Utility of Circulating Tumor DNA for Detection and Monitoring of Endometrial Cancer Recurrence and Progression

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Extraction and Quantitation of DNA
2.2. Targeted Next Generation Sequencing
2.3. Digital Droplet PCR
2.4. Whole Exome Sequencing and Personalized ctDNA Sequencing
2.5. Bioinformatic Analysis
3. Results
3.1. Sequencing of Primary Tumors
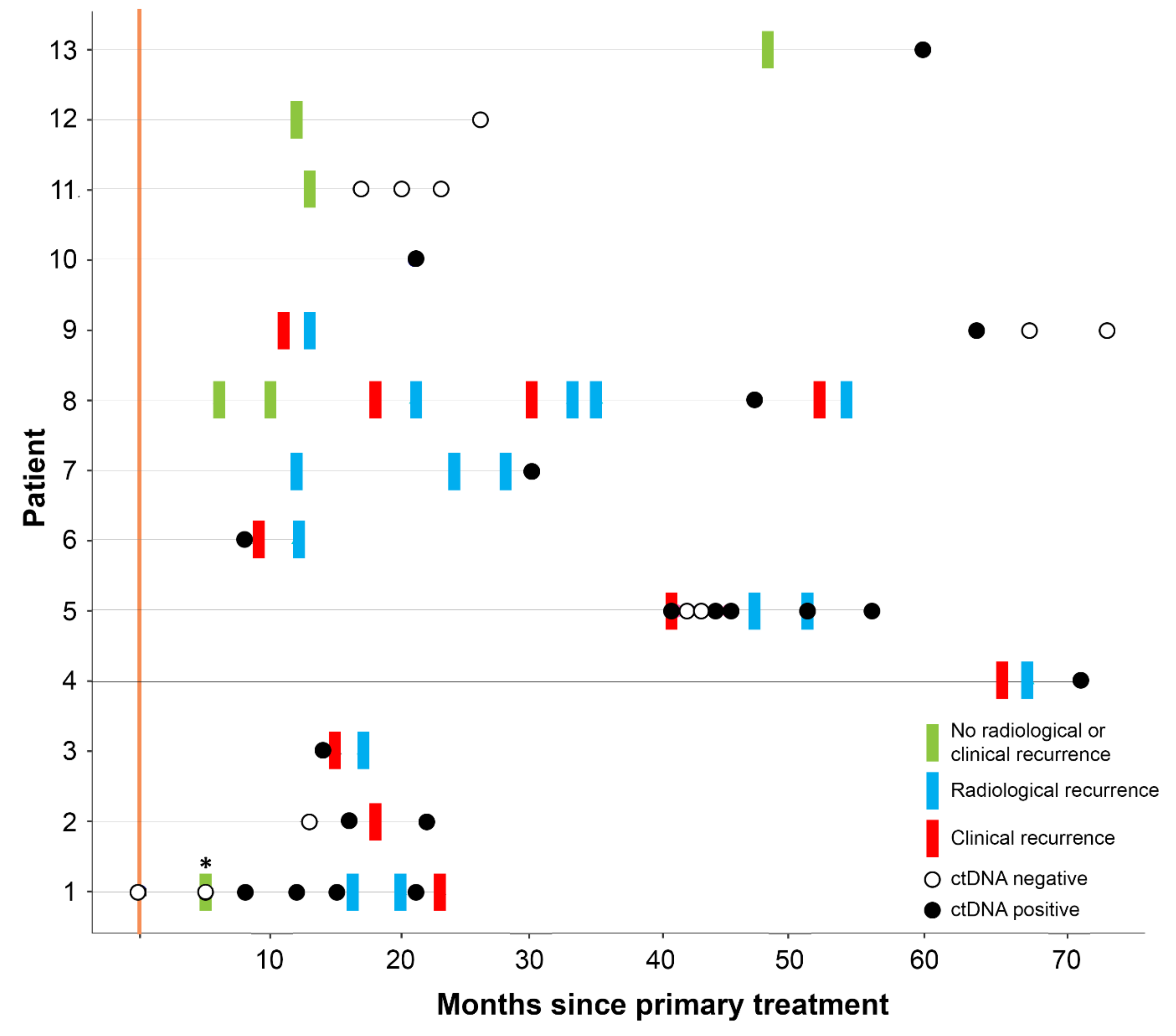
3.2. ctDNA Can Detect EC Recurrence and Progression Earlier than Scans
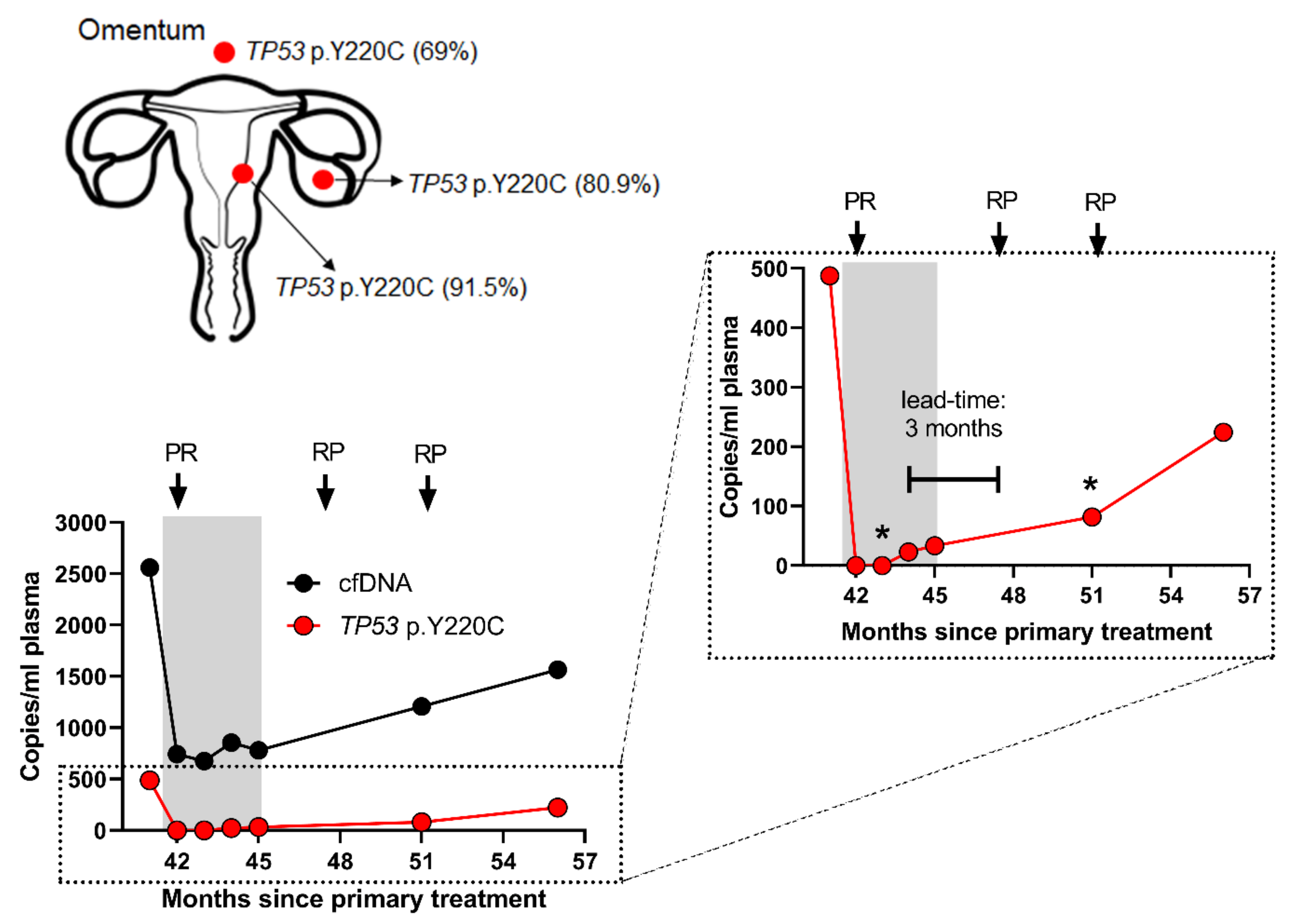
3.3. ctDNA Accurately Reflects EC Disease Kinetics during Treatment
3.4. Follow-Up of High-Risk EC Cases
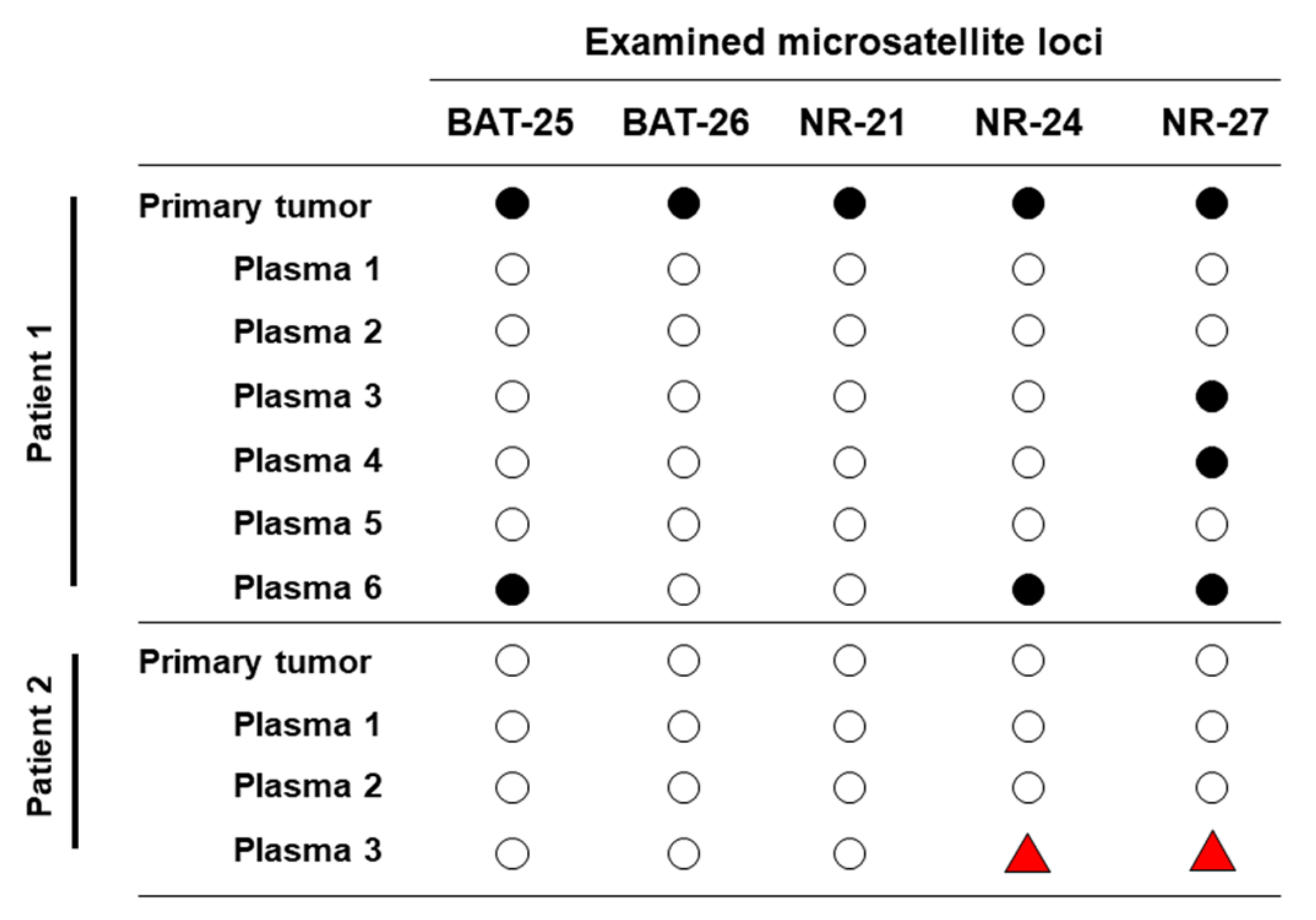
3.5. Longitudinal ctDNA Can Detect Acquired Microsatellite Instability
3.6. ctDNA is Reflective of EC Tumor Heterogeneity and Evolution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Preprint
References
- Miller, K.D.; Nogueira, L.; Mariotto, A.B.; Rowland, J.H.; Yabroff, K.R.; Alfano, C.M.; Jemal, A.; Kramer, J.L.; Siegel, R.L. Cancer treatment and survivorship statistics, 2019. CA Cancer J. Clin. 2019, 69, 363–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. Seer Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019. [Google Scholar]
- Makker, V.; Green, A.K.; Wenham, R.M.; Mutch, D.; Davidson, B.; Miller, D.S. New therapies for advanced, recurrent, and metastatic endometrial cancers. Gynecol. Oncol. Res. Pr. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Boer, S.M.; Powell, M.E.; Mileshkin, L.; Katsaros, D.; Bessette, P.; Haie-Meder, C.; Ottevanger, P.B.; Ledermann, J.A.; Khaw, P.; D’Amico, R.; et al. Adjuvant chemoradiotherapy versus radiotherapy alone in women with high-risk endometrial cancer (portec-3): Patterns of recurrence and post-hoc survival analysis of a randomised phase 3 trial. Lancet Oncol. 2019, 20, 1273–1285. [Google Scholar] [CrossRef] [Green Version]
- Abbink, K.; Zusterzeel, P.L.; Geurts-Moespot, A.J.; Herwaarden, A.E.V.; Pijnenborg, J.M.; Sweep, F.C.; Massuger, L.F. He4 is superior to ca125 in the detection of recurrent disease in high-risk endometrial cancer patients. Tumour Biol. 2018, 40, 1010428318757103. [Google Scholar] [CrossRef] [Green Version]
