
Murine Macrophages Modulate Their Inflammatory Profile in Response to Gas Plasma-Inactivated Pancreatic Cancer Cells

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Plasma Exposure Inhibited Pancreatic Cancer Cell Metabolism and Migration In Vitro
2.2. Plasma Exposure Inhibited PDA Growth via Apoptosis in the TUM-CAM Model In Ovo
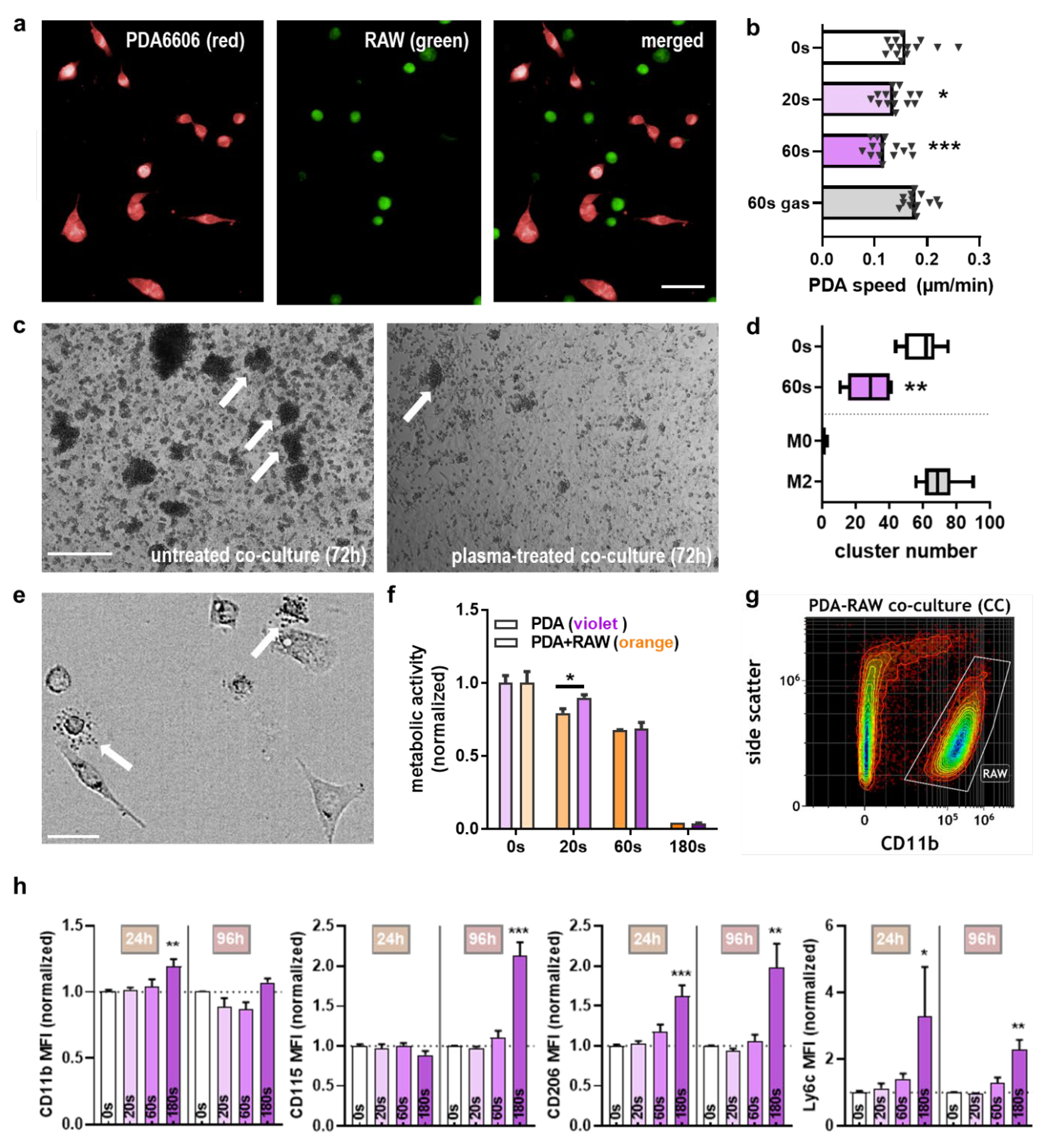
2.3. Macrophages Recognized Plasma-Treated PDA6606 Cells
2.4. Chemokine and Cytokine Profiles of Mono- and Co-Cultures with Plasma Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Plasma Treatment
4.3. ROS Analysis
4.4. Metabolic Activity and Viability
4.5. Cell Migration and Quantitative Microscopy
4.6. TUM-CAM Assay and Tumor Tissue Analysis
4.7. In Ovo Tumor Tissue Analysis
4.8. Flow Cytometry
4.9. ELISA
4.10. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of pancreatic cancer: Global trends, etiology and risk factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Esposito, I.; Kleeff, J.; Bergmann, F.; Reiser, C.; Herpel, E.; Friess, H.; Schirmacher, P.; Buchler, M.W. Most pancreatic cancer resections are r1 resections. Ann. Surg. Oncol. 2008, 15, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Zhang, Y.; Yu, X.; Yang, J.; LeBrun, D.G.; Chen, C.; Yao, Q.; Li, M. Overcoming drug resistance in pancreatic cancer. Expert Opin. Ther. Targets 2011, 15, 817–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellenrieder, V.; Adler, G.; Gress, T.M. Invasion and metastasis in pancreatic cancer. Ann. Oncol. 1999, 10, S46–S50. [Google Scholar] [CrossRef]
- Wang, H.C.; Hung, W.C.; Chen, L.T.; Pan, M.R. From friend to enemy: Dissecting the functional alteration of immunoregulatory components during pancreatic tumorigenesis. Int. J. Mol. Sci. 2018, 19, 3584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, R.; Yue, W.; Lattime, E.C.; Stein, M.N.; Xu, Q.; Tan, X.L. Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget 2016, 7, 50735–50754. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Cheng, L.; Qian, W.; Jiang, Z.; Sun, L.; Zhao, Y.; Zhou, Y.; Zhao, L.; Wang, P.; Duan, W.; et al. Itraconazole inhibits invasion and migration of pancreatic cancer cells by suppressing tgf-beta/smad2/3 signaling. Oncol. Rep. 2018, 39, 1573–1582. [Google Scholar] [CrossRef] [Green Version]
- Abdel Hadi, N.; Reyes-Castellanos, G.; Carrier, A. Targeting redox metabolism in pancreatic cancer. Int. J. Mol. Sci. 2021, 22, 1534. [Google Scholar] [CrossRef]
- Nakamura, T.; Fidler, I.J.; Coombes, K.R. Gene expression profile of metastatic human pancreatic cancer cells depends on the organ microenvironment. Cancer Res. 2007, 67, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Shields, M.A.; Dangi-Garimella, S.; Redig, A.J.; Munshi, H.G. Biochemical role of the collagen-rich tumour microenvironment in pancreatic cancer progression. Biochem. J. 2012, 441, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Graves, D.B. Mechanisms of plasma medicine: Coupling plasma physics, biochemistry, and biology. IEEE Trans. Radiat. Plasma Med. Sci. 2017, 1, 281–292. [Google Scholar] [CrossRef]
- Dubuc, A.; Monsarrat, P.; Virard, F.; Merbahi, N.; Sarrette, J.P.; Laurencin-Dalicieux, S.; Cousty, S. Use of cold-atmospheric plasma in oncology: A concise systematic review. Ther. Adv. Med. Oncol. 2018, 10, 1758835918786475. [Google Scholar] [CrossRef]
- Privat-Maldonado, A.; Schmidt, A.; Lin, A.; Weltmann, K.D.; Wende, K.; Bogaerts, A.; Bekeschus, S. Ros from physical plasmas: Redox chemistry for biomedical therapy. Oxid. Med. Cell. Longev. 2019, 2019, 9062098. [Google Scholar] [CrossRef] [Green Version]
- Brulle, L.; Vandamme, M.; Ries, D.; Martel, E.; Robert, E.; Lerondel, S.; Trichet, V.; Richard, S.; Pouvesle, J.M.; Le Pape, A. Effects of a non thermal plasma treatment alone or in combination with gemcitabine in a mia paca2-luc orthotopic pancreatic carcinoma model. PLoS ONE 2012, 7, e52653. [Google Scholar] [CrossRef]
