
Not Only Immune Escape—The Confusing Role of the TRP Metabolic Pathway in Carcinogenesis

 , , and
, , and 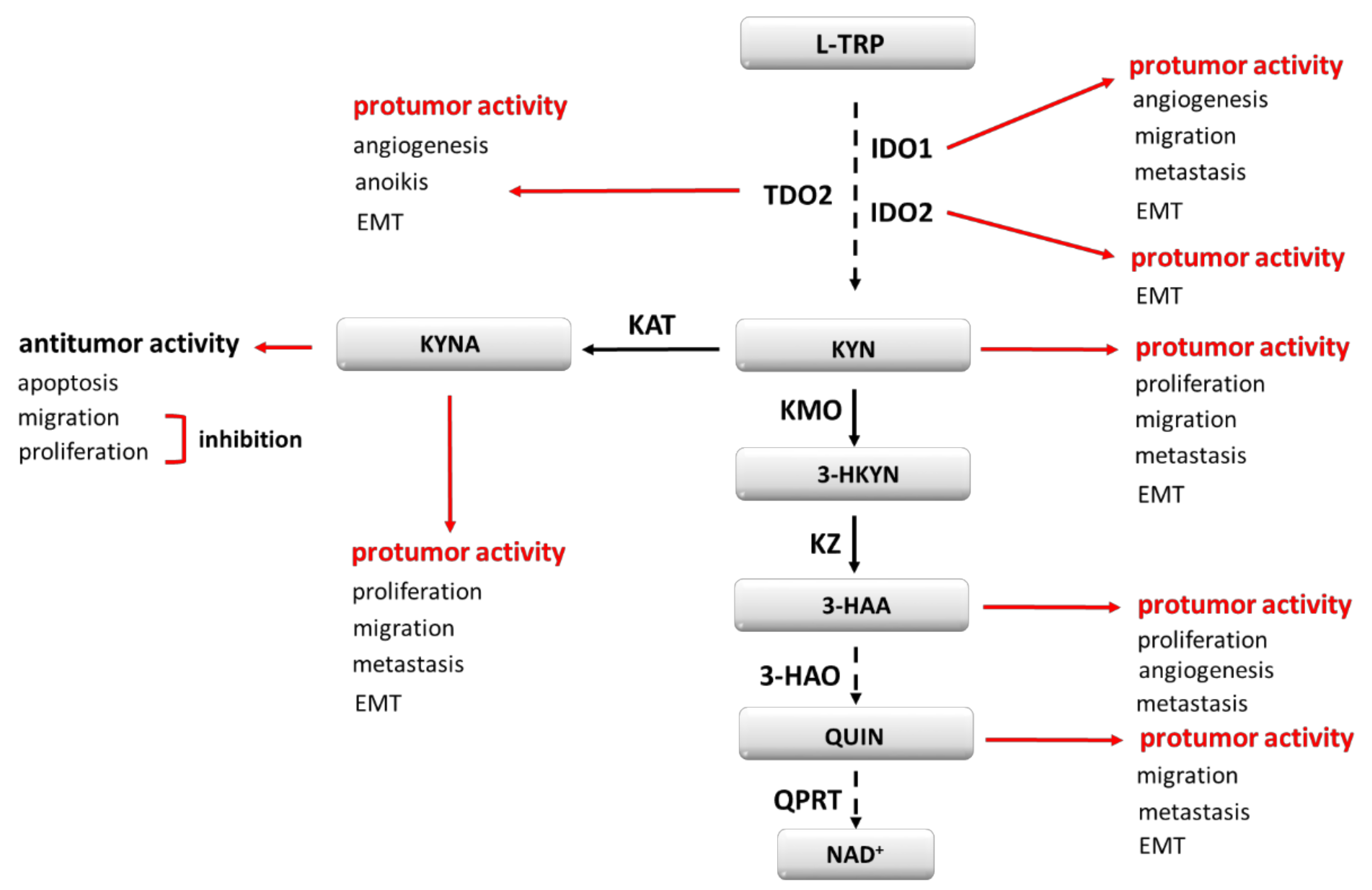{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
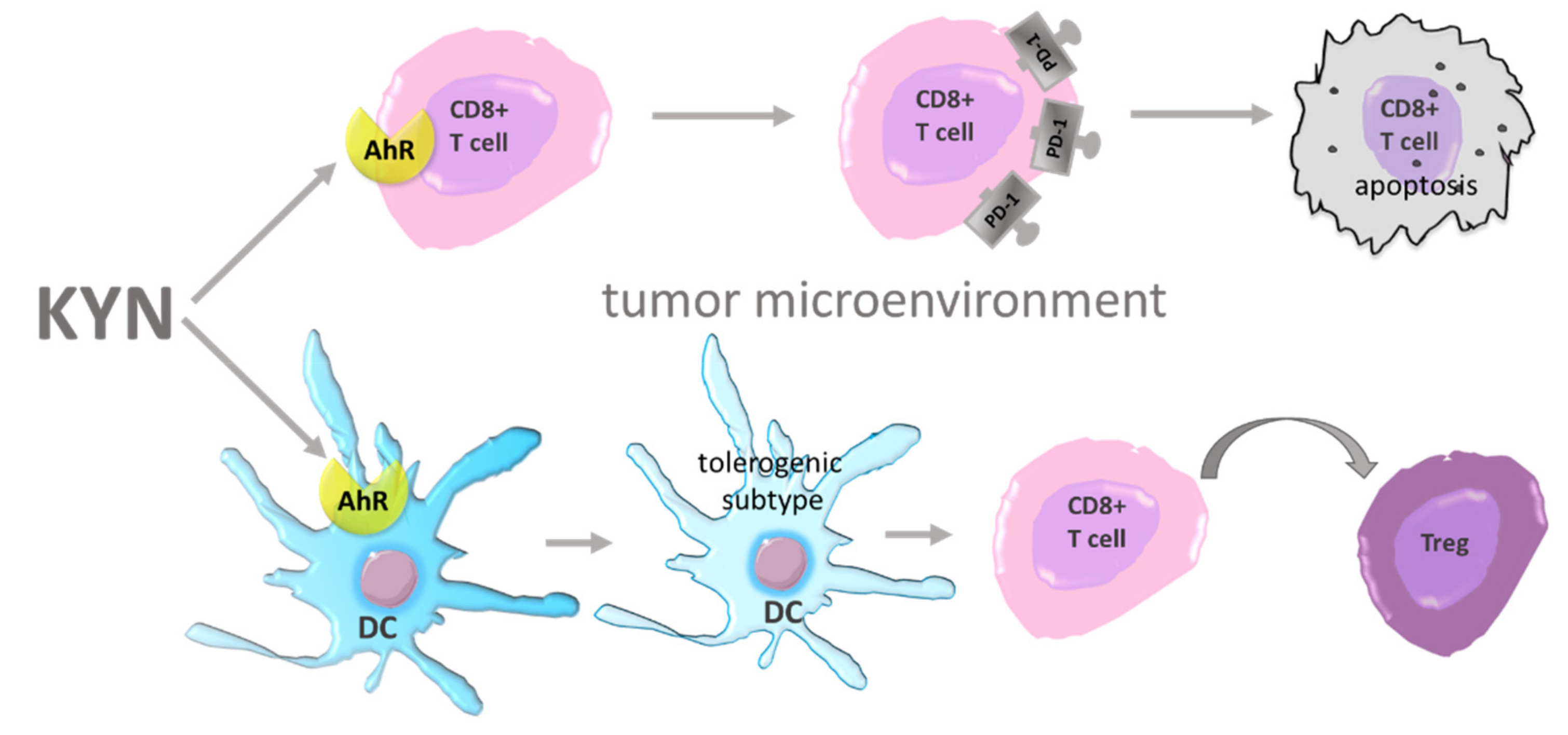
2. Mechanisms Involved in Immune Evasion
3. Tryptophan Metabolism and Its Modulators
4. IDO1 and Its Role in Cancer Development
5. TDO2, IDO2 and Their Role in Cancer Development
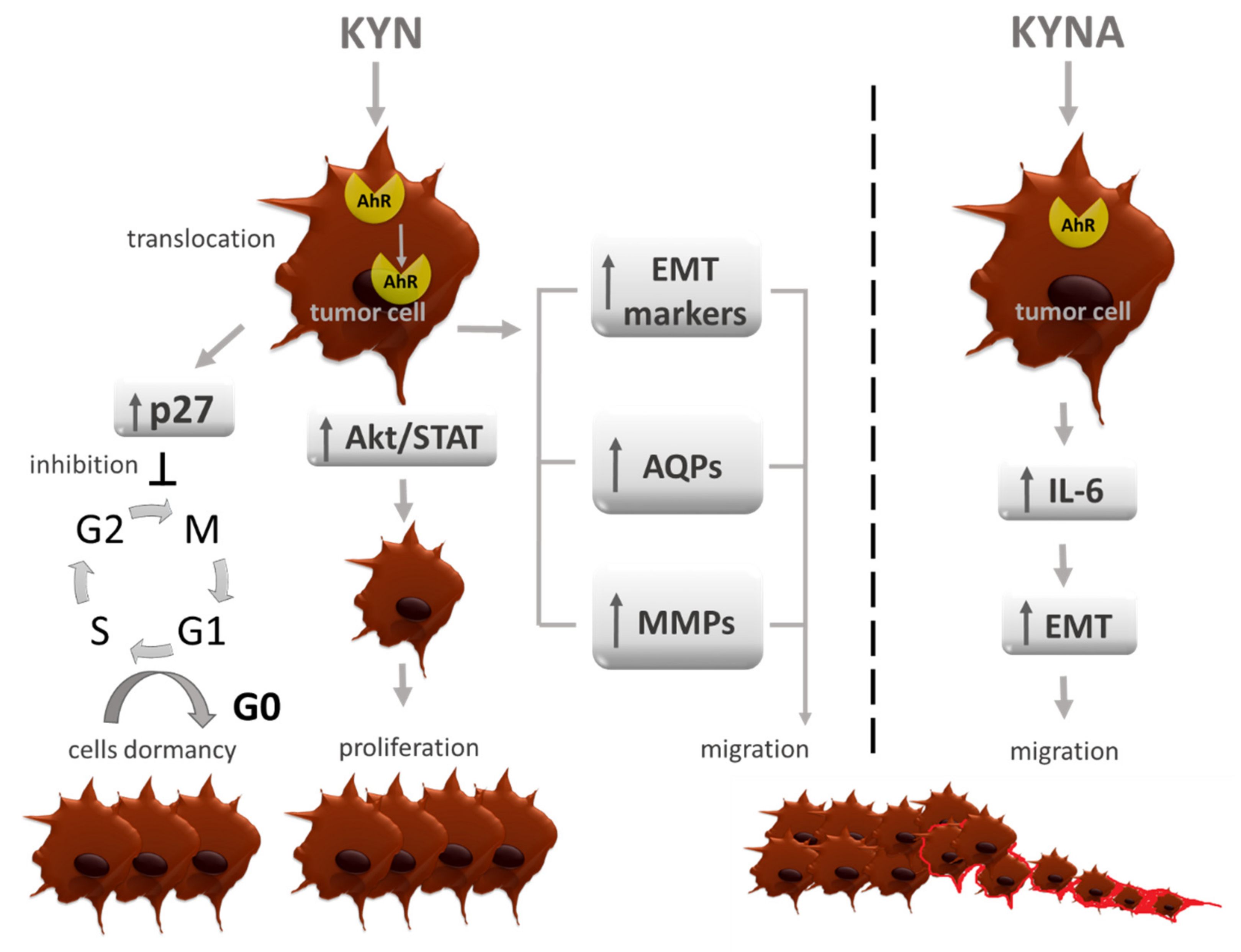
6. AhR Agonists—Carcinogenesis Modulators
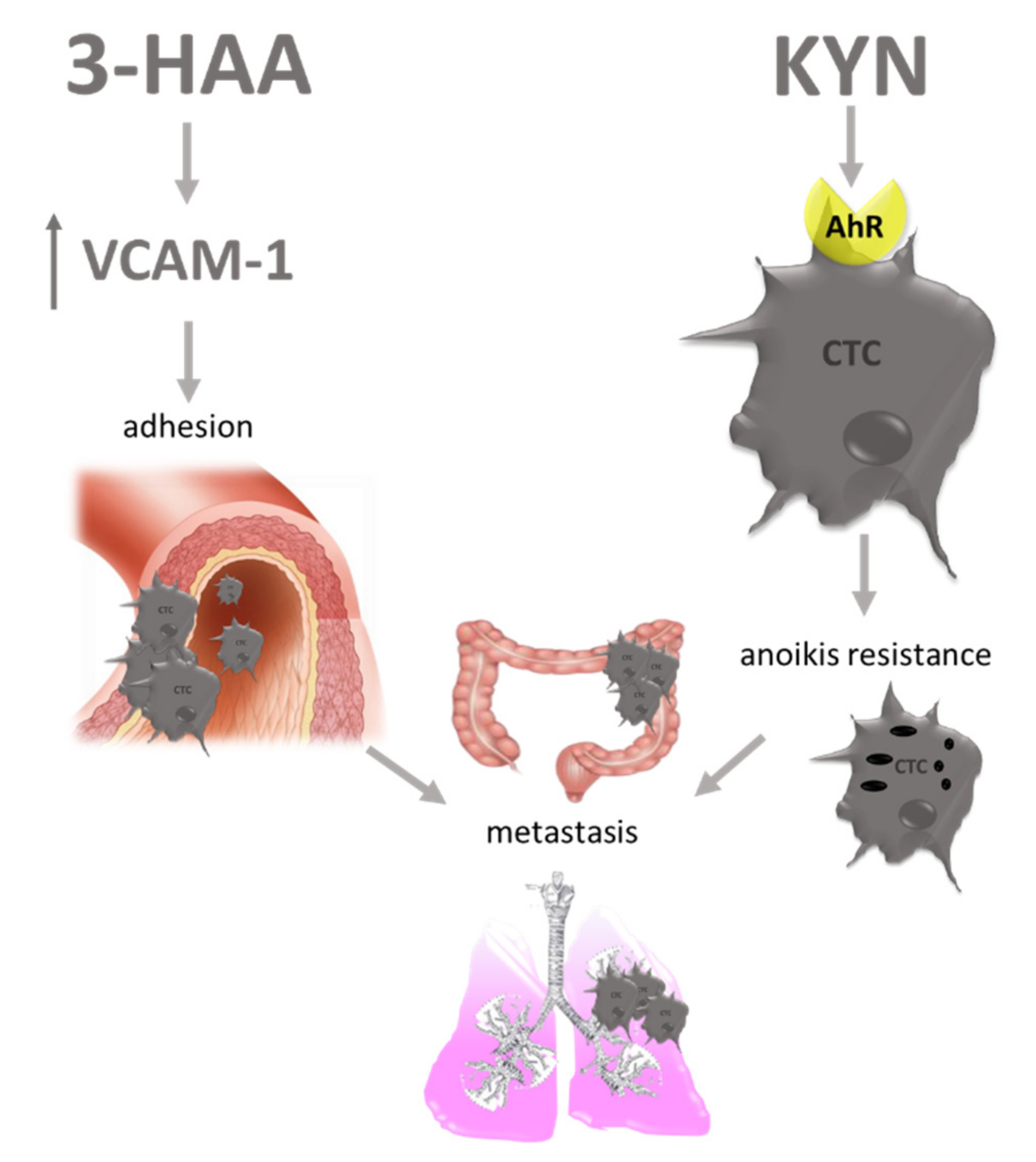
7. Other Carcinogenesis Modulating Effects of TRP Metabolites
8. Compounds Targeting Kynurenine Pathway in Clinical Trials
9. Other Aspects of TRP Metabolism
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kunimasa, K.; Goto, T. Immunosurveillance and Immunoediting of Lung Cancer: Current Perspectives and Challenges. Int. J. Mol. Sci. 2020, 21, 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lussier, D.M.; Schreiber, R.D. Cancer Immunosurveillance: Immunoediting. In Immunity to Pathogens and Tumors; Elsevier Inc.: Amsterdam, The Netherlands, 2016; Volume 4, pp. 396–405. [Google Scholar] [CrossRef]
- Teng, M.W.; Galon, J.; Fridman, W.H.; Smyth, M.J. From mice to humans: Developments in cancer immunoediting. J. Clin. Investig. 2015, 125, 3338–3346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.; Wen, T.; Dong, H. Bidirectional signals of PD-L1 in T cells that fraternize with cancer cells. Nat. Immunol. 2020, 21, 365–366. [Google Scholar] [CrossRef] [PubMed]
- Hermanowicz, J.; Sieklucka, B.; Nosek, K.; Pawlak, D. Intracellular mechanisms of tumor cells’ immunoresistance. Acta Biochim. Pol. 2020, 67, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J. Amino acid auxotrophy as a system of immunological control nodes. Nat. Immunol. 2016, 17, 132–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Hyun, H.; Li, L.T.; Wang, A.Z. Harnessing nanomedicine to overcome the immunosuppressive tumor microenvironment. Acta Pharmacol. Sin. 2020, 41, 970–985. [Google Scholar] [CrossRef]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Zhai, L.; Ladomersky, E.; Lenzen, A.; Nguyen, B.; Patel, R.; Lauing, K.L.; Wu, M.; Wainwright, D.A. IDO1 in cancer: A Gemini of immune checkpoints. Cell. Mol. Immunol. 2018, 15, 447–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkateswaran, N.; Lafita-Navarro, M.C.; Hao, Y.H.; Kilgore, J.A.; Perez-Castro, L.; Braverman, J.; Borenstein-Auerbach, N.; Kim, M.; Lesner, N.P.; Mishra, P.; et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019, 33, 1236–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balachandran, V.P.; Cavnar, M.J.; Zeng, S.; Bamboat, Z.M.; Ocuin, L.M.; Obaid, H.; Sorenson, E.C.; Popow, R.; Ariyan, C.; Rossi, F.; et al. Imatinib potentiates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat. Med. 2011, 17, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzada, T.; Lee, K.; Clarke, C.; Cooper, W.A.; Linton, A.; McCaughan, B.; Asher, R.; Clarke, S.; Reid, G.; Kao, S. High BIN1 expression has a favorable prognosis in malignant pleural mesothelioma and is associated with tumor infiltrating lymphocytes. Lung Cancer 2019, 130, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Correia, M.A. Heme: A regulator of rat hepatic tryptophan 2,3-dioxygenase? Arch. Biochem. Biophys. 2000, 377, 195–203. [Google Scholar] [CrossRef]
- Bender, D.A.; Laing, A.E.; Vale, J.A.; Papadaki, L.; Pugh, M. The effects of oestrogen administration on tryptophan metabolism in rats and in menopausal women receiving hormone replacement therapy. Biochem. Pharmacol. 1983, 32, 843–848. [Google Scholar] [CrossRef]
- Oxenkrug, G.F. Tryptophan kynurenine metabolism as a common mediator of genetic and environmental impacts in major depressive disorder: The serotonin hypothesis revisited 40 years later. Isr. J. Psychiatry Relat. Sci. 2010, 47, 56–63. [Google Scholar] [PubMed] [Green Version]
- Poulain-Godefroy, O.; Eury, E.; Leloire, A.; Hennart, B.; Guillemin, G.J.; Allorge, D.; Froguel, P. Induction of TDO2 and IDO2 in Liver by High-Fat Feeding in Mice: Discrepancies with Human Obesity. Int. J. Tryptophan Res. 2013, 6, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Ueland, P.M.; McCann, A.; Midttun, Ø.; Ulvik, A. Inflammation, vitamin B6 and related pathways. Mol. Asp. Med. 2017, 53, 10–27. [Google Scholar] [CrossRef]