- Coombes, R.C.; Page, K.; Salari, R.; Hastings, R.K.; Armstrong, A.; Ahmed, S.; Ali, S.; Cleator, S.; Kenny, L.; Stebbing, J.; et al. Personalized detection of circulating tumor DNA antedates breast cancer metastatic recurrence. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 4255–4263. [Google Scholar] [CrossRef] [Green Version]
- Cristofanilli, M.; Pierga, J.Y.; Reuben, J.; Rademaker, A.; Davis, A.A.; Peeters, D.J.; Fehm, T.; Nole, F.; Gisbert-Criado, R.; Mavroudis, D.; et al. The clinical use of circulating tumor cells (ctcs) enumeration for staging of metastatic breast cancer (mbc): International expert consensus paper. Crit. Rev. Oncol. Hematol. 2019, 134, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Page, K.; Shaw, J.A.; Guttery, D.S. The liquid biopsy: Towards standardisation in preparation for prime time. Lancet Oncol. 2019, 20, 758–760. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.A.; Guttery, D.S.; Hills, A.; Fernandez-Garcia, D.; Page, K.; Rosales, B.M.; Goddard, K.S.; Hastings, R.K.; Luo, J.; Ogle, O.; et al. Mutation analysis of cell-free DNA and single circulating tumor cells in metastatic breast cancer patients with high circulating tumor cell counts. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 88–96. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Garcia, D.; Hills, A.; Page, K.; Hastings, R.K.; Toghill, B.; Goddard, K.S.; Ion, C.; Ogle, O.; Boydell, A.R.; Gleason, K.; et al. Plasma cell-free DNA (cfdna) as a predictive and prognostic marker in patients with metastatic breast cancer. Breast Cancer Res. 2019, 21, 149. [Google Scholar] [CrossRef]
- Raimondi, C.; Gradilone, A.; Naso, G.; Cortesi, E.; Gazzaniga, P. Clinical utility of circulating tumor cell counting through cellsearch((r)): The dilemma of a concept suspended in limbo. Onco Targets Ther. 2014, 7, 619–625. [Google Scholar]
- Castro-Giner, F.; Aceto, N. Tracking cancer progression: From circulating tumor cells to metastasis. Genome Med. 2020, 12, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banko, P.; Lee, S.Y.; Nagygyorgy, V.; Zrinyi, M.; Chae, C.H.; Cho, D.H.; Telekes, A. Technologies for circulating tumor cell separation from whole blood. J. Hematol. Oncol. 2019, 12, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef]