- Liedtke, K.R.; Diedrich, S.; Pati, O.; Freund, E.; Flieger, R.; Heidecke, C.D.; Partecke, L.I.; Bekeschus, S. Cold physical plasma selectively elicits apoptosis in murine pancreatic cancer cells in vitro and in ovo. Anticancer Res. 2018, 38, 5655–5663. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Yamada, S.; Torii, K.; Takeda, S.; Nakamura, K.; Tanaka, H.; Kajiyama, H.; Kanda, M.; Fujii, T.; Nakayama, G.; et al. Effectiveness of plasma treatment on pancreatic cancer cells. Int. J. Oncol. 2015, 47, 1655–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partecke, L.I.; Gunther, C.; Hagemann, S.; Jacobi, C.; Merkel, M.; Sendler, M.; van Rooijen, N.; Kading, A.; Nguyen Trung, D.; Lorenz, E.; et al. Induction of m2-macrophages by tumour cells and tumour growth promotion by m2-macrophages: A quid pro quo in pancreatic cancer. Pancreatology 2013, 13, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Bekeschus, S.; Schmidt, A.; Weltmann, K.-D.; von Woedtke, T. The plasma jet kinpen–a powerful tool for wound healing. Clin. Plasma Med. 2016, 4, 19–28. [Google Scholar] [CrossRef]
- Liedtke, K.R.; Bekeschus, S.; Kaeding, A.; Hackbarth, C.; Kuehn, J.P.; Heidecke, C.D.; von Bernstorff, W.; von Woedtke, T.; Partecke, L.I. Non-thermal plasma-treated solution demonstrates antitumor activity against pancreatic cancer cells in vitro and in vivo. Sci. Rep. 2017, 7, 8319. [Google Scholar] [CrossRef]
- Bekeschus, S.; Freund, E.; Spadola, C.; Privat-Maldonado, A.; Hackbarth, C.; Bogaerts, A.; Schmidt, A.; Wende, K.; Weltmann, K.D.; von Woedtke, T.; et al. Risk assessment of kinpen plasma treatment of four human pancreatic cancer cell lines with respect to metastasis. Cancers 2019, 11, 1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Z.; Li, G.; Liu, J.; Xu, D.; Shi, X.; Zhang, G. Inhibitory effect of non-thermal plasma synergistic tegafur on pancreatic tumor cell line bxpc-3 proliferation. Plasma Process. Polym. 2019, 16, 1800165. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wyckoff, J.; Condeelis, J. Cell migration in tumors. Curr. Opin. Cell Biol. 2005, 17, 559–564. [Google Scholar] [CrossRef]
- Ren, B.; Cui, M.; Yang, G.; Wang, H.; Feng, M.; You, L.; Zhao, Y. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol. Cancer 2018, 17, 108. [Google Scholar] [CrossRef] [Green Version]
- Entschladen, F.; Drell, T.L.; Lang, K.; Joseph, J.; Zaenker, K.S. Tumour-cell migration, invasion, and metastasis: Navigation by neurotransmitters. Lancet Oncol. 2004, 5, 254–258. [Google Scholar] [CrossRef]
- Kim, C.H.; Kwon, S.; Bahn, J.H.; Lee, K.; Jun, S.I.; Rack, P.D.; Baek, S.J. Effects of atmospheric nonthermal plasma on invasion of colorectal cancer cells. Appl. Phys. Lett. 2010, 96, 243701. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Bleker, A.; Winter, J.; Bosel, A.; Reuter, S.; Weltmann, K.D. On the plasma chemistry of a cold atmospheric argon plasma jet with shielding gas device. Plasma Sources Sci. Technol. 2016, 25, 015005. [Google Scholar] [CrossRef]
- Kumar, N.; Perez-Novo, C.; Shaw, P.; Logie, E.; Privat-Maldonado, A.; Dewilde, S.; Smits, E.; Berghe, W.V.; Bogaerts, A. Physical plasma-derived oxidants sensitize pancreatic cancer cells to ferroptotic cell death. Free Radic. Biol. Med. 2021, 166, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Hara-Chikuma, M.; Papadopoulos, M.C. Aquaporins—New players in cancer biology. J. Mol. Med. 2008, 86, 523–529. [Google Scholar] [CrossRef] [Green Version]
- Bienert, G.P.; Moller, A.L.B.; Kristiansen, K.A.; Schulz, A.; Moller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Lin, L.; Gjika, E.; Cheng, X.; Canady, J.; Keidar, M. Selective treatment of pancreatic cancer cells by plasma-activated saline solutions. IEEE Trans. Radiat. Plasma Med Sci. 2018, 2, 116–120. [Google Scholar] [CrossRef]
