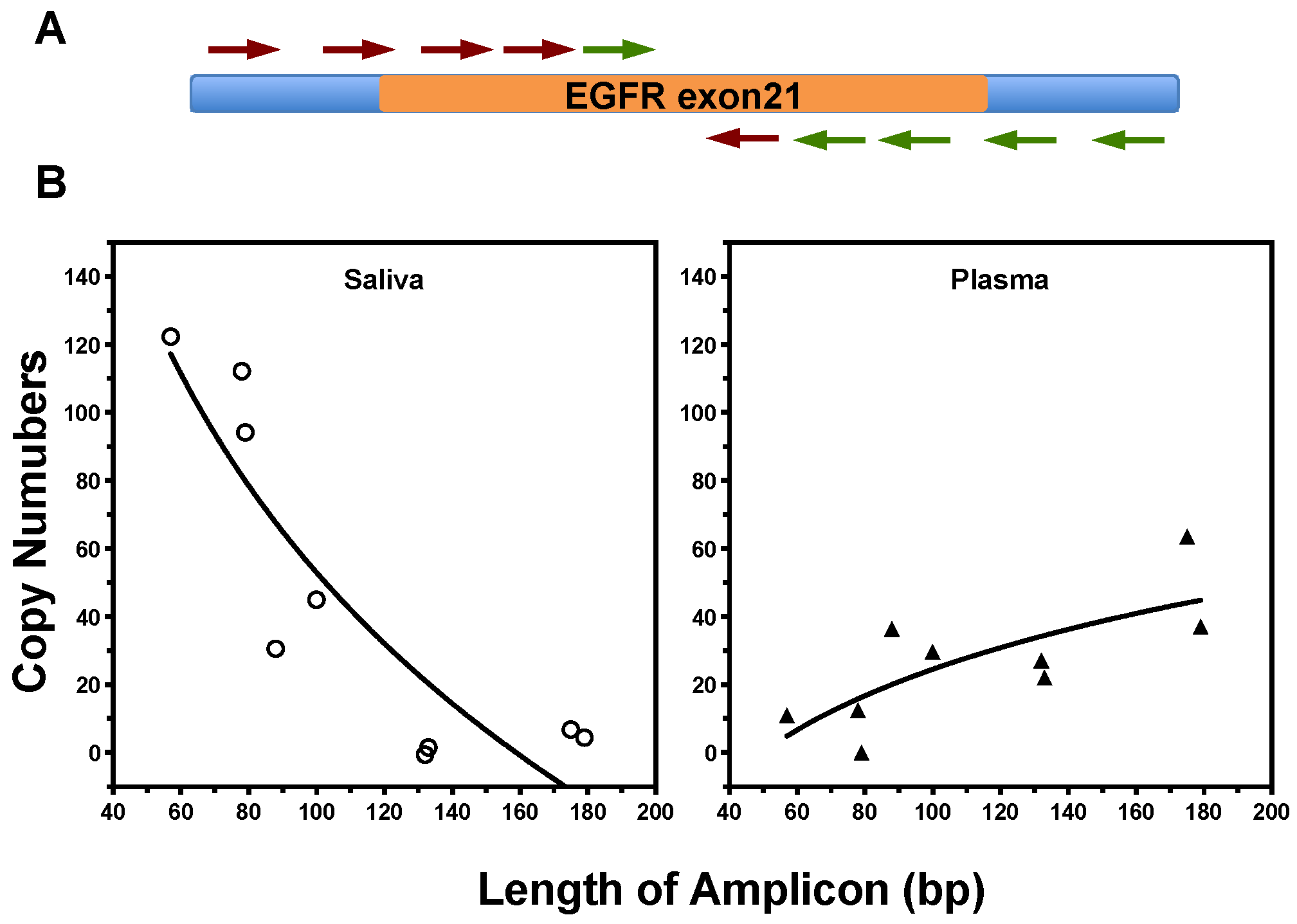
2.2. Size of ctDNA Fragments Impact EFIRM and ddPCR Detection
2.3. Characteristics of EGFR ctDNA with L858R Mutation in Plasma and Saliva of NSCLC Patients
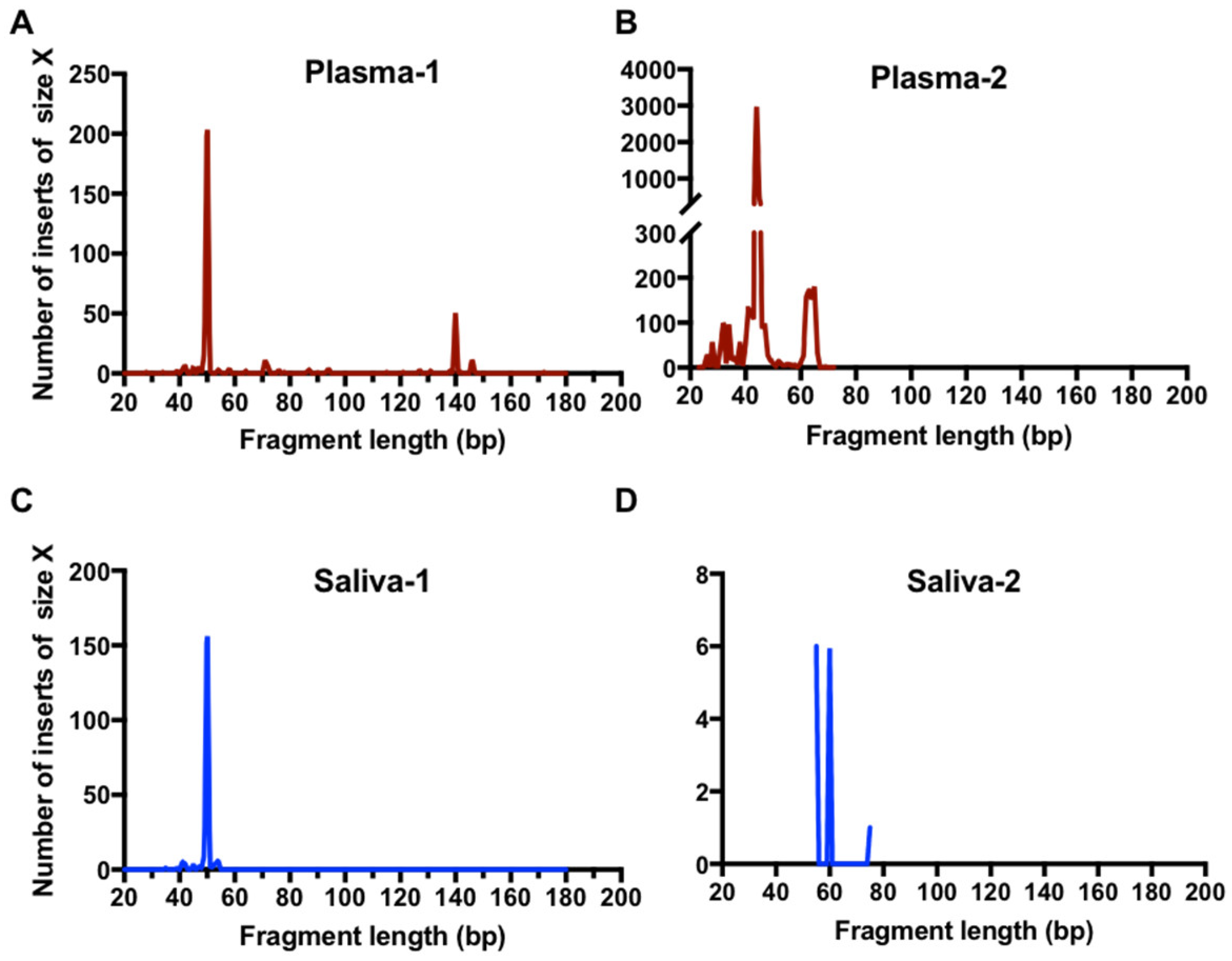
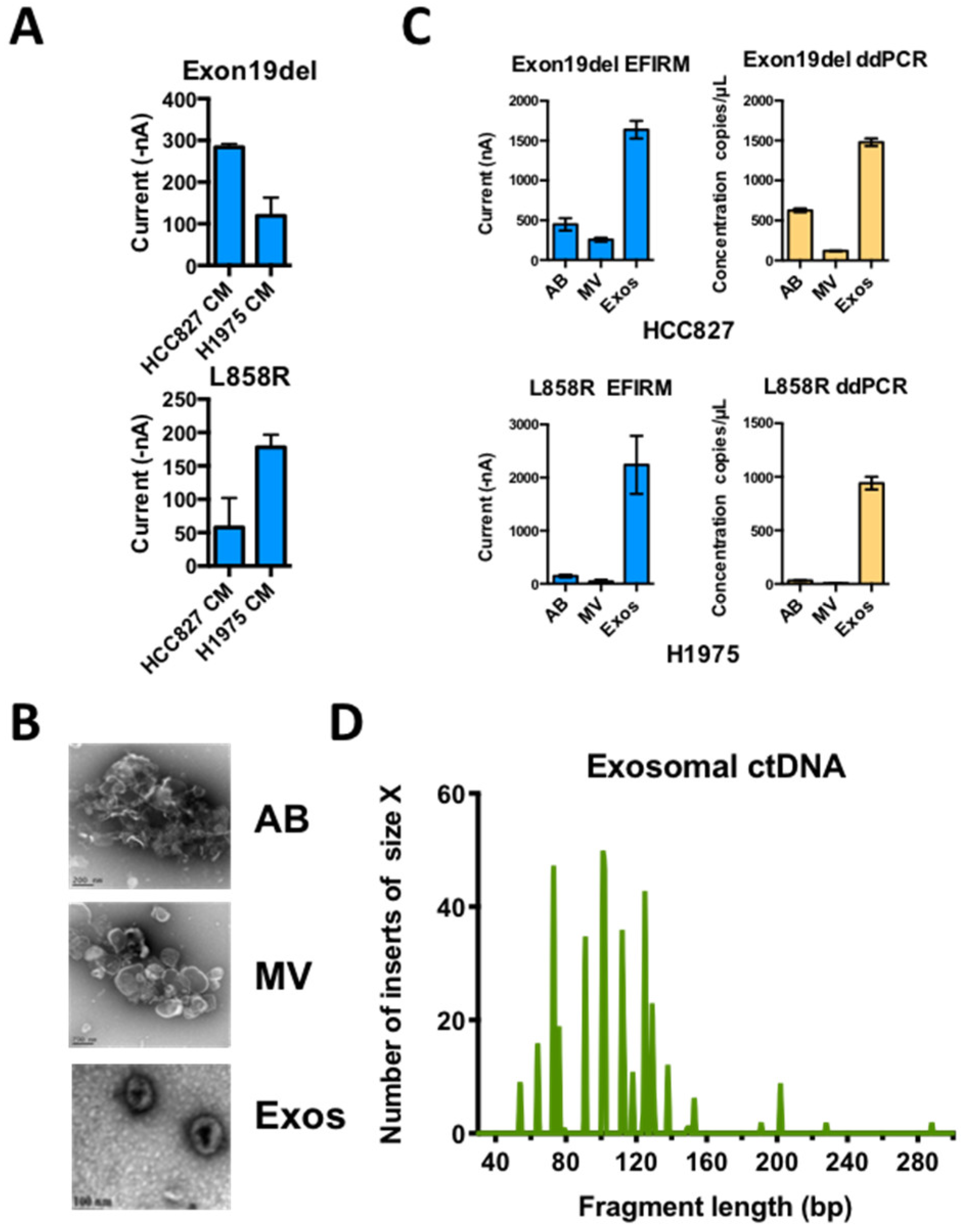
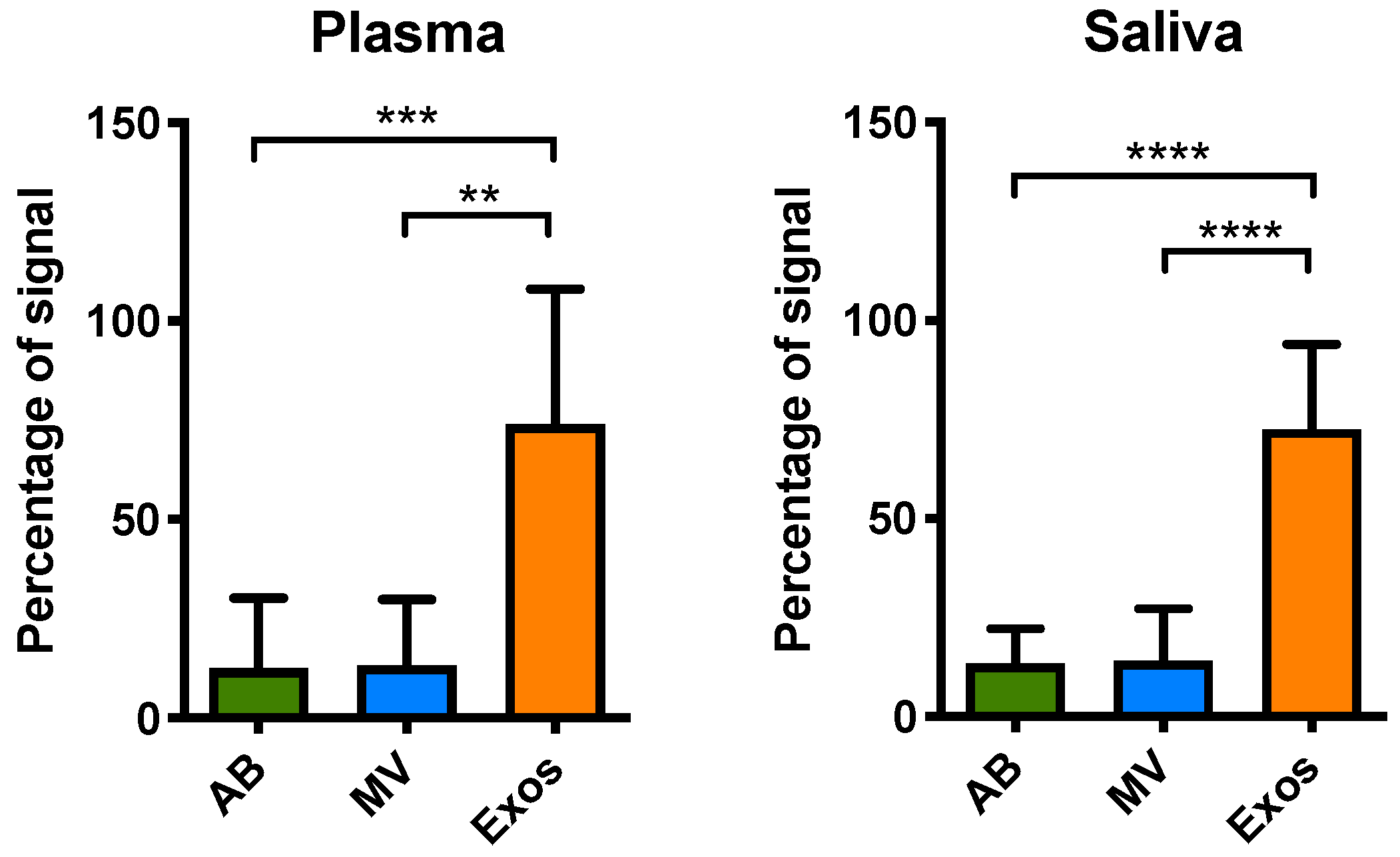
2.4. EGFR usctDNA are Predominately Located in Exosomes
3. Discussion
4. Materials and Methods
4.1. Patient Samples and Tissue Genotyping
4.2. Cell-free DNA Extraction and Quality Control
4.3. Detection of Mutated Sequences Using EFIRM Assays
4.4. Detection of Mutated Sequences Using ddPCR
4.5. Single-Stranded DNA Library Preparation and Deep Sequencing Analysis
4.6. Cell Culture and Extracellular Vesicle Isolation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reck, M.; Popat, S.; Reinmuth, N.; De Ruysscher, D.; Kerr, K.M.; Peters, S.; Group, E.G.W. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25 (Suppl. 3), iii27–iii39. [Google Scholar] [CrossRef] [PubMed]
- Sakurada, A.; Shepherd, F.A.; Tsao, M.S. Epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer: Impact of primary or secondary mutations. Clin. Lung Cancer 2006, 7 (Suppl. 4), S138–S144. [Google Scholar] [CrossRef] [PubMed]
- Ettinger, D.S.; Wood, D.E.; Aggarwal, C.; Aisner, D.L.; Akerley, W.; Bauman, J.R.; Bharat, A.; Bruno, D.S.; Chang, J.Y.; Chirieac, L.R.; et al. NCCN Guidelines Insights: Non-Small Cell Lung Cancer, Version 1.2020. J. Natl. Compr. Cancer Netw. 2019, 17, 1464–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, N.; Johnson, D.; Temin, S.; Baker, S., Jr.; Brahmer, J.; Ellis, P.M.; Giaccone, G.; Hesketh, P.J.; Jaiyesimi, I.; Leighl, N.B.; et al. Systemic Therapy for Stage IV Non-Small-Cell Lung Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 3484–3515. [Google Scholar] [CrossRef] [PubMed]