- Opitz, C.A.; Somarribas Patterson, L.F.; Mohapatra, S.R.; Dewi, D.L.; Sadik, A.; Platten, M.; Trump, S. The therapeutic potential of targeting tryptophan catabolism in cancer. Br. J. Cancer 2020, 122, 30–44. [Google Scholar] [CrossRef]
- Suciu-Foca, N.; Cortesini, R. Central role of ILT3 in the T suppressor cell cascade. Cell. Immunol. 2007, 248, 59–67. [Google Scholar] [CrossRef]
- Brenk, M.; Scheler, M.; Koch, S.; Neumann, J.; Takikawa, O.; Häcker, G.; Bieber, T.; von Bubnoff, D. Tryptophan deprivation induces inhibitory receptors ILT3 and ILT4 on dendritic cells favoring the induction of human CD4+CD25+ Foxp3+ T regulatory cells. J. Immunol. 2009, 183, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Manavalan, J.S.; Rossi, P.C.; Vlad, G.; Piazza, F.; Yarilina, A.; Cortesini, R.; Mancini, D.; Suciu-Foca, N. High expression of ILT3 and ILT4 is a general feature of tolerogenic dendritic cells. Transpl. Immunol. 2003, 11, 245–258. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, N.; Xue, Y.; Zhang, M.; Li, Y.; Si, Y.; Biao, X.; Jia, Y.; Wang, Y. Expression of immunoglobulin-like transcript (ILT)2 and ILT3 in human gastric cancer and its clinical significance. Mol. Med. Rep. 2012, 5, 910–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Wang, L.; Gao, W.; Li, L.; Cui, X.; Yang, H.; Lin, W.; Dang, Q.; Zhang, N.; Sun, Y. Inhibitory receptor immunoglobulin-like transcript 4 was highly expressed in primary ductal and lobular breast cancer and significantly correlated with IL-10. Diagn. Pathol. 2014, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gao, A.; Zhang, F.; Wang, S.; Wang, J.; Wang, J.; Han, S.; Yang, Z.; Chen, X.; Fang, Y.; et al. ILT3 promotes tumor cell motility and angiogenesis in non-small cell lung cancer. Cancer Lett. 2021, 501, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lu, C.X.; Zhang, F.; Lv, W.; Liu, C. Expression of ILT3 predicts poor prognosis and is inversely associated with infiltration of CD45RO+ T cells in patients with colorectal cancer. Pathol. Res. Pract. 2018, 214, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- García, M.; Palma, M.B.; Verine, J.; Miriuka, S.; Inda, A.M.; Errecalde, A.L.; Desgrandchamps, F.; Carosella, E.D.; Tronik-Le Roux, D. The immune-checkpoint HLA-G/ILT4 is involved in the regulation of VEGF expression in clear cell renal cell carcinoma. BMC Cancer 2020, 20, 1–11. [Google Scholar] [CrossRef]
- Courtney, A.H.; Lo, W.L.; Weiss, A. TCR Signaling: Mechanisms of Initiation and Propagation. Trends Biochem. Sci. 2018, 43, 108–123. [Google Scholar] [CrossRef]
- Colligan, S.H.; Tzetzo, S.L.; Abrams, S.I. Myeloid-driven mechanisms as barriers to antitumor CD8+ T cell activity. Mol. Immunol. 2020, 118, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Bhat, S.A.; Gogoi, D.; Gokhale, A.; Chiplunkar, S.V. Inhibition of Notch signalling has ability to alter the proximal and distal TCR signalling events in human CD3+ αβ T-cells. Mol. Immunol. 2017, 92, 116–124. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br. J. Cancer 2021, 124, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Tan, Y.; Qian, Y.; Xue, W.; Wang, Y.; Du, J.; Jin, L.; Ding, W. Clinicopathologic and prognostic significance of tumor-infiltrating CD8+ T cells in patients with hepatocellular carcinoma: A meta-analysis. Medicine 2019, 98, e13923. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Zhang, C.; Li, Q.; Dong, J.; Liu, Y.; Huang, Y.; Jiang, T.; Wu, A. Tumour-infiltrating CD4+ and CD8+ lymphocytes as predictors of clinical outcome in glioma. Br. J. Cancer 2014, 110, 2560–2568. [Google Scholar] [CrossRef] [PubMed]
- Dar, A.A.; Pradhan, T.N.; Kulkarni, D.P.; Shah, S.U.; Rao, K.V.; Chaukar, D.A.; D’Cruz, A.K.; Chiplunkar, S.V. Extracellular 2’5’-oligoadenylate synthetase 2 mediates T-cell receptor CD3-ζ chain down-regulation via caspase-3 activation in oral cancer. Immunology 2016, 147, 251–264. [Google Scholar] [CrossRef] [Green Version]