- Guttery, D.S.; Page, K.; Hills, A.; Woodley, L.; Marchese, S.D.; Rghebi, B.; Hastings, R.K.; Luo, J.; Pringle, J.H.; Stebbing, J.; et al. Noninvasive detection of activating estrogen receptor 1 (esr1) mutations in estrogen receptor-positive metastatic breast cancer. Clin. Chem. 2015, 61, 974–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsson, E.; Winter, C.; George, A.; Chen, Y.; Howlin, J.; Tang, M.H.; Dahlgren, M.; Schulz, R.; Grabau, D.; van Westen, D.; et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol. Med. 2015, 7, 1034–1047. [Google Scholar] [CrossRef] [PubMed]
- Page, K.; Guttery, D.S.; Fernandez-Garcia, D.; Hills, A.; Hastings, R.K.; Luo, J.; Goddard, K.; Shahin, V.; Woodley-Barker, L.; Rosales, B.M.; et al. Next generation sequencing of circulating cell-free DNA for evaluating mutations and gene amplification in metastatic breast cancer. Clin. Chem. 2017, 63, 532–541. [Google Scholar] [CrossRef] [Green Version]
- Shaw, J.A.; Page, K.; Blighe, K.; Hava, N.; Guttery, D.; Ward, B.; Brown, J.; Ruangpratheep, C.; Stebbing, J.; Payne, R.; et al. Genomic analysis of circulating cell-free DNA infers breast cancer dormancy. Genome Res. 2012, 22, 220–231. [Google Scholar] [CrossRef] [Green Version]
- Trigg, R.M.; Martinson, L.J.; Parpart-Li, S.; Shaw, J.A. Factors that influence quality and yield of circulating-free DNA: A systematic review of the methodology literature. Heliyon 2018, 4, e00699. [Google Scholar] [CrossRef] [Green Version]
- Gerber, T.; Taschner-Mandl, S.; Saloberger-Sindhoringer, L.; Popitsch, N.; Heitzer, E.; Witt, V.; Geyeregger, R.; Hutter, C.; Schwentner, R.; Ambros, I.M.; et al. Assessment of pre-analytical sample handling conditions for comprehensive liquid biopsy analysis. J. Mol. Diagn. 2020. [Google Scholar] [CrossRef]
- Markus, H.; Contente-Cuomo, T.; Farooq, M.; Liang, W.S.; Borad, M.J.; Sivakumar, S.; Gollins, S.; Tran, N.L.; Dhruv, H.D.; Berens, M.E.; et al. Evaluation of pre-analytical factors affecting plasma DNA analysis. Sci. Rep. 2018, 8, 7375. [Google Scholar] [CrossRef] [Green Version]
- Bos, M.K.; Angus, L.; Nasserinejad, K.; Jager, A.; Jansen, M.; Martens, J.W.M.; Sleijfer, S. Whole exome sequencing of cell-free DNA—A systematic review and bayesian individual patient data meta-analysis. Cancer Treat. Rev. 2020, 83, 101951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Chang, C.W.; Spoerke, J.M.; Yoh, K.E.; Kapoor, V.; Baudo, C.; Aimi, J.; Yu, M.; Liang-Chu, M.M.Y.; Suttmann, R.; et al. Low-pass whole-genome sequencing of circulating cell-free DNA demonstrates dynamic changes in genomic copy number in a squamous lung cancer clinical cohort. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 2254–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolivar, A.M.; Luthra, R.; Mehrotra, M.; Chen, W.; Barkoh, B.A.; Hu, P.; Zhang, W.; Broaddus, R.R. Targeted next-generation sequencing of endometrial cancer and matched circulating tumor DNA: Identification of plasma-based, tumor-associated mutations in early stage patients. Mod. Pathol. 2019, 32, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Murillas, I.; Chopra, N.; Comino-Mendez, I.; Beaney, M.; Tovey, H.; Cutts, R.J.; Swift, C.; Kriplani, D.; Afentakis, M.; Hrebien, S.; et al. Assessment of molecular relapse detection in early-stage breast cancer. JAMA Oncol. 2019, 5, 1473–1478. [Google Scholar] [CrossRef]
- Casas-Arozamena, C.; Diaz, E.; Moiola, C.P.; Alonso-Alconada, L.; Ferreiros, A.; Abalo, A.; Gil, C.L.; Oltra, S.S.; de Santiago, J.; Cabrera, S.; et al. Genomic profiling of uterine aspirates and cfdna as an integrative liquid biopsy strategy in endometrial cancer. J. Clin. Med. 2020, 9, 585. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.L.; Zhou, C.; Liam, C.K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced egfr mutation-positive non-small-cell lung cancer: Analyses from the phase iii, randomized, open-label, ensure study. Ann. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef]
- Andre, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for pik3ca-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctdna analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.T.; Tin, A.S.; et al. Analysis of plasma cell-free DNA by ultradeep sequencing in patients with stages i to iii colorectal cancer. JAMA Oncol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Vincent, J.J.; Mortimer, S.; Vowles, J.V.; Ulrich, B.C.; Banks, K.C.; Fairclough, S.R.; Zill, O.A.; Sikora, M.; Mokhtari, R.; et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 3539–3549. [Google Scholar] [CrossRef] [Green Version]
- Woodhouse, R.; Dennis, L.; Li, M.; Burns, C.; Ma, P.; Meng, W.; Dewal, N.; Vietz, C.; Hedge, P. Clinical and analytical validation of foundationone liquid cdx, a novel 324-gene blood-based comprehensive genomic profiling assay. J. Clin. Oncol. 