- Lindeman, N.I.; Cagle, P.T.; Aisner, D.L.; Arcila, M.E.; Beasley, M.B.; Bernicker, E.H.; Colasacco, C.; Dacic, S.; Hirsch, F.R.; Kerr, K.; et al. Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J. Thorac. Oncol. 2018, 13, 323–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heitzer, E.; Ulz, P.; Geigl, J.B. Circulating Tumor DNA as a Liquid Biopsy for Cancer. Clin. Chem. 2015, 61, 112–123. [Google Scholar] [CrossRef]
- Birkenkamp-Demtroder, K.; Nordentoft, I.; Christensen, E.; Hoyer, S.; Reinert, T.; Vang, S.; Borre, M.; Agerbaek, M.; Jensen, J.B.; Orntoft, T.F.; et al. Genomic Alterations in Liquid Biopsies from Patients with Bladder Cancer. Eur. Urol. 2016, 70, 75–82. [Google Scholar] [CrossRef]
- Wan, J.C.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Chan, K.C.; Jiang, P.; Zheng, Y.W.; Liao, G.J.; Sun, H.; Wong, J.; Siu, S.S.; Chan, W.C.; Chan, S.L.; Chan, A.T.; et al. Cancer genome scanning in plasma: Detection of tumor-associated copy number aberrations, single-nucleotide variants, and tumoral heterogeneity by massively parallel sequencing. Clin. Chem. 2013, 59, 211–224. [Google Scholar] [CrossRef] [Green Version]
- Yung, T.K.; Chan, K.C.; Mok, T.S.; Tong, J.; To, K.F.; Lo, Y.M. Single-molecule detection of epidermal growth factor receptor mutations in plasma by microfluidics digital PCR in non-small cell lung cancer patients. Clin. Cancer Res. 2009, 15, 2076–2084. [Google Scholar] [CrossRef] [Green Version]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A., Jr.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, I.H.; Lo, Y.M.; Zhang, J.; Liew, C.T.; Ng, M.H.; Wong, N.; Lai, P.B.; Lau, W.Y.; Hjelm, N.M.; Johnson, P.J. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 1999, 59, 71–73. [Google Scholar] [PubMed]