- Frydecka, I.; Kaczmarek, P.; Boćko, D.; Kosmaczewska, A.; Morilla, R.; Catovsky, D. Expression of signal-transducing zeta chain in peripheral blood T cells and natural killer cells in patients with Hodgkin’s disease in different phases of the disease. Leuk. Lymphoma 1999, 35, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Park, J.S.; Fatima, Y.; Kausar, M.; Park, J.H.; Jun, C.D. Potentiating the Antitumor Activity of Cytotoxic T Cells via the Transmembrane Domain of IGSF4 That Increases TCR Avidity. Front. Immunol. 2021, 11, 3667. [Google Scholar] [CrossRef]
- Bono, M.R.; Fernández, D.; Flores-Santibáñez, F.; Rosemblatt, M.; Sauma, D. CD73 and CD39 ectonucleotidases in T cell differentiation: Beyond immunosuppression. FEBS Lett. 2015, 589, 3454–3460. [Google Scholar] [CrossRef]
- Hajizadeh, F.; Masjedi, A.; Asl, S.H.; Kiani, F.K.; Peydaveisi, M.; Ghalamfarsa, G.; Jadidi-Niaragh, F.; Sevbitov, A. Adenosine and adenosine receptors in colorectal cancer. Int. Immunopharmacol. 2020, 87, 106853. [Google Scholar] [CrossRef]
- Shi, L.; Wu, Z.; Miao, J.; Du, S.; Ai, S.; Xu, E.; Feng, M.; Song, J.; Guan, W. Adenosine interaction with adenosine receptor A2a promotes gastric cancer metastasis by enhancing PI3K-AKT-mTOR signaling. Mol. Biol. Cell 2019, 30, 2527–2534. [Google Scholar] [CrossRef]
- Yan, A.; Joachims, M.L.; Thompson, L.F.; Miller, A.D.; Canoll, P.D.; Bynoe, M.S. CD73 Promotes Glioblastoma Pathogenesis and Enhances Its Chemoresistance via A2B Adenosine Receptor Signaling. J. Neurosci. 2019, 39, 4387–4402. [Google Scholar] [CrossRef] [Green Version]
- Janssen, L.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The immune system in cancer metastasis: Friend or foe? J. Immunother. Cancer 2017, 5, 1–14. [Google Scholar] [CrossRef]
- Najafi, M.; Farhood, B.; Mortezaee, K. Contribution of regulatory T cells to cancer: A review. J. Cell. Physiol. 2019, 234, 7983–7993. [Google Scholar] [CrossRef] [PubMed]
- Zoso, A.; Mazza, E.M.; Bicciato, S.; Mandruzzato, S.; Bronte, V.; Serafini, P.; Inverardi, L. Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via Treg-cell expansion. Eur. J. Immunol. 2014, 44, 3307–3319. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhou, S.; Wang, Y.; Li, R.L.; Zhong, C.; Liang, C.; Sun, Y. Higher intratumoral infiltrated Foxp3+ Treg numbers and Foxp3+/CD8+ ratio are associated with adverse prognosis in resectable gastric cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.Q.; Liu, F.F.; Zhang, X.M.; Guo, X.J.; Ren, M.J.; Fu, L. Tumor secretion of CCL22 activates intratumoral Treg infiltration and is independent prognostic predictor of breast cancer. PLoS ONE 2013, 8, e76379. [Google Scholar] [CrossRef]
- Liotta, F.; Gacci, M.; Frosali, F.; Querci, V.; Vittori, G.; Lapini, A.; Santarlasci, V.; Serni, S.; Cosmi, L.; Maggi, L.; et al. Frequency of regulatory T cells in peripheral blood and in tumour-infiltrating lymphocytes correlates with poor prognosis in renal cell carcinoma. BJU Int. 2011, 107, 1500–1506. [Google Scholar] [CrossRef]
- Wang, D.; Yang, L.; Yu, W.; Wu, Q.; Lian, J.; Li, F.; Liu, S.; Li, A.; He, Z.; Liu, J.; et al. Colorectal cancer cell-derived CCL20 recruits regulatory T cells to promote chemoresistance via FOXO1/CEBPB/NF-κB signaling. J. Immunother. Cancer 2019, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velaei, K.; Samadi, N.; Barazvan, B.; Rad, J.S. Tumor microenvironment-mediated chemoresistance in breast cancer. Breast 2016, 30, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Erdman, S.E.; Rao, V.P.; Olipitz, W.; Taylor, C.L.; Jackson, E.A.; Levkovich, T.; Lee, C.W.; Horwitz, B.H.; Fox, J.G.; Ge, Z.; et al. Unifying roles for regulatory T cells and inflammation in cancer. Int. J. Cancer 2010, 126, 1651–1665. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. What are regulatory T cells (Treg) regulating in cancer and why? Semin. Cancer Biol. 2012, 22, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Jordanova, E.S.; Gorter, A.; Ayachi, O.; Prins, F.; Durrant, L.G.; Kenter, G.G.; van der Burg, S.H.; Fleuren, G.J. Human leukocyte antigen class I, MHC class I chain-related molecule A, and CD8+/regulatory T-cell ratio: Which variable determines survival of cervical cancer patients? Clin. Cancer Res. 2008, 14, 2028–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, D.A.A.; Kim, H.J.; Zhou, P.; Holderried, T.A.; Wang, X.; Dranoff, G.; Cantor, H. Disruption of CD8+ Treg activity results in expansion of T follicular helper cells and enhanced antitumor immunity. Cancer Immunol. Res. 2014, 2, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Mao, S.; Shi, D.; Zhang, J.; Zhang, Z.; Guo, Y.; Wu, Y.; Wang, R.; Wang, L.; Huang, Y.; et al. MicroRNA-153 Decreases Tryptophan Catabolism and Inhibits Angiogenesis in Bladder Cancer by Targeting Indoleamine 2,3-Dioxygenase 1. Front. Oncol. 2019, 9, 619. [Google Scholar] [CrossRef]
- Pan, J.; Yuan, K.; Peng, S.; Huang, Y.; Zhang, Y.; Hu, Y.; Feng, Y.; Shi, Y.; Liu, Y.; Wang, H.; et al. Gene silencing of indoleamine 2,3-dioxygenase hinders tumor growth through angiogenesis inhibition. Int. J. Oncol. 2017, 50, 2136–2144. [Google Scholar] [CrossRef] [PubMed]
- Mondal, A.; Smith, C.; DuHadaway, J.B.; Sutanto-Ward, E.; Prendergast, G.C.; Bravo-Nuevo, A.; Muller, A.J. IDO1 is an Integral Mediator of Inflammatory Neovascularization. EBioMedicine 2016, 14, 74–82. [Google Scholar] [CrossRef] [Green Version]
- Terai, M.; Londin, E.; Rochani, A.; Link, E.; Lam, B.; Kaushal, G.; Bhushan, A.; Orloff, M.; Sato, T. Expression of Tryptophan 2,3-Dioxygenase in Metastatic Uveal Melanoma. Cancers 2020, 12, 405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhai, J.; Kong, X.; Wang, X.; Wang, Z.; Fang, Y.; Wang, J. Comprehensive Analysis of the Expressionand Prognosis for TDO2 in Breast Cancer. Mol. Ther. Oncolytics 2020, 17, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Hung, J.Y.; Chiang, S.Y.; Jian, S.F.; Wu, C.Y.; Lin, Y.S.; Tsai, Y.M.; Chou, S.H.; Tsai, M.J.; Kuo, P.L. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget 2016, 7, 27584–27598. [Google Scholar] [CrossRef] [Green Version]
- Venmar, K.T.; Carter, K.J.; Hwang, D.G.; Dozier, E.A.; Fingleton, B. IL4 receptor ILR4α regulates metastatic colonization by mammary tumors through multiple signaling pathways. Cancer Res. 2014, 74, 4329–4340. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Cheng, Q.; Zhang, B.; Zhang, M. IL-13 induces the expression of 11βHSD2 in IL-13Rα2 dependent manner and promotes the malignancy of colorectal cancer. Am. J. Transl. Res. 2016, 8, 1064–1072. [Google Scholar]
- Suzuki, A.; Leland, P.; Joshi, B.H.; Puri, R.K. Targeting of IL-4 and IL-13 receptors for cancer therapy. Cytokine 2015, 75, 79–88. [Google Scholar] [CrossRef]
- Guruprasath, P.; Kim, J.; Gunassekaran, G.R.; Chi, L.; Kim, S.; Park, R.W.; Kim, S.H.; Baek, M.C.; Bae, S.M.; Kim, S.Y.; et al. Interleukin-4 receptor-targeted delivery of Bcl-xL siRNA sensitizes tumors to chemotherapy and inhibits tumor growth. Biomaterials 2017, 142, 101–111. [Google Scholar] [CrossRef]
- Hallett, M.A.; Venmar, K.T.; Fingleton, B. Cytokine stimulation of epithelial cancer cells: The similar and divergent functions of IL-4 and IL-13. Cancer Res. 2012, 72, 6338–6343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venmar, K.T.; Kimmel, D.W.; Cliffel, D.E.; Fingleton, B. IL4 receptor α mediates enhanced glucose and glutamine metabolism to support breast cancer growth. Biochim. Biophys. Acta 2015, 1853, 1219–1228. [Google Scholar] [CrossRef] [Green Version]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Wu, D.; Zhou, S.; Wan, F.; Liu, H.; Xu, X.; Xu, X.; Zhao, Y.; Tang, M. The pancreatic cancer secreted REG4 promotes macrophage polarization to M2 through EGFR/AKT/CREB pathway. Oncol. Rep. 2016, 35, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Che, X.; Qiu, X.; Li, Z.; Yang, B.; Wang, S.; Hou, K.; Fan, Y.; Qu, X.; Liu, Y. M2 macrophage infiltration into tumor islets leads to poor prognosis in non-small-cell lung cancer. Cancer Manag. Res. 2019, 11, 6125–6138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Hang, J.J.; Han, T.; Zhuo, M.; Jiao, F.; Wang, L.W. The M2 phenotype of tumor-associated macrophages in the stroma confers a poor prognosis in pancreatic cancer. Tumour Biol. 2016, 37, 8657–8664. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, J.; Li, D.; Mao, Y.; Mo, F.; Du, W.; Ma, X. Prognostic significance of tumor-associated macrophages in ovarian cancer: A meta-analysis. Gynecol. Oncol. 2017, 147, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Räihä, M.R.; Puolakkainen, P.A. Tumor-associated macrophages (TAMs) as biomarkers for gastric cancer: A review. Chronic Dis. Transl. Med. 2018, 4, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Salmaninejad, A.; Valilou, S.F.; Soltani, A.; Ahmadi, S.; Abarghan, Y.J.; Rosengren, R.J.; Sahebkar, A. Tumor-associated macrophages: Role in cancer development and therapeutic implications. Cell. Oncol. 2019, 42, 591–608. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.Q.; Du, W.L.; Cai, M.H.; Yao, J.Y.; Zhao, Y.Y.; Mou, X.Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell. Immunol. 2020, 353, 104119. [Google Scholar] [CrossRef]
- Komohara, Y.; Horlad, H.; Ohnishi, K.; Fujiwara, Y.; Bai, B.; Nakagawa, T.; Suzu, S.; Nakamura, H.; Kuratsu, J.; Takeya, M. Importance of direct macrophage-tumor cell interaction on progression of human glioma. Cancer Sci. 2012, 103, 2165–2172. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Hasita, H.; Ohnishi, K.; Fujiwara, Y.; Suzu, S.; Eto, M.; Takeya, M. Macrophage infiltration and its prognostic relevance in clear cell renal cell carcinoma. Cancer Sci. 2011, 102, 1424–1431. [Google Scholar] [CrossRef]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; He, M.Y.; Zhu, L.F.; Yang, C.C.; Zhou, M.L.; Wang, Q.; Zhang, W.; Zheng, Y.Y.; Wang, D.M.; Xu, Z.Q.; et al. Tumor-associated macrophages correlate with the clinicopathological features and poor outcomes via inducing epithelial to mesenchymal transition in oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinnakota, K.; Zhang, Y.; Selvanesan, B.C.; Topi, G.; Salim, T.; Sand-Dejmek, J.; Jönsson, G.; Sjölander, A. M2-like macrophages induce colon cancer cell invasion via matrix metalloproteinases. J. Cell. Physiol. 2017, 232, 3468–3480. [Google Scholar] [CrossRef] [PubMed]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [Green Version]
- Fields, G.B. Mechanisms of Action of Novel Drugs Targeting Angiogenesis-Promoting Matrix Metalloproteinases. Front. Immunol. 2019, 10, 1278. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Sun, L.; Xu, F.; Liu, L.; Hu, F.; Song, D.; Hou, Z.; Wu, W.; Luo, X.; Wang, J.; et al. M2 Macrophage-Derived Exosomes Promote Cell Migration and Invasion in Colon Cancer. Cancer Res. 2019, 79, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Luo, Q.; Wang, W.; Li, J.; Wang, T.; Wang, P.; Chen, L.; Zhang, P.; Chen, H.; Liu, Y.; et al. Tumor-associated macrophages-derived exosomes promote the migration of gastric cancer cells by transfer of functional Apolipoprotein E. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gao, W.; Tang, Q.; Yu, Y.; You, W.; Wu, Z.; Fan, Y.; Zhang, L.; Wu, C.; Han, G.; et al. M2 macrophage-derived exosomes facilitate hepatocarcinoma metastasis by transferring αM β2 integrin to tumor cells. Hepatology 2020. [Google Scholar] [CrossRef]
- Ireland, L.V.; Mielgo, A. Macrophages and Fibroblasts, Key Players in Cancer Chemoresistance. Front. Cell Dev. Biol. 2018, 6, 131. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Yang, Q. MiR-21 modulates the polarization of macrophages and increases the effects of M2 macrophages on promoting the chemoresistance of ovarian cancer. Life Sci. 2020, 242, 117162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Yin, W.; Wu, A.; Tang, Y.; Wang, J.; Pan, Z.; Lin, T.; Zhang, M.; Chen, B.; Duan, Y.; et al. Dual-targeting to Cancer Cells and M2 macrophages via Biomimetic Delivery of Mannosylated Albumine Nanoparticles for Drug-Resistant Cancer Therapy. Adv. Funct. Mater. 2017, 27, 1700403. [Google Scholar] [CrossRef]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’Alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef] [Green Version]
- Mottahedeh, J.; Haffner, M.C.; Grogan, T.R.; Hashimoto, T.; Crowell, P.D.; Beltran, H.; Sboner, A.; Bareja, R.; Esopi, D.; Isaacs, W.B.; et al. CD38 is methylated in prostate cancer and regulates extracellular NAD. Cancer Metab. 2018, 6, 1–17. [Google Scholar] [CrossRef]