2020, 38, e13685. [Google Scholar] [CrossRef]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Gurioli, G.; Brighi, N.; Lolli, C.; Schepisi, G.; Casadei, C.; Burgio, S.L.; Gargiulo, S.; Ravaglia, G.; Rossi, L.; et al. Plasma androgen receptor in prostate cancer. Cancers 2019, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, E.; Camacho-Vanegas, O.; Anand, S.; Sebra, R.; Catalina Camacho, S.; Garnar-Wortzel, L.; Nair, N.; Moshier, E.; Wooten, M.; Uzilov, A.; et al. Personalized circulating tumor DNA biomarkers dynamically predict treatment response and survival in gynecologic cancers. PLoS ONE 2015, 10, e0145754. [Google Scholar] [CrossRef]
- Page, K.; Guttery, D.S.; Zahra, N.; Primrose, L.; Elshaw, S.R.; Pringle, J.H.; Blighe, K.; Marchese, S.D.; Hills, A.; Woodley, L.; et al. Influence of plasma processing on recovery and analysis of circulating nucleic acids. PLoS ONE 2013, 8, e77963. [Google Scholar] [CrossRef] [Green Version]
- Page, K.; Powles, T.; Slade, M.J.; MT, D.E.B.; Walker, R.A.; Coombes, R.C.; Shaw, J.A. The importance of careful blood processing in isolation of cell-free DNA. Ann. N. Y. Acad. Sci. 2006, 1075, 313–317. [Google Scholar] [CrossRef]
- Greytak, S.R.; Engel, K.B.; Parpart-Li, S.; Murtaza, M.; Bronkhorst, A.J.; Pertile, M.D.; Moore, H.M. Harmonizing cell-free DNA collection and processing practices through evidence-based guidance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3104–3109. [Google Scholar] [CrossRef] [Green Version]
- Van Ginkel, J.H.; van den Broek, D.A.; van Kuik, J.; Linders, D.; de Weger, R.; Willems, S.M.; Huibers, M.M.H. Preanalytical blood sample workup for cell-free DNA analysis using droplet digital pcr for future molecular cancer diagnostics. Cancer Med. 2017, 6, 2297–2307. [Google Scholar] [CrossRef]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Ulz, P.; Thallinger, G.G.; Auer, M.; Graf, R.; Kashofer, K.; Jahn, S.W.; Abete, L.; Pristauz, G.; Petru, E.; Geigl, J.B.; et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat. Genet. 2016, 48, 1273–1278. [Google Scholar] [CrossRef]
- Streubel, A.; Stenzinger, A.; Stephan-Falkenau, S.; Kollmeier, J.; Misch, D.; Blum, T.G.; Bauer, T.; Landt, O.; Am Ende, A.; Schirmacher, P.; et al. Comparison of different semi-automated cfdna extraction methods in combination with umi-based targeted sequencing. Oncotarget 2019, 10, 5690–5702. [Google Scholar] [CrossRef] [PubMed]