- Bekeschus, S.; Kading, A.; Schroder, T.; Wende, K.; Hackbarth, C.; Liedtke, K.R.; van der Linde, J.; von Woedtke, T.; Heidecke, C.D.; Partecke, L.I. Cold physical plasma-treated buffered saline solution as effective agent against pancreatic cancer cells. Anticancer Agents Med. Chem. 2018, 18, 824–831. [Google Scholar] [CrossRef]
- Sato, Y.; Yamada, S.; Takeda, S.; Hattori, N.; Nakamura, K.; Tanaka, H.; Mizuno, M.; Hori, M.; Kodera, Y. Effect of plasma-activated lactated ringer’s solution on pancreatic cancer cells in vitro and in vivo. Ann. Surg. Oncol. 2018, 25, 299–307. [Google Scholar] [CrossRef]
- Azzariti, A.; Iacobazzi, R.M.; Di Fonte, R.; Porcelli, L.; Gristina, R.; Favia, P.; Fracassi, F.; Trizio, I.; Silvestris, N.; Guida, G.; et al. Plasma-activated medium triggers cell death and the presentation of immune activating danger signals in melanoma and pancreatic cancer cells. Sci. Rep. 2019, 9, 4099. [Google Scholar] [CrossRef] [PubMed]
- Van Loenhout, J.; Flieswasser, T.; Freire Boullosa, L.; De Waele, J.; Van Audenaerde, J.; Marcq, E.; Jacobs, J.; Lin, A.; Lion, E.; Dewitte, H.; et al. Cold atmospheric plasma-treated pbs eliminates immunosuppressive pancreatic stellate cells and induces immunogenic cell death of pancreatic cancer cells. Cancers 2019, 11, 1597. [Google Scholar] [CrossRef] [Green Version]
- Itakura, J.; Ishiwata, T.; Friess, H.; Fujii, H.; Matsumoto, Y.; Büchler, M.W.; Korc, M. Enhanced expression of vascular endothelial growth factor in human pancreatic cancer correlates with local disease progression. Clin. Cancer Res. 1997, 3, 1309–1316. [Google Scholar]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to tumor angiogenesis from innate immune cells within the tumor microenvironment: Implications for immunotherapy. Front. Immunol. 2018, 9, 527. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.Y.; Zheng, M.; Li, Y.M.; Fan, X.Y.; Wang, J.C.; Li, Z.C.; Yang, H.J.; Yu, J.M.; Cui, J.; Jiang, J.L.; et al. Rip3 promotes colitis-associated colorectal cancer by controlling tumor cell proliferation and cxcl1-induced immune suppression. Theranostics 2019, 9, 3659–3673. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Tao, G.Q.; Zhang, Y.; Cai, B.; Sun, J.; Tian, Z.Q. Tgf-beta in pancreatic cancer initiation and progression: Two sides of the same coin. Cell Biosci. 2017, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Ling, L.; van Dam, H.; Zhou, F.; Zhang, L. Tgf-beta signaling in cancer metastasis. Acta Biochim. Biophys. Sin. 2018, 50, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland-Goldsmith, M.A.; Maruyama, H.; Matsuda, K.; Idezawa, T.; Ralli, M.; Ralli, S.; Korc, M. Soluble type ii transforming growth factor-β receptor attenuates expression of metastasis-associated genes and suppresses pancreatic cancer cell metastasis. Mol. Cancer Ther. 2002, 1, 161–167. [Google Scholar]
- Melisi, D.; Ishiyama, S.; Sclabas, G.M.; Fleming, J.B.; Xia, Q.; Tortora, G.; Abbruzzese, J.L.; Chiao, P.J. Ly2109761, a novel transforming growth factor beta receptor type i and type ii dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol. Cancer Ther. 2008, 7, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, Y.; Takeyama, H.; Guha, S. Cytokine network: New targeted therapy for pancreatic cancer. Curr. Pharm. Des. 2012, 18, 2416–2419. [Google Scholar] [CrossRef]
- Goumas, F.A.; Holmer, R.; Egberts, J.H.; Gontarewicz, A.; Heneweer, C.; Geisen, U.; Hauser, C.; Mende, M.M.; Legler, K.; Rocken, C.; et al. Inhibition of il-6 signaling significantly reduces primary tumor growth and recurrencies in orthotopic xenograft models of pancreatic cancer. Int. J. Cancer 2015, 137, 1035–1046. [Google Scholar] [CrossRef]
- Holmer, R.; Goumas, F.A.; Waetzig, G.H.; Rose-John, S.; Kalthoff, H. Interleukin-6: A villain in the drama of pancreatic cancer development and progression. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 371–380. [Google Scholar] [CrossRef]
- Huang, L.; Hu, B.; Ni, J.; Wu, J.; Jiang, W.; Chen, C.; Yang, L.; Zeng, Y.; Wan, R.; Hu, G.; et al. Transcriptional repression of socs3 mediated by il-6/stat3 signaling via dnmt1 promotes pancreatic cancer growth and metastasis. J. Exp. Clin. Cancer Res. 2016, 35, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshani, R.; McCarthy, F.; Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014, 345, 157–163. [Google Scholar] [CrossRef]
- Lesina, M.; Wormann, S.M.; Neuhofer, P.; Song, L.; Algul, H. Interleukin-6 in inflammatory and malignant diseases of the pancreas. Semin. Immunol. 2014, 26, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Chen, Y.J.; Chang, W.A.; Jian, S.F.; Fan, H.L.; Wang, J.Y.; Kuo, P.L. Interaction between tumor-associated dendritic cells and colon cancer cells contributes to tumor progression via cxcl1. Int. J. Mol. Sci. 2018, 19, 2427. [Google Scholar] [CrossRef] [Green Version]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A cxcl1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via cxcl1 and mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor cell-intrinsic factors underlie heterogeneity of immune cell infiltration and response to immunotherapy. Immunity 2018, 49, 178–193.e177. [Google Scholar] [CrossRef] [Green Version]
- Sockolosky, J.T.; Dougan, M.; Ingram, J.R.; Ho, C.C.; Kauke, M.J.; Almo, S.C.; Ploegh, H.L.; Garcia, K.C. Durable antitumor responses to cd47 blockade require adaptive immune stimulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2646–E2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.; Razzokov, J.; Verswyvel, H.; Privat-Maldonado, A.; De Backer, J.; Yusupov, M.; Cardenas De La Hoz, E.; Ponsaerts, P.; Smits, E.; Bogaerts, A. Oxidation of innate immune checkpoint cd47 on cancer cells with non-thermal plasma. Cancers 2021, 13, 579. [Google Scholar] [CrossRef] [PubMed]
- Clemen, R.; Freund, E.; Mrochen, D.; Miebach, L.; Schmidt, A.; Rauch, B.H.; Lackmann, J.W.; Martens, U.; Wende, K.; Lalk, M.; et al. Gas plasma technology augments ovalbumin immunogenicity and ot-ii t cell activation conferring tumor protection in mice. Adv. Sci. 2021, 2003395. [Google Scholar] [CrossRef]
- Clemen, R.; Bekeschus, S. Oxidatively modified proteins: Cause and control of diseases. Appl. Sci. 2020, 10, 6419. [Google Scholar] [CrossRef]
- Bruno, G.; Heusler, T.; Lackmann, J.-W.; von Woedtke, T.; Weltmann, K.-D.; Wende, K. Cold physical plasma-induced oxidation of cysteine yields reactive sulfur species (rss). Clin. Plasma Med. 2019, 14, 100083. [Google Scholar] [CrossRef]
- Heusler, T.; Bruno, G.; Bekeschus, S.; Lackmann, J.-W.; von Woedtke, T.; Wende, K. Can the effect of cold physical plasma-derived oxidants be transported via thiol group oxidation? Clin. Plasma Med. 2019, 14, 100086. [Google Scholar] [CrossRef]
- Lackmann, J.W.; Wende, K.; Verlackt, C.; Golda, J.; Volzke, J.; Kogelheide, F.; Held, J.; Bekeschus, S.; Bogaerts, A.; Schulz-von der Gathen, V.; et al. Chemical fingerprints of cold physical plasmas-an experimental and computational study using cysteine as tracer compound. Sci. Rep. 2018, 8, 7736. [Google Scholar] [CrossRef]
- Itano, N.; Zhuo, L.; Kimata, K. Impact of the hyaluronan-rich tumor microenvironment on cancer initiation and progression. Cancer Sci. 2008, 99, 1720–1725. [Google Scholar] [CrossRef]
- Yusupov, M.; Privat-Maldonado, A.; Cordeiro, R.M.; Verswyvel, H.; Shaw, P.; Razzokov, J.; Smits, E.; Bogaerts, A. Oxidative damage to hyaluronan–cd44 interactions as an underlying mechanism of action of oxidative stress-inducing cancer therapy. Redox Biol. 2021, 43, 101968. [Google Scholar] [CrossRef] [PubMed]
- Bekeschus, S.; Clemen, R.; Niessner, F.; Sagwal, S.K.; Freund, E.; Schmidt, A. Medical gas plasma jet technology targets murine melanoma in an immunogenic fashion. Adv. Sci. 2020, 7, 1903438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekeschus, S.; Ressel, V.; Freund, E.; Gelbrich, N.; Mustea, A.; Stope, M.B. Gas plasma-treated prostate cancer cells augment myeloid cell activity and cytotoxicity. Antioxidants 2020, 9, 323. [Google Scholar] [CrossRef]
- Clemen, R.; Heirman, P.; Lin, A.; Bogaerts, A.; Bekeschus, S. Physical plasma-treated skin cancer cells amplify tumor cytotoxicity of human natural killer (nk) cells. Cancers 2020, 12, 3575. [Google Scholar] [CrossRef]
- Mahdikia, H.; Saadati, F.; Freund, E.; Gaipl, U.S.; Majidzadeh, A.K.; Shokri, B.; Bekeschus, S. Gas plasma irradiation of breast cancers promotes immunogenicity, tumor reduction, and an abscopal effect in vivo. Oncoimmunology 2021, 10, 1859731. [Google Scholar] [CrossRef]
- Pasqual-Melo, G.; Sagwal, S.K.; Freund, E.; Gandhirajan, R.K.; Frey, B.; von Woedtke, T.; Gaipl, U.; Bekeschus, S. Combination of gas plasma and radiotherapy has immunostimulatory potential and additive toxicity in murine melanoma cells in vitro. Int. J. Mol. Sci. 2020, 21, 1379. [Google Scholar] [CrossRef] [Green Version]
- Bekeschus, S.; Clemen, R.; Metelmann, H.-R. Potentiating anti-tumor immunity with physical plasma. Clin. Plasma Med. 2018, 12, 17–22. [Google Scholar] [CrossRef]
- Khalili, M.; Daniels, L.; Lin, A.; Krebs, F.C.; Snook, A.E.; Bekeschus, S.; Bowne, W.B.; Miller, V. Non-thermal plasma-induced immunogenic cell death in cancer: A topical review. J. Phys. D Appl. Phys. 2019, 52, 423001. [Google Scholar] [CrossRef] [PubMed]
- Bekeschus, S.; Seebauer, C.; Wende, K.; Schmidt, A. Physical plasma and leukocytes-immune or reactive? Biol. Chem. 2018, 400, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Bekeschus, S.; Winterbourn, C.C.; Kolata, J.; Masur, K.; Hasse, S.; Broker, B.M.; Parker, H.A. Neutrophil extracellular trap formation is elicited in response to cold physical plasma. J. Leukoc. Biol. 2016, 100, 791–799. [Google Scholar] [CrossRef]
- Bekeschus, S.; Meyer, D.; Arlt, K.; von Woedtke, T.; Miebach, L.; Freund, E.; Clemen, R. Argon plasma exposure augments costimulatory ligands and cytokine release in human monocyte-derived dendritic cells. Int. J. Mol. Sci. 2021, 22, 3790. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. Cxcr2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Sano, M.; Ijichi, H.; Takahashi, R.; Miyabayashi, K.; Fujiwara, H.; Yamada, T.; Kato, H.; Nakatsuka, T.; Tanaka, Y.; Tateishi, K.; et al. Blocking cxcls-cxcr2 axis in tumor-stromal interactions contributes to survival in a mouse model of pancreatic ductal adenocarcinoma through reduced cell invasion/migration and a shift of immune-inflammatory microenvironment. Oncogenesis 2019, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Sangaletti, S.; Tripodo, C.; Sandri, S.; Torselli, I.; Vitali, C.; Ratti, C.; Botti, L.; Burocchi, A.; Porcasi, R.; Tomirotti, A.; et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. 2014, 74, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Giraldo, N.A.; Sanchez-Salas, R.; Peske, J.D.; Vano, Y.; Becht, E.; Petitprez, F.; Validire, P.; Ingels, A.; Cathelineau, X.; Fridman, W.H.; et al. The clinical role of the tme in solid cancer. Br. J. Cancer 2019, 120, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Pausch, T.M.; Aue, E.; Wirsik, N.M.; Freire Valls, A.; Shen, Y.; Radhakrishnan, P.; Hackert, T.; Schneider, M.; Schmidt, T. Metastasis-associated fibroblasts promote angiogenesis in metastasized pancreatic cancer via the cxcl8 and the ccl2 axes. Sci. Rep. 2020, 10, 5420. [Google Scholar] [CrossRef]
- Bekeschus, S.; Schmidt, A.; Kramer, A.; Metelmann, H.R.; Adler, F.; von Woedtke, T.; Niessner, F.; Weltmann, K.D.; Wende, K. High throughput image cytometry micronucleus assay to investigate the presence or absence of mutagenic effects of cold physical plasma. Environ. Mol. Mutagen. 2018, 59, 268–277. [Google Scholar] [CrossRef]
- Kluge, S.; Bekeschus, S.; Bender, C.; Benkhai, H.; Sckell, A.; Below, H.; Stope, M.B.; Kramer, A. Investigating the mutagenicity of a cold argon-plasma jet in an het-mn model. PLoS ONE 2016, 11, e0160667. [Google Scholar] [CrossRef] [Green Version]
- Lobrich, M.; Shibata, A.; Beucher, A.; Fisher, A.; Ensminger, M.; Goodarzi, A.A.; Barton, O.; Jeggo, P.A. Gammah2ax foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimization. Cell Cycle 2010, 9, 662–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, S.; Wacker, E.; Li, Y.F.; Shimizu, T.; Thomas, H.M.; Morfill, G.E.; Karrer, S.; Zimmermann, J.L.; Bosserhoff, A.K. Cold atmospheric plasma, a new strategy to induce senescence in melanoma cells. Exp. Dermatol. 2013, 22, 284–289. [Google Scholar] [CrossRef]
- Chang, J.W.; Kang, S.U.; Shin, Y.S.; Kim, K.I.; Seo, S.J.; Yang, S.S.; Lee, J.S.; Moon, E.; Baek, S.J.; Lee, K.; et al. Non-thermal atmospheric pressure plasma induces apoptosis in oral cavity squamous cell carcinoma: Involvement of DNA-damage-triggering sub-g(1) arrest via the atm/p53 pathway. Arch Biochem. Biophys. 2014, 545, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Plewa, J.M.; Yousfi, M.; Frongia, C.; Eichwald, O.; Ducommun, B.; Merbahi, N.; Lobjois, V. Low-temperature plasma-induced antiproliferative effects on multi-cellular tumor spheroids. New J. Phys. 2014, 16, 043027. [Google Scholar] [CrossRef]
- Bekeschus, S.; Schutz, C.S.; Niessner, F.; Wende, K.; Weltmann, K.D.; Gelbrich, N.; von Woedtke, T.; Schmidt, A.; Stope, M.B. Elevated h2ax phosphorylation observed with kinpen plasma treatment is not caused by ros-mediated DNA damage but is the consequence of apoptosis. Oxid. Med. Cell. Longev. 2019, 2019, 8535163. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Woedtke, T.V.; Stenzel, J.; Lindner, T.; Polei, S.; Vollmar, B.; Bekeschus, S. One year follow-up risk assessment in skh-1 mice and wounds treated with an argon plasma jet. Int. J. Mol. Sci. 2017, 18, 868. [Google Scholar] [CrossRef]
- Metelmann, H.-R.; Vu, T.T.; Do, H.T.; Le, T.N.B.; Hoang, T.H.A.; Phi, T.T.T.; Luong, T.M.L.; Doan, V.T.; Nguyen, T.T.H.; Nguyen, T.H.M.; et al. Scar formation of laser skin lesions after cold atmospheric pressure plasma (cap) treatment: A clinical long term observation. Clin. Plasma Med. 2013, 1, 30–35. [Google Scholar] [CrossRef]
- Bekeschus, S.; Liebelt, G.; Menz, J.; Berner, J.; Sagwal, S.K.; Wende, K.; Weltmann, K.D.; Boeckmann, L.; von Woedtke, T.; Metelmann, H.R.; et al. Tumor cell metabolism correlates with resistance to gas plasma treatment: The evaluation of three dogmas. Free Radic. Biol. Med. 2021, 167, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Freund, E.; Spadola, C.; Schmidt, A.; Privat-Maldonado, A.; Bogaerts, A.; von Woedtke, T.; Weltmann, K.D.; Heidecke, C.D.; Partecke, L.I.; Kading, A.; et al. Risk evaluation of emt and inflammation in metastatic pancreatic cancer cells following plasma treatment. Front. Phys. 2020, 8, 9618. [Google Scholar] [CrossRef]
- Sun, H.; Miao, C.; Liu, W.; Qiao, X.; Yang, W.; Li, L.; Li, C. Tgf-beta1/tbetarii/smad3 signaling pathway promotes vegf expression in oral squamous cell carcinoma tumor-associated macrophages. Biochem. Biophys. Res. Commun. 2018, 497, 583–590. [Google Scholar] [CrossRef]
- Wang, W.; Marinis, J.M.; Beal, A.M.; Savadkar, S.; Wu, Y.; Khan, M.; Taunk, P.S.; Wu, N.; Su, W.; Wu, J.; et al. Rip1 kinase drives macrophage-mediated adaptive immune tolerance in pancreatic cancer. Cancer Cell 2018, 34, 757–774.e757. [Google Scholar] [CrossRef] [Green Version]
- Rebelo, S.P.; Pinto, C.; Martins, T.R.; Harrer, N.; Estrada, M.F.; Loza-Alvarez, P.; Cabecadas, J.; Alves, P.M.; Gualda, E.J.; Sommergruber, W.; et al. 3d-3-culture: A tool to unveil macrophage plasticity in the tumour microenvironment. Biomaterials 2018, 163, 185–197. [Google Scholar] [CrossRef]
- Ruffell, D.; Mourkioti, F.; Gambardella, A.; Kirstetter, P.; Lopez, R.G.; Rosenthal, N.; Nerlov, C. A creb-c/ebpbeta cascade induces m2 macrophage-specific gene expression and promotes muscle injury repair. Proc. Natl. Acad. Sci. USA 2009, 106, 17475–17480. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Shapiro, B.; Vucic, E.A.; Vogt, S.; Bar-Sagi, D. Tumor cell-derived il1beta promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. 2020, 80, 1088–1101. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, P.; Karyampudi, L.; Shreeder, B.; Krempski, J.; Bahr, D.; Daum, J.; Kalli, K.R.; Goode, E.L.; Block, M.S.; Cannon, M.J.; et al. Il10 release upon pd-1 blockade sustains immunosuppression in ovarian cancer. Cancer Res. 2017, 77, 6667–6678. [Google Scholar] [CrossRef] [Green Version]
- Borgoni, S.; Iannello, A.; Cutrupi, S.; Allavena, P.; D’Incalci, M.; Novelli, F.; Cappello, P. Depletion of tumor-associated macrophages switches the epigenetic profile of pancreatic cancer infiltrating t cells and restores their anti-tumor phenotype. Oncoimmunology 2018, 7, e1393596. [Google Scholar] [CrossRef]
- Fallon, J.; Tighe, R.; Kradjian, G.; Guzman, W.; Bernhardt, A.; Neuteboom, B.; Lan, Y.; Sabzevari, H.; Schlom, J.; Greiner, J.W. The immunocytokine nhs-il12 as a potential cancer therapeutic. Oncotarget 2014, 5, 1869–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkayyal, A.A.; Tai, L.H.; Kennedy, M.A.; de Souza, C.T.; Zhang, J.; Lefebvre, C.; Sahi, S.; Ananth, A.A.; Mahmoud, A.B.; Makrigiannis, A.P.; et al. Nk-cell recruitment is necessary for eradication of peritoneal carcinomatosis with an il12-expressing maraba virus cellular vaccine. Cancer Immunol. Res. 2017, 5, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Peron, J.M.; Bureau, C.; Gourdy, P.; Lulka, H.; Souque, A.; Calippe, B.; Selves, J.; Al Saati, T.; Bernad, J.; Cordelier, P.; et al. Treatment of experimental murine pancreatic peritoneal carcinomatosis with fibroblasts genetically modified to express il12: A role for peritoneal innate immunity. Gut 2007, 56, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Benner, B.; Scarberry, L.; Suarez-Kelly, L.P.; Duggan, M.C.; Campbell, A.R.; Smith, E.; Lapurga, G.; Jiang, K.; Butchar, J.P.; Tridandapani, S.; et al. Generation of monocyte-derived tumor-associated macrophages using tumor- conditioned media provides a novel method to study tumor-associated macrophages in vitro. J. Immunother. Cancer 2019, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khabipov, A.; Kading, A.; Liedtke, K.R.; Freund, E.; Partecke, L.I.; Bekeschus, S. Raw 264.7 macrophage polarization by pancreatic cancer cells-a model for studying tumour-promoting macrophages. Anticancer Res. 2019, 39, 2871–2882. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Brouchet, A.; Gilhodes, J.; Acker, N.V.; Brion, R.; Bouvier, C.; Assemat, P.; Gaspar, N.; Aubert, S.; Guinebretiere, J.M.; Marie, B.; et al. Characterization of macrophages and osteoclasts in the osteosarcoma tumor microenvironment at diagnosis: New perspective for osteosarcoma treatment? Cancers 2021, 13, 423. [Google Scholar] [CrossRef]
- Liedtke, K.R.; Freund, E.; Hackbarth, C.; Heidecke, C.-D.; Partecke, L.-I.; Bekeschus, S. A myeloid and lymphoid infiltrate in murine pancreatic tumors exposed to plasma-treated medium. Clin. Plasma Med. 2018, 11, 10–17. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Yu, J.; Feng, L.; Hou, S.; Liu, Y.; Guo, M.; Xie, Y.; Meng, J.; Zhang, H.; et al. Targeting the shift from m1 to m2 macrophages in experimental autoimmune encephalomyelitis mice treated with fasudil. PLoS ONE 2013, 8, e54841. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Qi, Q.; Wang, P.; Chen, H.; Chen, Z.; Meng, Z.; Liu, L. Serum level of ccl2 predicts outcome of patients with pancreatic cancer. Acta Gastroenterol. Belg. 2020, 83, 295–299. [Google Scholar] [PubMed]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the ccl2/ccr2 axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, L.A.; Doherty, G.A.; Sheahan, K.; Ryan, E.J. Human tumor-infiltrating myeloid cells: Phenotypic and functional diversity. Front. Immunol. 2017, 8, 86. [Google Scholar] [CrossRef]