- Lv, H.; Lv, G.; Chen, C.; Zong, Q.; Jiang, G.; Ye, D.; Cui, X.; He, Y.; Xiang, W.; Han, Q.; et al. NAD+ Metabolism Maintains Inducible PD-L1 Expression to Drive Tumor Immune Evasion. Cell Metab. 2021, 33, 110–127.e5. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Hamanishi, J.; Abiko, K.; Matsumura, N.; Baba, T.; Konishi, I. Dual Faces of IFNγ in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin. Cancer Res. 2016, 22, 2329–2334. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Lang, J. Soluble PD-1 and PD-L1: Predictive and prognostic significance in cancer. Oncotarget 2017, 8, 97671–97682. [Google Scholar] [CrossRef] [Green Version]
- Ock, C.Y.; Kim, S.; Keam, B.; Kim, M.; Kim, T.M.; Kim, J.H.; Jeon, Y.K.; Lee, J.S.; Kwon, S.K.; Hah, J.H.; et al. PD-L1 expression is associated with epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 15901–15914. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Koh, J.; Kim, M.Y.; Kwon, D.; Go, H.; Kim, Y.A.; Jeon, Y.K.; Chung, D.H. PD-L1 expression is associated with epithelial-to-mesenchymal transition in adenocarcinoma of the lung. Hum. Pathol. 2016, 58, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiong, Y.; Li, J.; Zheng, X.; Zhou, Q.; Turner, A.; Wu, C.; Lu, B.; Jiang, J. PD-L1 Expression Promotes Epithelial to Mesenchymal Transition in Human Esophageal Cancer. Cell. Physiol. Biochem. 2017, 42, 2267–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, H.; Zhao, Q.; Xia, Y.; Hu, X.; Guo, J. PD-L1 induces epithelial-to-mesenchymal transition via activating SREBP-1c in renal cell carcinoma. Med. Oncol. 2015, 32, 1–7. [Google Scholar] [CrossRef]
- Clark, C.A.; Gupta, H.B.; Sareddy, G.; Pandeswara, S.; Lao, S.; Yuan, B.; Drerup, J.M.; Padron, A.; Conejo-Garcia, J.; Murthy, K.; et al. Tumor-Intrinsic PD-L1 Signals Regulate Cell Growth, Pathogenesis, and Autophagy in Ovarian Cancer and Melanoma. Cancer Res. 2016, 76, 6964–6974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, X.Y.; Hu, D.X.; Chen, W.Q.; Chen, R.Q.; Qian, S.R.; Li, C.Y.; Li, Y.J.; Xiong, X.X.; Liu, D.; Pan, F.; et al. PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochimica et biophysica acta. Mol. Basis Dis. 2018, 1864, 1754–1769. [Google Scholar] [CrossRef]
- Mandarano, M.; Bellezza, G.; Belladonna, M.L.; Vannucci, J.; Gili, A.; Ferri, I.; Lupi, C.; Ludovini, V.; Falabella, G.; Metro, G.; et al. Indoleamine 2,3-Dioxygenase 2 Immunohistochemical Expression in Resected Human Non-small Cell Lung Cancer: A Potential New Prognostic Tool. Front. Immunol. 2020, 11, 839. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.B.; Zhu, S.P.; Liu, T.P.; Zhao, H.; Chen, P.F.; Duan, Y.J.; Hu, R. Cancer Associated Fibroblasts Promote Renal Cancer Progression Through a TDO/Kyn/AhR Dependent Signaling Pathway. Front. Oncol. 2021, 11, 905. [Google Scholar] [CrossRef]
- Li, L.; Wang, T.; Li, S.; Chen, Z.; Wu, J.; Cao, W.; Wo, Q.; Qin, X.; Xu, J. TDO2 Promotes the EMT of Hepatocellular Carcinoma Through Kyn-AhR Pathway. Front. Oncol. 2021, 10, 3008. [Google Scholar] [CrossRef]
- Du, L.; Xing, Z.; Tao, B.; Li, T.; Yang, D.; Li, W.; Zheng, Y.; Kuang, C.; Yang, Q. Both IDO1 and TDO contribute to the malignancy of gliomas via the Kyn-AhR-AQP4 signaling pathway. Signal Transduct. Target. Ther. 2020, 5, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadopoulos, M.C.; Saadoun, S.; Verkman, A.S. Aquaporins and cell migration. Pflug. Arch. 2008, 456, 693–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanowicz, J.M.; Kwiatkowska, I.; Pawlak, D. Important players in carcinogenesis as potential targets in cancer therapy: An update. Oncotarget 2020, 11, 3078–3101. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Ribatti, D. Aquaporins in tumor growth and angiogenesis. Cancer Lett. 2010, 294, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Ma, Y.; Li, W.; Liu, X.; Ying, G.; Fu, L.; Gu, F. Role of aquaporin-4 in the regulation of migration and invasion of human glioma cells. Int. J. Oncol. 2011, 38, 1521–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekki, K.; Vogel, H.; Li, W.; Ito, T.; Sweeney, C.; Haarmann-Stemmann, T.; Matsumura, F.; Vogel, C.F. The aryl hydrocarbon receptor (AhR) mediates resistance to apoptosis induced in breast cancer cells. Pestic. Biochem. Physiol. 