- Erger, F.; Norling, D.; Borchert, D.; Leenen, E.; Habbig, S.; Wiesener, M.S.; Bartram, M.P.; Wenzel, A.; Becker, C.; Toliat, M.R.; et al. Cfnome—A single assay for comprehensive epigenetic analyses of cell-free DNA. Genome Med. 2020, 12, 54. [Google Scholar] [CrossRef]
- Lin, L.H.; Chang, K.W.; Kao, S.Y.; Cheng, H.W.; Liu, C.J. Increased plasma circulating cell-free DNA could be a potential marker for oral cancer. Int. J. Mol. Sci. 2018, 19, 3303. [Google Scholar] [CrossRef] [Green Version]
- Ungerer, V.; Bronkhorst, A.J.; Holdenrieder, S. Preanalytical variables that affect the outcome of cell-free DNA measurements. Crit. Rev. Clin. Lab. Sci. 2020, 1–24. [Google Scholar] [CrossRef]
- Johansson, G.; Andersson, D.; Filges, S.; Li, J.; Muth, A.; Godfrey, T.E.; Stahlberg, A. Considerations and quality controls when analyzing cell-free tumor DNA. Biomol. Detect. Quantif. 2019, 17, 100078. [Google Scholar] [CrossRef] [PubMed]
- Rimmer, A.; Phan, H.; Mathieson, I.; Iqbal, Z.; Twigg, S.R.F.; Consortium, W.G.S.; Wilkie, A.O.M.; McVean, G.; Lunter, G. Integrating mapping-, assembly- and haplotype-based approaches for calling variants in clinical sequencing applications. Nat. Genet. 2014, 46, 912–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Niu, B.; Ye, K.; Zhang, Q.; Lu, C.; Xie, M.; McLellan, M.D.; Wendl, M.C.; Ding, L. Msisensor: Microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics 2014, 30, 1015–1016. [Google Scholar] [CrossRef] [Green Version]
- Lolli, C.; De Lisi, D.; Conteduca, V.; Gurioli, G.; Scarpi, E.; Schepisi, G.; Ravaglia, G.; Menna, C.; Farolfi, A.; Altavilla, A.; et al. Testosterone levels and androgen receptor copy number variations in castration-resistant prostate cancer treated with abiraterone or enzalutamide. Prostate 2019, 79, 1211–1220. [Google Scholar] [CrossRef]
- Parkinson, C.A.; Gale, D.; Piskorz, A.M.; Biggs, H.; Hodgkin, C.; Addley, H.; Freeman, S.; Moyle, P.; Sala, E.; Sayal, K.; et al. Exploratory analysis of tp53 mutations in circulating tumour DNA as biomarkers of treatment response for patients with relapsed high-grade serous ovarian carcinoma: A retrospective study. PLoS Med. 2016, 13, e1002198. [Google Scholar] [CrossRef] [PubMed]
- Pectasides, D.; Xiros, N.; Papaxoinis, G.; Pectasides, E.; Sykiotis, C.; Koumarianou, A.; Psyrri, A.; Gaglia, A.; Kassanos, D.; Gouveris, P.; et al. Carboplatin and paclitaxel in advanced or metastatic endometrial cancer. Gynecol. Oncol. 2008, 109, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, A.; Durham, J.N.; Keefer, L.A.; Bartlett, B.R.; Zielonka, M.; Murphy, D.; White, J.R.; Lu, S.; Verner, E.L.; Ruan, F.; et al. Noninvasive detection of microsatellite instability and high tumor mutation burden in cancer patients treated with pd-1 blockade. Clin. Cancer Res. 2019, 25, 7024–7034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namlos, H.M.; Boye, K.; Mishkin, S.J.; Baroy, T.; Lorenz, S.; Bjerkehagen, B.; Stratford, E.W.; Munthe, E.; Kudlow, B.A.; Myklebost, O.; et al. Noninvasive detection of ctdna reveals intratumor heterogeneity and is associated with tumor burden in gastrointestinal stromal tumor. Mol. Cancer Ther. 2018, 17, 2473–2480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abida, W.; Cheng, M.L.; Armenia, J.; Middha, S.; Autio, K.A.; Vargas, H.A.; Rathkopf, D.; Morris, M.J.; Danila, D.C.; Slovin, S.F.; et al. Analysis of the prevalence of microsatellite instability in prostate cancer and response to immune checkpoint blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to pd-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Bang, Y.J.; Berton-Rigaud, D.; Elez, E.; Pishvaian, M.J.; Rugo, H.S.; Puzanov, I.; Mehnert, J.M.; Aung, K.L.; Lopez, J.; et al. Safety and antitumor activity of pembrolizumab in advanced programmed death ligand 1-positive endometrial cancer: Results from the keynote-028 study. J. Clin. Oncol. 2017, 35, 2535–2541. [Google Scholar] [CrossRef]
- Arend, R.C.; Jones, B.A.; Martinez, A.; Goodfellow, P. Endometrial cancer: Molecular markers and management of advanced stage disease. Gynecol. Oncol. 2018, 150, 569–580. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Ott, P.A. Pd-1 inhibitors in endometrial cancer. Oncotarget 2017, 8, 106169–106170. [Google Scholar] [CrossRef]
- Barrington, D.A.; Dilley, S.E.; Smith, H.J.; Straughn, J.M., Jr. Pembrolizumab in advanced recurrent endometrial cancer: A cost-effectiveness analysis. Gynecol. Oncol. 2019, 153, 381–384. [Google Scholar] [CrossRef]
- Relton, A.; Collins, A.; Guttery, D.S.; Gorsia, D.; McDermott, H.; Moss, E.L. Patient acceptability of ctdna testing in endometrial cancer follow-up. medRxiv 2019. [Google Scholar] [CrossRef]


 = alleilic shift not detected;
= alleilic shift not detected;  = alleilic shift detected;