- Helm, O.; Held-Feindt, J.; Grage-Griebenow, E.; Reiling, N.; Ungefroren, H.; Vogel, I.; Kruger, U.; Becker, T.; Ebsen, M.; Rocken, C.; et al. Tumor-associated macrophages exhibit pro- and anti-inflammatory properties by which they impact on pancreatic tumorigenesis. Int. J. Cancer 2014, 135, 843–861. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of m2-polarized tumor-associated macrophage in pancreatic cancer. J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef]
- Reuter, S.; von Woedtke, T.; Weltmann, K.D. The kinpen-a review on physics and chemistry of the atmospheric pressure plasma jet and its applications. J. Phys. D Appl. Phys. 2018, 51, 233001. [Google Scholar] [CrossRef] [Green Version]
- Freund, E.; Liedtke, K.R.; Miebach, L.; Wende, K.; Heidecke, A.; Kaushik, N.K.; Choi, E.H.; Partecke, L.I.; Bekeschus, S. Identification of two kinase inhibitors with synergistic toxicity with low-dose hydrogen peroxide in colorectal cancer cells in vitro. Cancers 2020, 12, 122. [Google Scholar] [CrossRef] [Green Version]
- Seebauer, C.; Freund, E.; Hasse, S.; Miller, V.; Segebarth, M.; Lucas, C.; Kindler, S.; Dieke, T.; Metelmann, H.R.; Daeschlein, G.; et al. Effects of cold physical plasma on oral lichen planus: An in-vitro study. Oral Dis. 2021. [Google Scholar] [CrossRef]



 ) or significant trend for linearity (↑ or ↓). CCL = CC-chemokine ligand; CXCL = (C-X-C motif) ligand; IL = interleukin; IFN = interferon; MCP = macrophage-chemoattractant protein; TGF = transforming growth factor; TNF = tumor necrosis factor; VEGF = vascular endothelial growth factor.
) or significant trend for linearity (↑ or ↓). CCL = CC-chemokine ligand; CXCL = (C-X-C motif) ligand; IL = interleukin; IFN = interferon; MCP = macrophage-chemoattractant protein; TGF = transforming growth factor; TNF = tumor necrosis factor; VEGF = vascular endothelial growth factor.
) or significant trend for linearity (↑ or ↓). CCL = CC-chemokine ligand; CXCL = (C-X-C motif) ligand; IL = interleukin; IFN = interferon; MCP = macrophage-chemoattractant protein; TGF = transforming growth factor; TNF = tumor necrosis factor; VEGF = vascular endothelial growth factor.
) or significant trend for linearity (↑ or ↓). CCL = CC-chemokine ligand; CXCL = (C-X-C motif) ligand; IL = interleukin; IFN = interferon; MCP = macrophage-chemoattractant protein; TGF = transforming growth factor; TNF = tumor necrosis factor; VEGF = vascular endothelial growth factor.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khabipov, A.; Freund, E.; Liedtke, K.R.; Käding, A.; Riese, J.; van der Linde, J.; Kersting, S.; Partecke, L.-I.; Bekeschus, S. Murine Macrophages Modulate Their Inflammatory Profile in Response to Gas Plasma-Inactivated Pancreatic Cancer Cells. Cancers 2021, 13, 2525. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112525
Khabipov A, Freund E, Liedtke KR, Käding A, Riese J, van der Linde J, Kersting S, Partecke L-I, Bekeschus S. Murine Macrophages Modulate Their Inflammatory Profile in Response to Gas Plasma-Inactivated Pancreatic Cancer Cells. Cancers. 2021; 13(11):2525. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112525
Chicago/Turabian StyleKhabipov, Aydar, Eric Freund, Kim Rouven Liedtke, Andre Käding, Janik Riese, Julia van der Linde, Stephan Kersting, Lars-Ivo Partecke, and Sander Bekeschus. 2021. "Murine Macrophages Modulate Their Inflammatory Profile in Response to Gas Plasma-Inactivated Pancreatic Cancer Cells" Cancers 13, no. 11: 2525. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112525