2015, 120, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Liang, X.; Yin, X.; Lv, J.; Tang, K.; Ma, J.; Ji, T.; Zhang, H.; Dong, W.; Jin, X.; et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wand, Q.; Yang, L.; Liu, Q.; Ju, R.; Guo, L.; Ye, C.; Zhang, D. Blockade of IDO-kynurenine-AhR pathway promotes cell apoptosis in carboxyamidotriazole induced tumor cell dormancy apoptosis oscillation. ResearchSquare 2020. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, X.; Dong, W.; Fang, Y.; Lv, J.; Zhang, T.; Fiskesund, R.; Xie, J.; Liu, J.; Yin, X.; et al. Tumor-Repopulating Cells Induce PD-1 Expression in CD8+ T Cells by Transferring Kynurenine and AhR Activation. Cancer Cell 2018, 33, 480–494.e7. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Harden, J.L.; Anderson, C.D.; Egilmez, N.K. Tolerogenic Phenotype of IFN-γ-Induced IDO+ Dendritic Cells Is Maintained via an Autocrine IDO-Kynurenine/AhR-IDO Loop. J. Immunol. 2016, 197, 962–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- DiNatale, B.C.; Murray, I.A.; Schroeder, J.C.; Flaveny, C.A.; Lahoti, T.S.; Laurenzana, E.M.; Omiecinski, C.J.; Perdew, G.H. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010, 115, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef]
- Ortiz-Montero, P.; Londoño-Vallejo, A.; Vernot, J.P. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 2017, 15, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.Y.; Yu, I.S.; Lin, Y.C.; Chang, Y.T.; Chen, C.C.; Lin, K.H.; Tseng, T.H.; Kargren, M.; Tai, Y.L.; Shen, T.L.; et al. Activation of Aryl Hydrocarbon Receptor by Kynurenine Impairs Progression and Metastasis of Neuroblastoma. Cancer Res. 2019, 79, 5550–5562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Boivin, G.P.; Knudsen, E.S.; Nebert, D.W.; Xia, Y.; Puga, A. The aryl hydrocarbon receptor functions as a tumor suppressor of liver carcinogenesis. Cancer Res. 2010, 70, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, M.; Hankinson, O. 2,3,7,8 tetrachlorodibenzo p dioxin suppresses the growth of human colorectal cancer cells in vitro: Implication of the aryl hydrocarbon receptor signaling. Int. J. Oncol. 2019, 54, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Contador-Troca, M.; Alvarez-Barrientos, A.; Barrasa, E.; Rico-Leo, E.M.; Catalina-Fernández, I.; Menacho-Márquez, M.; Bustelo, X.R.; García-Borrón, J.C.; Gómez-Durán, A.; Sáenz-Santamaría, J.; et al. The dioxin receptor has tumor suppressor activity in melanoma growth and metastasis. Carcinogenesis 2013, 34, 2683–2693. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Feng, Y.; Wang, Y.; An, R. Aryl hydrocarbon receptor enhances the expression of miR-150-5p to suppress in prostate cancer progression by regulating MAP3K12. Arch. Biochem. Biophys. 2018, 654, 47–54. [Google Scholar] [CrossRef]
- Huang, J.Y.; Larose, T.L.; Luu, H.N.; Wang, R.; Fanidi, A.; Alcala, K.; Stevens, V.L.; Weinstein, S.J.; Albanes, D.; Caporaso, N.E.; et al. Circulating markers of cellular immune activation in prediagnostic blood sample and lung cancer risk in the Lung Cancer Cohort Consortium (LC3). Int. J. Cancer 2020, 146, 2394–2405. [Google Scholar] [CrossRef]
- Cheng, X.; Liu, X.; Liu, X.; Guo, Z.; Sun, H.; Zhang, M.; Ji, Z.; Sun, W. Metabolomics of Non-muscle Invasive Bladder Cancer: Biomarkers for Early Detection of Bladder Cancer. Front. Oncol. 2018, 8, 494. [Google Scholar] [CrossRef]
- Hiraku, Y.; Inoue, S.; Oikawa, S.; Yamamoto, K.; Tada, S.; Nishino, K.; Kawanishi, S. Metal-mediated oxidative damage to cellular and isolated DNA by certain tryptophan metabolites. Carcinogenesis 1995, 16, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, I.; Hermanowicz, J.M.; Mysliwiec, M.; Pawlak, D. Oxidative Storm Induced by Tryptophan Metabolites: Missing Link between Atherosclerosis and Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2020, 2020, 6656033. [Google Scholar] [CrossRef]