= alleilic shift detected;  = alleilic shift has changed from negative to positive.
= alleilic shift not detected; = alleilic shift detected; = alleilic shift has changed from negative to positive.
= alleilic shift has changed from negative to positive.
= alleilic shift not detected; = alleilic shift detected; = alleilic shift has changed from negative to positive.

{kind=link}
{kind=link}
{kind=link}
| Patient | Histology | Stage at Diagnosis | Recurrence | Site of Recurrence | Outcome |
|---|---|---|---|---|---|
| 1 | Endometrioid G1 | IB | Yes | Pelvic | Alive with disease |
| 2 | Endometrioid G3 | IB | Yes | Distant | Died with disease |
| 3 | Serous | IA | Yes | Distant | Alive with disease |
| 4 | Endometrioid G1 | IB | Yes | Pelvic, distant | Alive with disease |
| 5 | Endometrioid G3 | IVB | Yes | Distant | Alive with disease |
| 6 | Endometrioid G2 | IVB | Yes | Distant | Died with disease |
| 7 | Endometrioid G1 | IVB | Yes | Distant | Alive with disease |
| 8 | Carcinosarcoma + Serous | IVB | Yes | Pelvic, distant | Alive with disease |
| 9 | Endometrioid G1 | IA | Yes | Vaginal vault | Alive no disease |
| 10 | Endometrioid G1 | I | No | NA | Died no disease |
| 11 | Serous | IIIC1 | No | NA | Alive no disease |
| 12 | Clear Cell | IIIB | No | NA | Alive no disease |
| 13 | Carcinosarcoma | IIIA | No | NA | Alive no disease |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moss, E.L.; Gorsia, D.N.; Collins, A.; Sandhu, P.; Foreman, N.; Gore, A.; Wood, J.; Kent, C.; Silcock, L.; Guttery, D.S. Utility of Circulating Tumor DNA for Detection and Monitoring of Endometrial Cancer Recurrence and Progression. Cancers 2020, 12, 2231. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082231
Moss EL, Gorsia DN, Collins A, Sandhu P, Foreman N, Gore A, Wood J, Kent C, Silcock L, Guttery DS. Utility of Circulating Tumor DNA for Detection and Monitoring of Endometrial Cancer Recurrence and Progression. Cancers. 2020; 12(8):2231. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082231
Chicago/Turabian StyleMoss, Esther L., Diviya N. Gorsia, Anna Collins, Pavandeep Sandhu, Nalini Foreman, Anupama Gore, Joey Wood, Christopher Kent, Lee Silcock, and David S. Guttery. 2020. "Utility of Circulating Tumor DNA for Detection and Monitoring of Endometrial Cancer Recurrence and Progression" Cancers 12, no. 8: 2231. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12082231