- Chan, K.C.; Jiang, P.; Chan, C.W.; Sun, K.; Wong, J.; Hui, E.P.; Chan, S.L.; Chan, W.C.; Hui, D.S.; Ng, S.S.; et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. Proc. Natl. Acad. Sci. USA 2013, 110, 18761–18768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rolfo, C.; Mack, P.C.; Scagliotti, G.V.; Baas, P.; Barlesi, F.; Bivona, T.G.; Herbst, R.S.; Mok, T.S.; Peled, N.; Pirker, R.; et al. Liquid Biopsy for Advanced Non-Small Cell Lung Cancer (NSCLC): A Statement Paper from the IASLC. J. Thorac. Oncol. 2018, 13, 1248–1268. [Google Scholar] [CrossRef] [Green Version]
- Underhill, H.R.; Kitzman, J.O.; Hellwig, S.; Welker, N.C.; Daza, R.; Baker, D.N.; Gligorich, K.M.; Rostomily, R.C.; Bronner, M.P.; Shendure, J. Fragment Length of Circulating Tumor DNA. PLoS Genet. 2016, 12, e1006162. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Bidard, F.C.; Madic, J.; Mariani, P.; Piperno-Neumann, S.; Rampanou, A.; Servois, V.; Cassoux, N.; Desjardins, L.; Milder, M.; Vaucher, I.; et al. Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int. J. Cancer 2014, 134, 1207–1213. [Google Scholar] [CrossRef]
- Martignetti, J.A.; Camacho-Vanegas, O.; Priedigkeit, N.; Camacho, C.; Pereira, E.; Lin, L.; Garnar-Wortzel, L.; Miller, D.; Losic, B.; Shah, H.; et al. Personalized ovarian cancer disease surveillance and detection of candidate therapeutic drug target in circulating tumor DNA. Neoplasia 2014, 16, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Chan, C.W.; Chan, K.C.; Cheng, S.H.; Wong, J.; Wong, V.W.; Wong, G.L.; Chan, S.L.; Mok, T.S.; Chan, H.L.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef] [Green Version]
- Schwarzenbach, H.; Eichelser, C.; Kropidlowski, J.; Janni, W.; Rack, B.; Pantel, K. Loss of heterozygosity at tumor suppressor genes detectable on fractionated circulating cell-free tumor DNA as indicator of breast cancer progression. Clin. Cancer Res. 2012, 18, 5719–5730. [Google Scholar] [CrossRef] [Green Version]
- Mouliere, F.; Piskorz, A.M.; Chandrananda, D.; Moore, E.; Morris, J.; Smith, C.G.; Goranova, T.; Heider, K.; Mair, R.; Supernat, A.; et al. Selecting Short DNA Fragments In Plasma Improves Detection Of Circulating Tumour DNA. BioRxiv 2017, 134437. [Google Scholar] [CrossRef]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahlert, C.; Melo, S.A.; Protopopov, A.; Tang, J.; Seth, S.; Koch, M.; Zhang, J.; Weitz, J.; Chin, L.; Futreal, A.; et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J. Biol. Chem. 2014, 289, 3869–3875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Che, S.P.; Kurywchak, P.; Tavormina, J.L.; Gansmo, L.B.; Correa de Sampaio, P.; Tachezy, M.; Bockhorn, M.; Gebauer, F.; Haltom, A.R.; et al. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol. Ther. 2017, 18, 158–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alix-Panabieres, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Lin, C.C.; Joon, A.; Feng, Z.D.; Troche, G.; Lira, M.E.; Chia, D.; Mao, M.; Ho, C.L.; Su, W.C.; et al. Noninvasive Saliva-based EGFR Gene Mutation Detection in Patients with Lung Cancer. Am. J. Respir. Crit. Care Med. 2014, 190, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Pu, D.; Liang, H.; Wei, F.; Akin, D.; Feng, Z.; Yan, Q.; Li, Y.; Zhen, Y.; Xu, L.; Dong, G.; et al. Evaluation of a novel saliva-based epidermal growth factor receptor mutation detection for lung cancer: A pilot study. Thorac. Cancer 2016, 7, 428–436. [Google Scholar] [CrossRef]
- Wei, F.; Strom, C.M.; Cheng, J.; Lin, C.C.; Hsu, C.Y.; Soo Hoo, G.W.; Chia, D.; Kim, Y.; Li, F.; Elashoff, D.; et al. Electric Field-Induced Release and Measurement Liquid Biopsy for Noninvasive Early Lung Cancer Assessment. J. Mol. Diagn. 2018, 20, 738–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, F.; Yang, J.P.; Wong, D.T.W. Detection of exosomal biomarker by electric field-induced release and measurement (EFIRM). Biosens. Bioelectron. 2013, 44, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Douillard, J.Y.; Ostoros, G.; Cobo, M.; Ciuleanu, T.; Cole, R.; McWalter, G.; Walker, J.; Dearden, S.; Webster, A.; Milenkova, T.; et al. Gefitinib Treatment in EGFR Mutated Caucasian NSCLC Circulating-Free Tumor DNA as a Surrogate for Determination of EGFR Status. J. Thorac. Oncol. 2014, 9, 1345–1353. [Google Scholar] [CrossRef] [Green Version]
- Sebastien, S.; Vaca-Paniagua, F.; Villar, S.; Oliver, J.; Schuster, T.; Blanche, H.; Girard, N.; Tredaniel, J.; Guilleminault, L.; Gervais, R.; et al. Noninvasive Diagnosis of Actionable Mutations by Deep Sequencing of Circulating Free DNA in Lung Cancer from Never-Smokers: A Proof-of-Concept Study from BioCAST/IFCT-1002. Clin. Cancer Res. 2014, 20, 4613–4624. [Google Scholar] [CrossRef] [Green Version]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.A.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive Detection of Response and Resistance in EGFR-Mutant Lung Cancer Using Quantitative Next-Generation Genotyping of Cell-Free Plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacher, A.G.; Paweletz, C.; Dahlberg, S.E.; Alden, R.S.; O’Connell, A.; Feeney, N.; Mach, S.L.; Janne, P.A.; Oxnard, G.R. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016, 2, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mok, T.; Wu, Y.L.; Lee, J.S.; Yu, C.J.; Sriuranpong, V.; Sandoval-Tan, J.; Ladrera, G.; Thongprasert, S.; Srimuninnimit, V.; Liao, M.L.; et al. Detection and Dynamic Changes of EGFR Mutations from Circulating Tumor DNA as a Predictor of Survival Outcomes in NSCLC Patients Treated with First-line Intercalated Erlotinib and Chemotherapy. Clin. Cancer Res. 2015, 21, 3196–3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Qing, X.; Xiumin, W.; Yali, B.; Chi, S.; Bak, S.H.; Lee, H.Y.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; et al. Longitudinal monitoring of EGFR mutations in plasma predicts outcomes of NSCLC patients treated with EGFR TKIs: Korean Lung Cancer Consortium (KLCC-12-02). Oncotarget 2016, 7, 6984–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sholl, L.M.; Aisner, D.L.; Allen, T.C.; Beasley, M.B.; Cagle, P.T.; Capelozzi, V.L.; Dacic, S.; Hariri, L.P.; Kerr, K.M.; Lantuejoul, S.; et al. Liquid Biopsy in Lung Cancer: A Perspective From Members of the Pulmonary Pathology Society. Arch. Pathol. Lab. Med. 2016, 140, 825–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnham, P.; Kim, M.S.; Agbor-Enoh, S.; Luikart, H.; Valantine, H.A.; Khush, K.K.; De Vlaminck, I. Single-stranded DNA library preparation uncovers the origin and diversity of ultrashort cell-free DNA in plasma. Sci. Rep. 2016, 6, 27859. [Google Scholar] [CrossRef]
- Snyder, M.W.; Kircher, M.; Hill, A.J.; Daza, R.M.; Shendure, J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016, 164, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224. [Google Scholar] [CrossRef] [Green Version]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef]
- Tu, M.; Cheng, J.; Chen, Y.L.; Jea, W.C.; Chen, W.L.; Chen, C.J.; Ho, C.L.; Huang, W.L.; Lin, C.C.; Su, W.C.; et al. Electric Field-Induced Release and Measurement (EFIRM): Characterization and Technical Validation of a Novel Liquid Biopsy Platform in Plasma and Saliva. J. Mol. Diagn. 2020. [Google Scholar] [CrossRef]
- Reclusa, P.; Laes, J.-F.; Malapelle, U.; Valentino, A.; Rocco, D.; Gil-Bazo, I.; Rolfo, C. EML4-ALK translocation identification in RNA exosomal cargo (ExoALK) in NSCLC patients: A novel role for liquid biopsy. Transl. Cancer Res. 2018, 8 (Suppl. 1), S76–S78. [Google Scholar] [CrossRef]
- Krug, A.K.; Enderle, D.; Karlovich, C.; Priewasser, T.; Bentink, S.; Spiel, A.; Brinkmann, K.; Emenegger, J.; Grimm, D.G.; Castellanos-Rizaldos, E.; et al. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Ann. Oncol. 2018, 29, 700–706. [Google Scholar] [CrossRef] [Green Version]
- Leighl, N.B.; Page, R.D.; Raymond, V.M.; Daniel, D.B.; Divers, S.G.; Reckamp, K.L.; Villalona-Calero, M.A.; Dix, D.; Odegaard, J.I.; Lanman, R.B.; et al. Clinical Utility of Comprehensive Cell-free DNA Analysis to Identify Genomic Biomarkers in Patients with Newly Diagnosed Metastatic Non-small Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4691–4700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, C.; Thompson, J.C.; Black, T.A.; Katz, S.I.; Fan, R.; Yee, S.S.; Chien, A.L.; Evans, T.L.; Bauml, J.M.; Alley, E.W.; et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 5, 173–180. [Google Scholar] [CrossRef]
- Mack, P.C.; Banks, K.C.; Espenschied, C.R.; Burich, R.A.; Zill, O.A.; Lee, C.E.; Riess, J.W.; Mortimer, S.A.; Talasaz, A.; Lanman, R.B.; et al. Spectrum of driver mutations and clinical impact of circulating tumor DNA analysis in non-small cell lung cancer: Analysis of over 8000 cases. Cancer 2020, 126, 3219–3228. [Google Scholar] [CrossRef] [PubMed]
- Remon, J.; Swalduz, A.; Planchard, D.; Ortiz-Cuaran, S.; Mezquita, L.; Lacroix, L.; Jovelet, C.; Rouleau, E.; Leonce, C.; De Kievit, F.; et al. Outcomes in oncogenic-addicted advanced NSCLC patients with actionable mutations identified by liquid biopsy genomic profiling using a tagged amplicon-based NGS assay. PLoS ONE 2020, 15, e0234302. [Google Scholar] [CrossRef]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating genomic features for non-invasive early lung cancer detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef]
- Lennon, A.M.; Buchanan, A.H.; Kinde, I.; Warren, A.; Honushefsky, A.; Cohain, A.T.; Ledbetter, D.H.; Sanfilippo, F.; Sheridan, K.; Rosica, D.; et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020, 369. [Google Scholar] [CrossRef]