- Song, H.; Park, H.; Kim, Y.S.; Kim, K.D.; Lee, H.K.; Cho, D.H.; Yang, J.W.; Hur, D.Y. L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int. Immunopharmacol. 2011, 11, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Guillerey, C.; Smyth, M.J. NK Cells and Cancer Immunoediting. Curr. Top. Microbiol. Immunol. 2016, 395, 115–145. [Google Scholar] [CrossRef]
- Chockley, P.J.; Chen, J.; Chen, G.; Beer, D.G.; Standiford, T.J.; Keshamouni, V.G. Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer. J. Clin. Investig. 2018, 128, 1384–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimaoka, H.; Takeno, S.; Maki, K.; Sasaki, T.; Hasegawa, S.; Yamashita, Y. A cytokine signal inhibitor for rheumatoid arthritis enhances cancer metastasis via depletion of NK cells in an experimental lung metastasis mouse model of colon cancer. Oncol. Lett. 2017, 14, 3019–3027. [Google Scholar] [CrossRef] [Green Version]
- Aydin, E.; Johansson, J.; Nazir, F.H.; Hellstrand, K.; Martner, A. Role of NOX2-Derived Reactive Oxygen Species in NK Cell-Mediated Control of Murine Melanoma Metastasis. Cancer Immunol. Res. 2017, 5, 804–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luis-García, E.R.; Limón-Pacheco, J.H.; Serrano-García, N.; Hernández-Pérez, A.D.; Pedraza-Chaverri, J.; Orozco-Ibarra, M. Sulforaphane prevents quinolinic acid-induced mitochondrial dysfunction in rat striatum. J. Biochem. Mol. Toxicol. 2017, 31, e21837. [Google Scholar] [CrossRef]
- Guerra, F.; Guaragnella, N.; Arbini, A.A.; Bucci, C.; Giannattasio, S.; Moro, L. Mitochondrial Dysfunction: A Novel Potential Driver of Epithelial-to-Mesenchymal Transition in Cancer. Front. Oncol. 2017, 7, 295. [Google Scholar] [CrossRef] [Green Version]
- Bishnupuri, K.S.; Alvarado, D.M.; Khouri, A.N.; Shabsovich, M.; Chen, B.; Dieckgraefe, B.K.; Ciorba, M.A. IDO1 and Kynurenine Pathway Metabolites Activate PI3K-Akt Signaling in the Neoplastic Colon Epithelium to Promote Cancer Cell Proliferation and Inhibit Apoptosis. Cancer Res. 2019, 79, 1138–1150. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.X.; Zhang, W.N.; Sun, Y.Y.; Li, Y.H.; Xu, Z.M.; Fu, W.N. CREB promotes laryngeal cancer cell migration via MYCT1/NAT10 axis. OncoTargets Ther. 2018, 11, 1323–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ren, Y.; Zhuang, H.; Meng, X.; Huang, S.; Li, Y.; Hehir, M.; Wang, P. Decrease of phosphorylated proto-oncogene CREB at Ser 133 site inhibits growth and metastatic activity of renal cell cancer. Expert Opin. Ther. Targets 2015, 19, 985–995. [Google Scholar] [CrossRef]
- Kankanamalage, S.G.; Karra, A.S.; Cobb, M.H. WNK pathways in cancer signaling networks. Cell Commun. Signal. 2018, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Simonavicius, N.; Wu, X.; Swaminath, G.; Reagan, J.; Tian, H.; Ling, L. Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J. Biol. Chem. 2006, 281, 22021–22028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.; Hu, H.; Fang, Y. Tyrphostin analogs are GPR35 agonists. FEBS Lett. 2011, 585, 1957–1962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.J.; Zhou, Y.J.; Yang, X.L.; Shao, Z.M.; Ou, Z.L. The role and clinical significance of the CXCL17-CXCR8 (GPR35) axis in breast cancer. Biochem. Biophys. Res. Commun. 2017, 493, 1159–1167. [Google Scholar] [CrossRef]
- Ali, H.; AbdelMageed, M.; Olsson, L.; Israelsson, A.; Lindmark, G.; Hammarström, M.L.; Hammarström, S.; Sitohy, B. Utility of G protein-coupled receptor 35 expression for predicting outcome in colon cancer. Tumour Biol. 2019, 41, 1010428319858885. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Li, D.; Zhang, Y.; Chen, J.; Zhang, Y.; Liao, C.; Qin, S.; Tian, Y.; Zhang, Z.; Xu, F. Functional metabolomics reveal the role of AHR/GPR35 mediated kynurenic acid gradient sensing in chemotherapy-induced intestinal damage. Acta Pharm. Sin. B 2021, 11, 763–780. [Google Scholar] [CrossRef]
- Schneditz, G.; Elias, J.E.; Pagano, E.; Cader, M.Z.; Saveljeva, S.; Long, K.; Mukhopadhyay, S.; Arasteh, M.; Lawley, T.D.; Dougan, G.; et al. GPR35 promotes glycolysis, proliferation, and oncogenic signaling by engaging with the sodium potassium pump. Sci. Signal. 2019, 12, eaau9048. [Google Scholar] [CrossRef] [PubMed]
- Pagano, E.; Elias, J.E.; Schneditz, G.; Saveljeva, S.; Holland, L.M.; Borrelli, F.; Karlsen, T.H.; