Fibronectin-Expressing Mesenchymal Tumor Cells Promote Breast Cancer Metastasis

 , , , , , and
, , , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
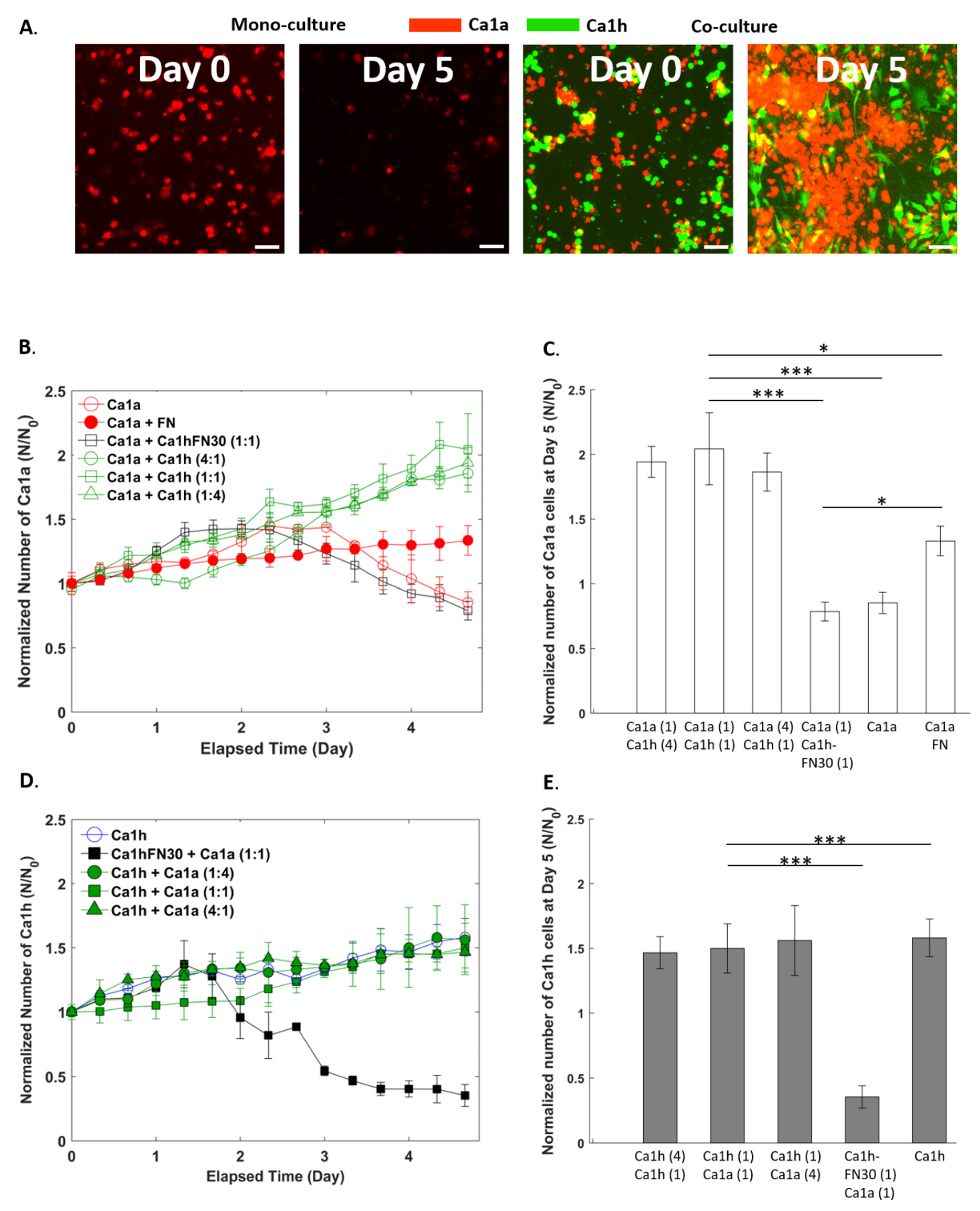
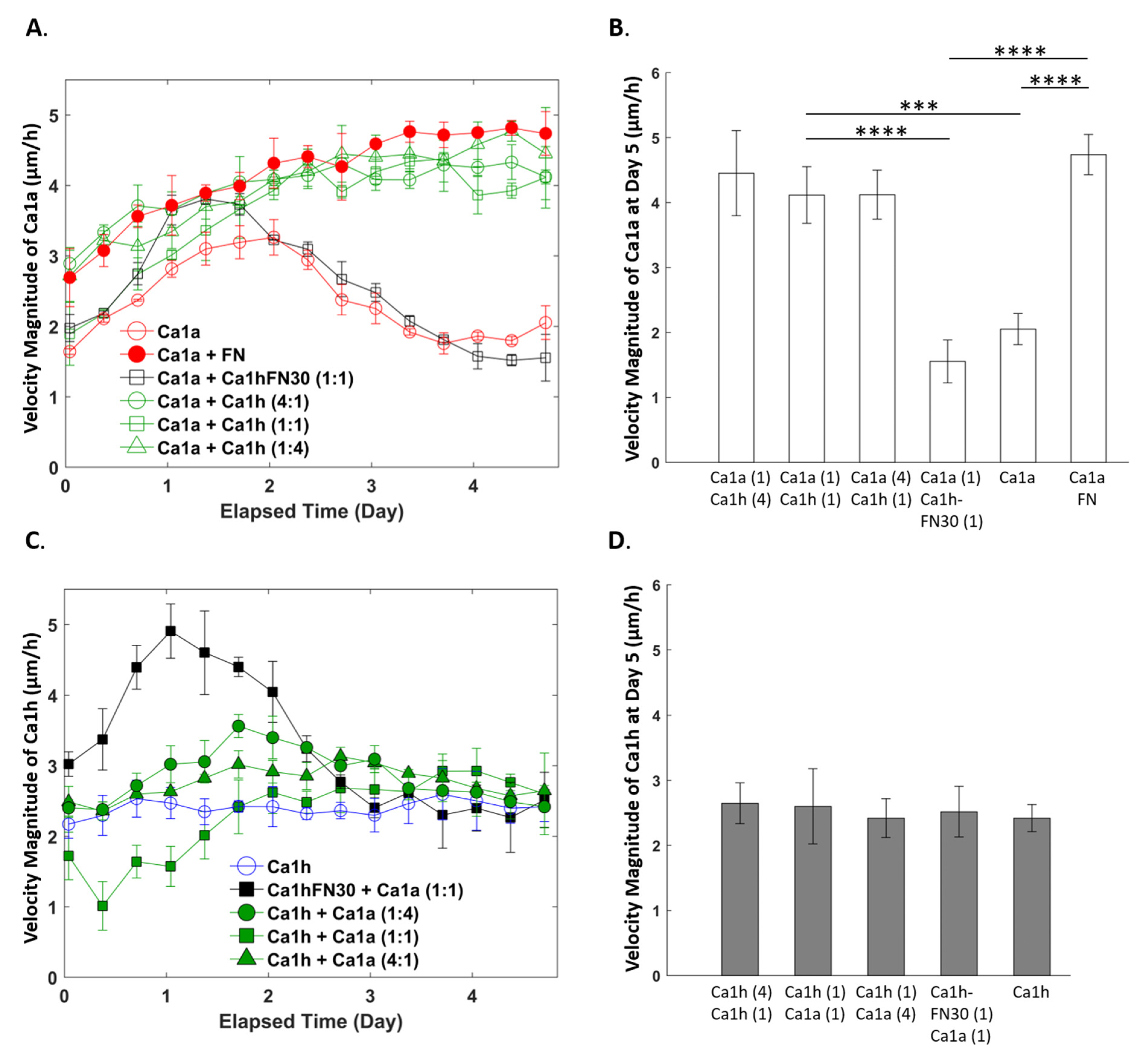
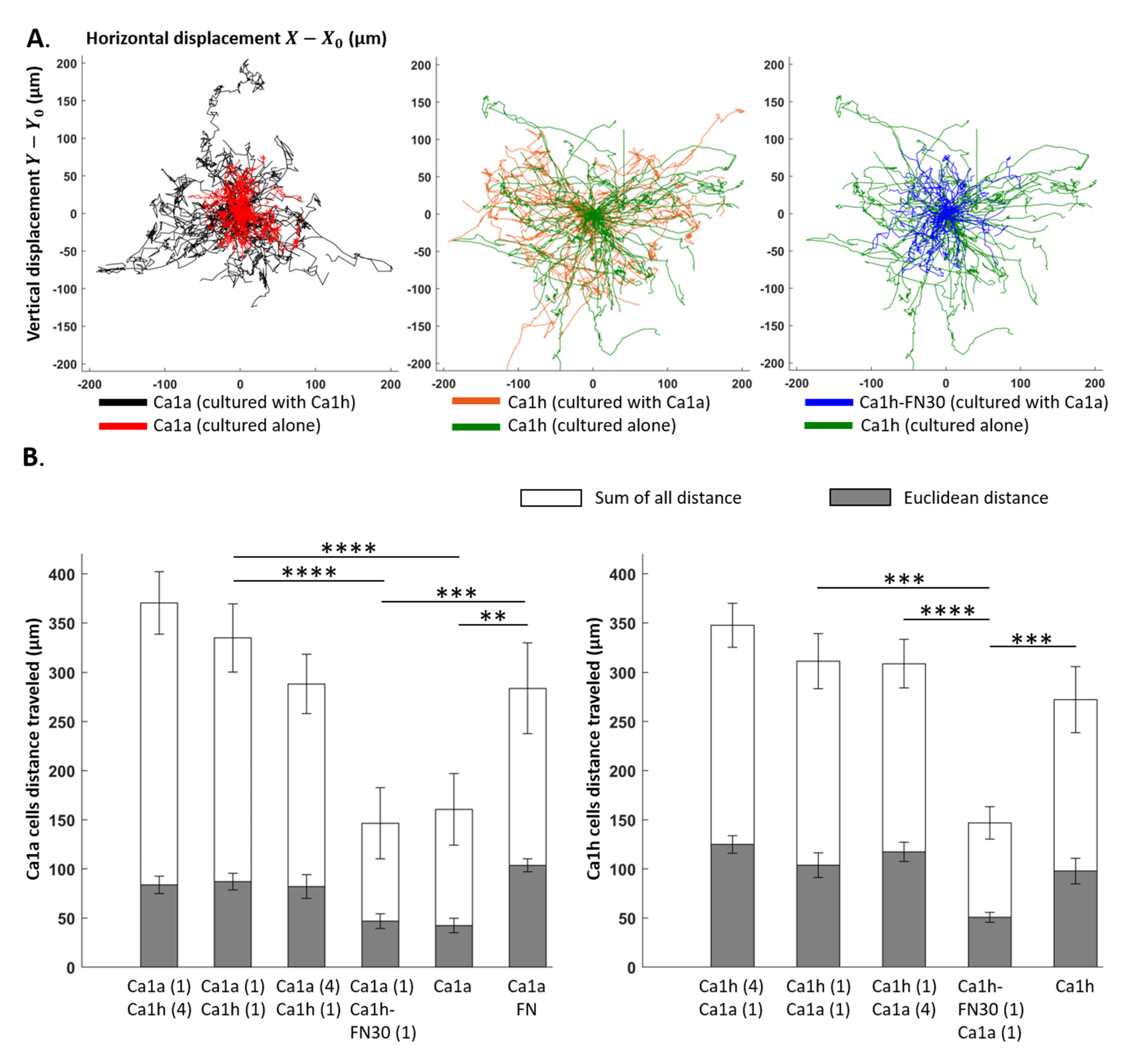
2.1. Comparison of Measurements within Culture Groups
2.2. Effect of Co-Culture on Cell Proliferation
2.3. Effect of Co-Culture on Cell Migration Velocity
2.4. Effect of Co-Culture on Trajectory
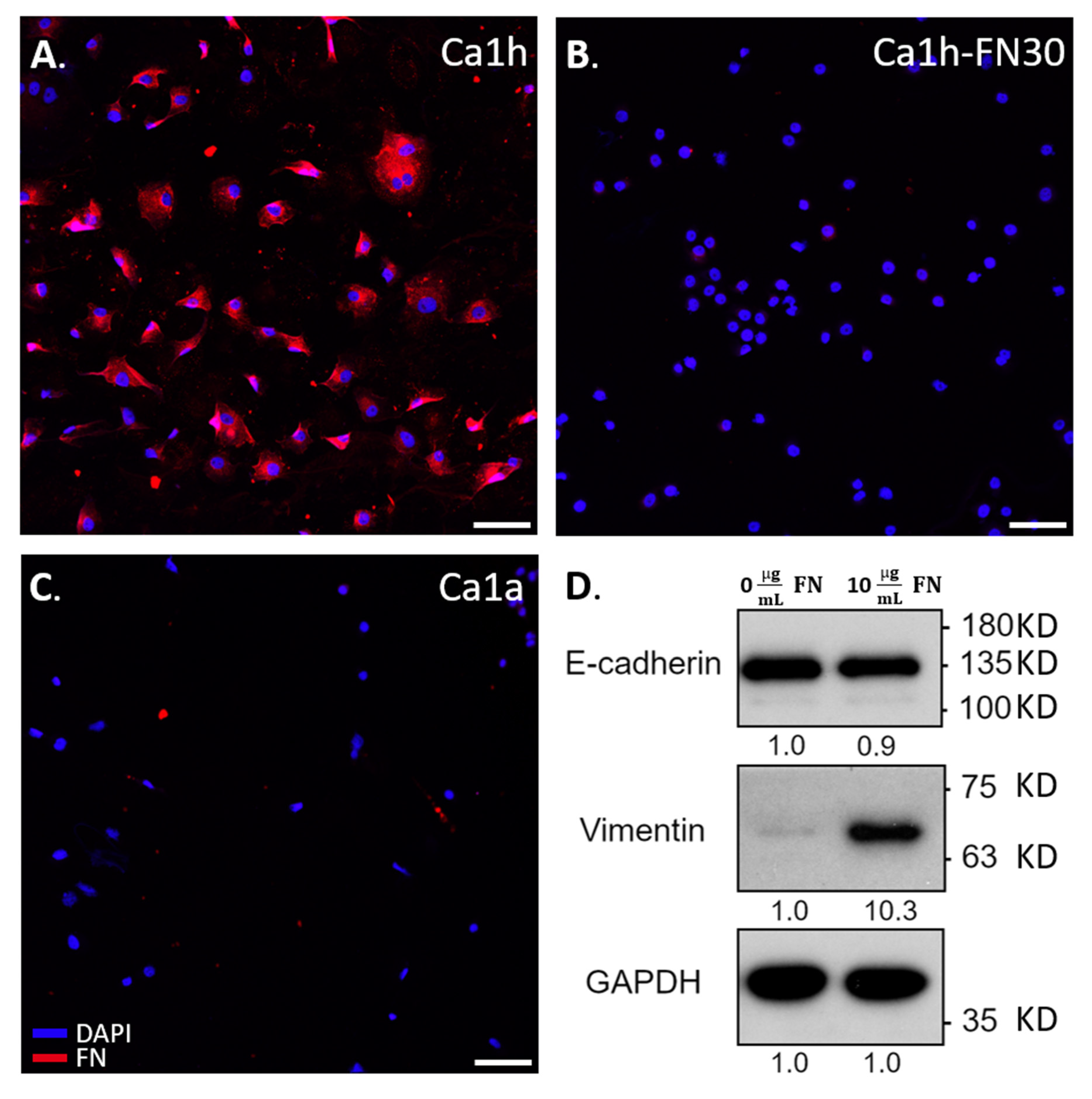
2.5. Immunohistochemical Staining and Immunoblotting
3. Discussion
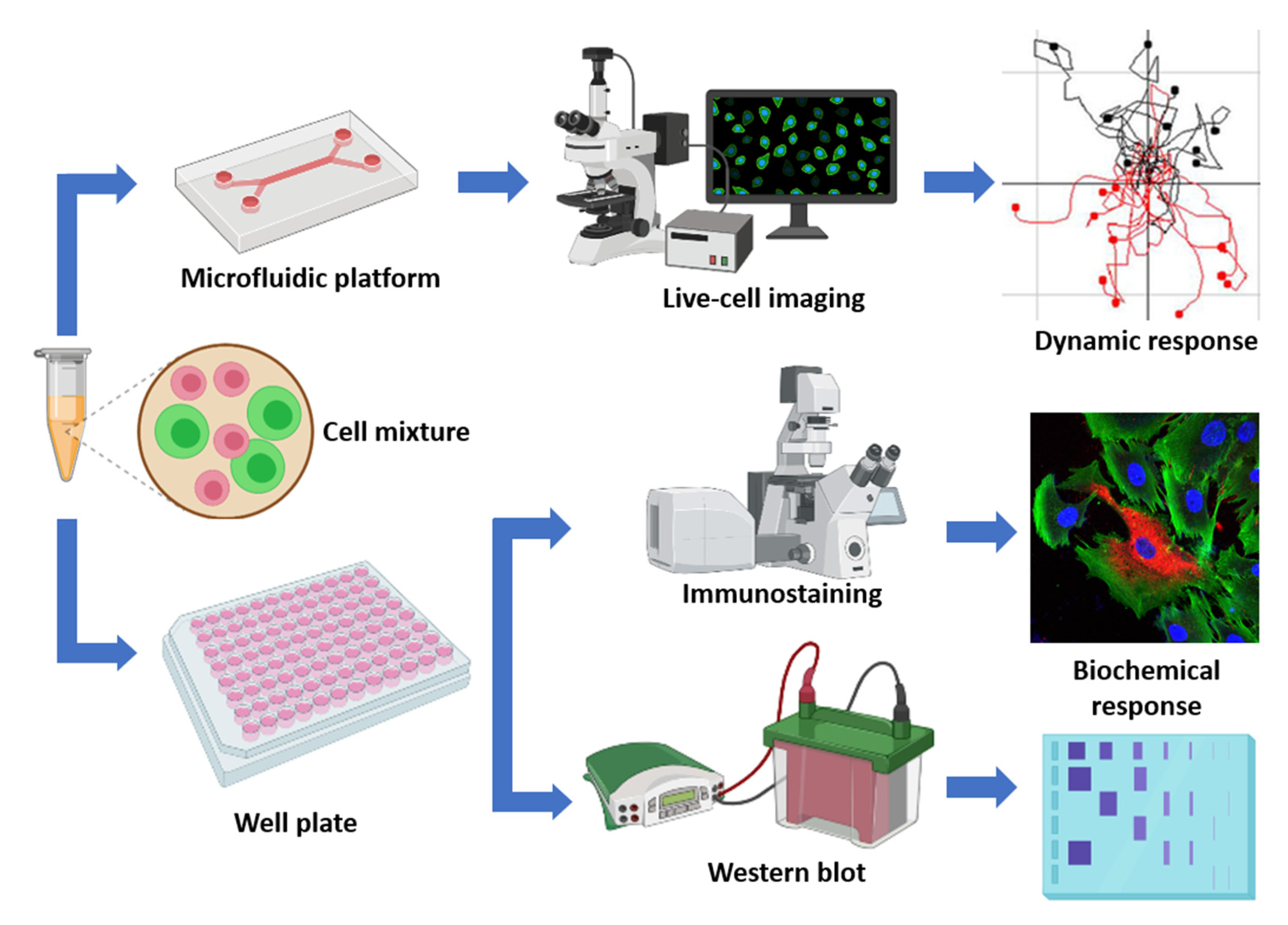
4. Materials and Methods
4.1. Cell Culture
4.2. Microfluidic 3D Hydrogel Assays
4.3. Time-lapse Imaging
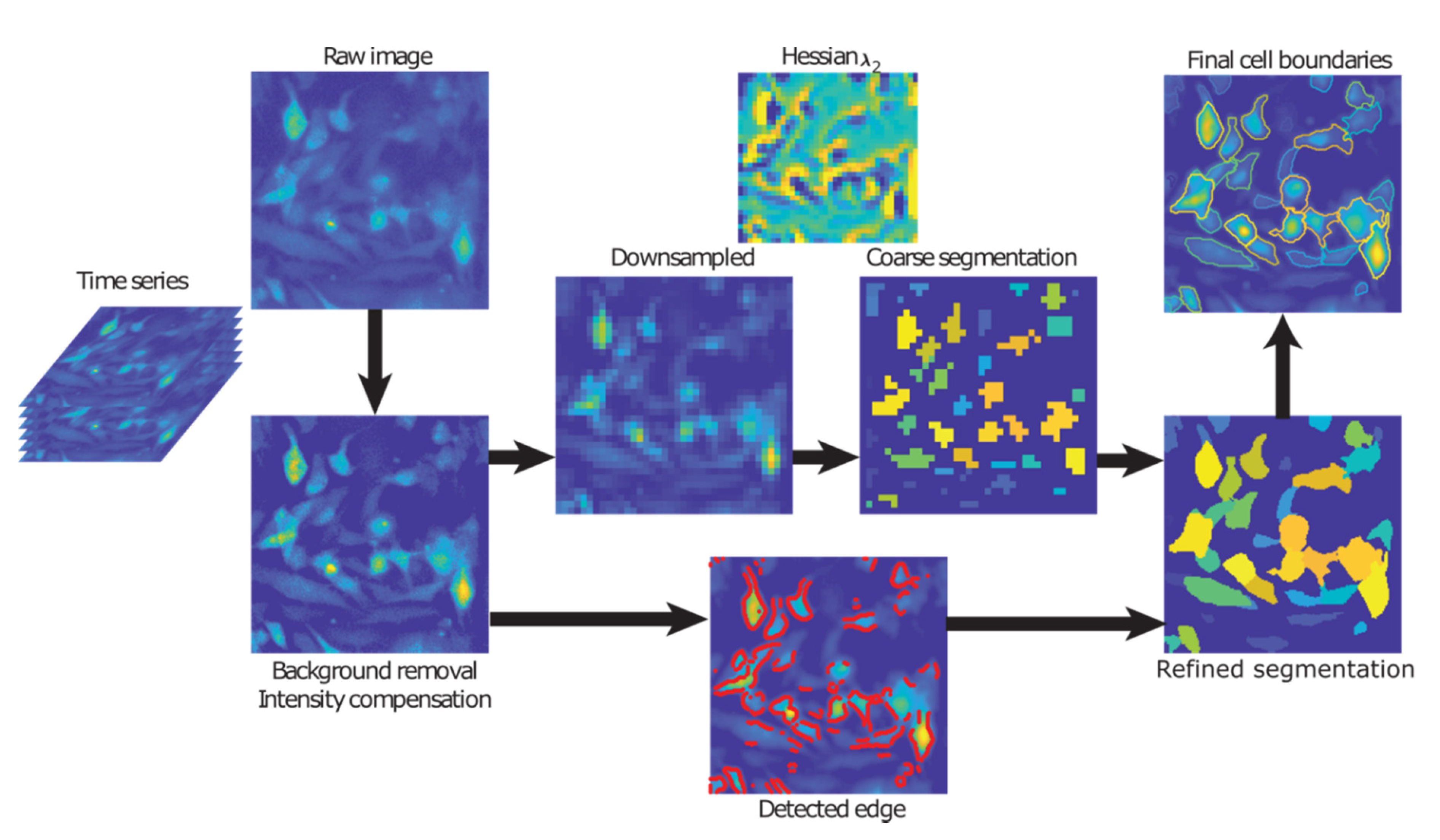
4.4. Cell Segmentation
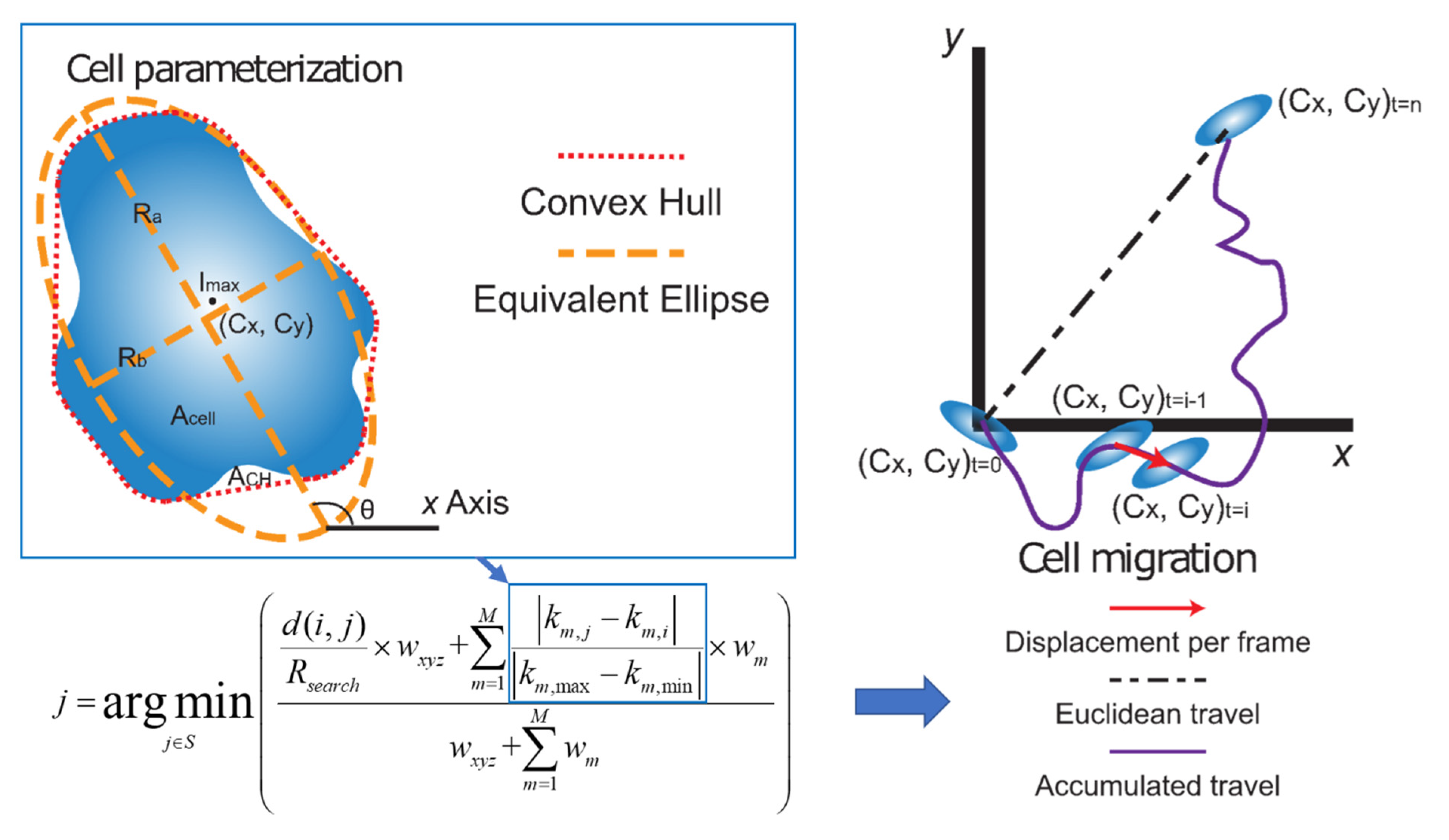
4.5. Cell Parameterization
4.6. Quantifying Cell Growth and Motility
4.7. Immunohistochemical Staining and Immunoblotting
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Niikura, N.; Liu, J.; Hayashi, N.; Mittendorf, E.A.; Gong, Y.; Palla, S.L.; Tokuda, Y.; Gonzalez-Angulo, A.M.; Hortobagyi, G.N.; Ueno, N.T. Loss of Human Epidermal Growth Factor Receptor 2 (HER2) Expression in Metastatic Sites of HER2-Overexpressing Primary Breast Tumors. J. Clin. Oncol. 2012, 30, 593–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röcken, M. Early tumor dissemination, but late metastasis: Insights into tumor dormancy. J. Clin. Investig. 2010, 120, 1800–1803. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Weinberg, R.A. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, C.E.; Ma, J.; Bryan, L.; Jemal, A. Breast cancer statistics, 2013. CA A Cancer J. Clin. 2013, 64, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 215. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.R.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Talmadge, J.E. Clonal Selection of Metastasis within the Life History of a Tumor. Cancer Res. 2007, 67, 11471–11475. [Google Scholar] [CrossRef] [Green Version]
- Welch, D.R.; Hurst, D.R. Defining the Hallmarks of Metastasis. Cancer Res. 2019, 79, 3011–3027. [Google Scholar] [CrossRef]
- Shinde, A.; Libring, S.; Alpsoy, A.; Abdullah, A.; Schaber, J.A.; Solorio, L.; Wendt, M.K. Autocrine Fibronectin Inhibits Breast Cancer Metastasis. Mol. Cancer Res. 2018, 16, 1579–1589. [Google Scholar] [CrossRef] [Green Version]
- Balanis, N.; Wendt, M.K.; Schiemann, B.J.; Wang, Z.; Schiemann, W.P.; Carlin, C. Epithelial to Mesenchymal Transition Promotes Breast Cancer Progression via a Fibronectin-dependent STAT3 Signaling Pathway*. J. Biol. Chem. 2013, 288, 17954–17967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savagner, P. The epithelial–mesenchymal transition (EMT) phenomenon. Ann. Oncol. 2010, 21, vii89–vii92. [Google Scholar] [CrossRef]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Heterogeneity in breast cancer. J. Clin. Investig. 2011, 121, 3786–3788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinde, A.; Paez, J.S.; Libring, S.; Hopkins, K.; Solorio, L.; Wendt, M.K. Transglutaminase-2 facilitates extracellular vesicle-mediated establishment of the metastatic niche. Oncogenesis 2020, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Libring, S.M.; Shinde, A.; Chanda, M.K.; Nuru, M.; George, H.; Saleh, A.M.; Abdullah, A.; Kinzer-Ursem, T.L.; Calve, S.; Wendt, M.K.; et al. The Dynamic Relationship of Breast Cancer Cells and Fibroblasts in Fibronectin Accumulation at Primary and Metastatic Tumor Sites. Cancers 2020, 12, 1270. [Google Scholar] [CrossRef] [PubMed]
- Lochter, A.; Bissell, M.J. Involvement of extracellular matrix constituents in breast cancer. Semin. Cancer Biol. 1995, 6, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Ardekani, A.M.; Vlachos, P.P. Microscale, scanning defocusing volumetric particle-tracking velocimetry. Exp. Fluids 2019, 60, 89. [Google Scholar] [CrossRef]
- Cardwell, N.D.; Vlachos, P.P.; Thole, K.A. A multi-parametric particle-pairing algorithm for particle tracking in single and multiphase flows. Meas. Sci. Technol. 2011, 22, 105406. [Google Scholar] [CrossRef]
- Shaw, P.G.; Chaerkady, R.; Wang, T.; Vasilatos, S.; Huang, Y.; Van Houten, B.; Pandey, A.; Davidson, N.E. Integrated Proteomic and Metabolic Analysis of Breast Cancer Progression. PLoS ONE 2013, 8, e76220. [Google Scholar] [CrossRef] [Green Version]
- Marx, E.; Mueller-Klieser, W.; Vaupel, P. Lactate-induced inhibition of tumor cell proliferation. Int. J. Radiat. Oncol. 1988, 14, 947–955. [Google Scholar] [CrossRef]
- Evdokimova, V.; Tognon, C.; Ng, T.; Sorensen, P.H. Reduced proliferation and enhanced migration: Two sides of the same coin? Molecular mechanisms of metastatic progression by YB-1. Cell Cycle 2009, 8, 2901–2906. [Google Scholar] [CrossRef] [PubMed]
- Floor, S.; Van Staveren, W.C.G.; Larsimont, D.; Dumont, J.E.; Maenhaut, C. Cancer cells in epithelial-to-mesenchymal transition and tumor-propagating–cancer stem cells: Distinct, overlapping or same populations. Oncogene 2011, 30, 4609–4621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2013, 33, 1649–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraley, S.I.; Wu, P.-H.; He, L.; Feng, Y.; Krisnamurthy, R.; Longmore, G.D.; Wirtz, D. Three-dimensional matrix fiber alignment modulates cell migration and MT1-MMP utility by spatially and temporally directing protrusions. Sci. Rep. 2015, 5, 14580. [Google Scholar] [CrossRef] [Green Version]
- Tomášová, L.; Guttenberg, Z.; Hoffmann, B.; Merkel, R. Advanced 2D/3D cell migration assay for faster evaluation of chemotaxis of slow-moving cells. PLoS ONE 2019, 14, e0219708. [Google Scholar] [CrossRef] [Green Version]
- Zantl, R.; Horn, E. Chemotaxis of Slow Migrating Mammalian Cells Analysed by Video Microscopy. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2011; Volume 769, pp. 191–203. [Google Scholar]
- Zicha, D.; Dunn, G.A.; Brown, A.F. A new direct-viewing chemotaxis chamber. J. Cell Sci. 1991, 99, 769–775. [Google Scholar]
- Zigmond, S.H. Ability of polymorphonuclear leukocytes to orient in gradients of chemotactic factors. J. Cell Biol. 1977, 75, 606–616. [Google Scholar] [CrossRef] [Green Version]
- Boyden, S. The chemotactic effect of mixtures of antibody and antigen on polymorphonuclear leucocytes. J. Exp. Med. 1962, 115, 453–466. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Basheer, H.A.; Ayuso, J.M.; Ahmet, D.; Mazzini, M.; Patel, R.; Shnyder, S.D.; Vinader, V.; Afarinkia, K. Agarose Spot as a Comparative Method for in situ Analysis of Simultaneous Chemotactic Responses to Multiple Chemokines. Sci. Rep. 2017, 7, 1075. [Google Scholar] [CrossRef]
- Canny, J. A Computational Approach to Edge Detection. IEEE Trans. Pattern Anal. Mach. Intell. 1986, 8, 679–698. [Google Scholar] [CrossRef] [PubMed]
- Ulman, V.; Maška, M.; Magnusson, K.E.G.; Ronneberger, O.; Haubold, C.; Harder, N.; Matula, P.; Matula, P.; Svoboda, D.; Radojevic, M.; et al. An objective comparison of cell-tracking algorithms. Nat. Methods 2017, 14, 1141–1152. [Google Scholar] [CrossRef]
- Sbalzarini, I.F.; Koumoutsakos, P. Feature point tracking and trajectory analysis for video imaging in cell biology. J. Struct. Biol. 2005, 151, 182–195. [Google Scholar] [CrossRef]
- Rosin, P.L. COMPUTING GLOBAL SHAPE MEASURES. In Handbook of Pattern Recognition and Computer Vision; World Scientific Pub Co Pte Ltd.: Singapore, 2005; pp. 177–196. [Google Scholar]
- Angelucci, C.; Maulucci, G.; Lama, G.; Proietti, G.; Colabianchi, A.; Papi, M.; Maiorana, A.; De Spirito, M.; Micera, A.; Balzamino, B.O.; et al. Epithelial-Stromal Interactions in Human Breast Cancer: Effects on Adhesion, Plasma Membrane Fluidity and Migration Speed and Directness. PLoS ONE 2012, 7, e50804. [Google Scholar] [CrossRef] [PubMed]
- Semmling, V.; Lukacs-Kornek, V.; Thaiss, C.A.; Quast, T.; Hochheiser, K.; Panzer, U.; Rossjohn, J.; Perlmutter, P.; Cao, J.; Godfrey, D.I.; et al. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell–licensed DCs. Nat. Immunol. 2010, 11, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Zengel, P.; Nguyen-Hoang, A.; Schildhammer, C.; Zantl, R.; Kahl, V.; Horn, E. μ-Slide Chemotaxis: A new chamber for long-term chemotaxis studies. BMC Cell Biol. 2011, 12, 21. [Google Scholar] [CrossRef] [Green Version]
- Margiotta, A.; Progida, C.; Bakke, O.; Bucci, C. Characterization of the role of RILP in cell migration. Eur. J. Histochem. 2017, 61, 2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jun, B.H.; Guo, T.; Libring, S.; Chanda, M.K.; Paez, J.S.; Shinde, A.; Wendt, M.K.; Vlachos, P.P.; Solorio, L. Fibronectin-Expressing Mesenchymal Tumor Cells Promote Breast Cancer Metastasis. Cancers 2020, 12, 2553. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092553
Jun BH, Guo T, Libring S, Chanda MK, Paez JS, Shinde A, Wendt MK, Vlachos PP, Solorio L. Fibronectin-Expressing Mesenchymal Tumor Cells Promote Breast Cancer Metastasis. Cancers. 2020; 12(9):2553. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092553
Chicago/Turabian StyleJun, Brian H., Tianqi Guo, Sarah Libring, Monica K. Chanda, Juan Sebastian Paez, Aparna Shinde, Michael K. Wendt, Pavlos P. Vlachos, and Luis Solorio. 2020. "Fibronectin-Expressing Mesenchymal Tumor Cells Promote Breast Cancer Metastasis" Cancers 12, no. 9: 2553. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers12092553