- Devonshire, A.S.; Whale, A.S.; Gutteridge, A.; Jones, G.; Cowen, S.; Foy, C.A.; Huggett, J.F. Towards standardisation of cell-free DNA measurement in plasma: Controls for extraction efficiency, fragment size bias and quantification. Anal. Bioanal. Chem. 2014, 406, 6499–6512. [Google Scholar] [CrossRef] [Green Version]
- Mauger, F.; Dulary, C.; Daviaud, C.; Deleuze, J.F.; Tost, J. Comprehensive evaluation of methods to isolate, quantify, and characterize circulating cell-free DNA from small volumes of plasma. Anal. Bioanal. Chem. 2015, 407, 6873–6878. [Google Scholar] [CrossRef] [PubMed]
- Gansauge, M.T.; Meyer, M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 2013, 8, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Bahn, J.H.; Zhang, Q.; Li, F.; Chan, T.M.; Lin, X.; Kim, Y.; Wong, D.T.; Xiao, X. The landscape of microRNA, Piwi-interacting RNA, and circular RNA in human saliva. Clin. Chem. 2015, 61, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, F.; Wei, F.; Huang, W.-L.; Lin, C.-C.; Li, L.; Shen, M.M.; Yan, Q.; Liao, W.; Chia, D.; Tu, M.; et al. Ultra-Short Circulating Tumor DNA (usctDNA) in Plasma and Saliva of Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers 2020, 12, 2041. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082041
Li F, Wei F, Huang W-L, Lin C-C, Li L, Shen MM, Yan Q, Liao W, Chia D, Tu M, et al. Ultra-Short Circulating Tumor DNA (usctDNA) in Plasma and Saliva of Non-Small Cell Lung Cancer (NSCLC) Patients. Cancers. 2020; 12(8):2041. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082041
Chicago/Turabian StyleLi, Feng, Fang Wei, Wei-Lun Huang, Chien-Chung Lin, Liang Li, Macy M. Shen, Qingxiang Yan, Wei Liao, David Chia, Michael Tu, and et al. 2020. "Ultra-Short Circulating Tumor DNA (usctDNA) in Plasma and Saliva of Non-Small Cell Lung Cancer (NSCLC) Patients" Cancers 12, no. 8: 2041. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082041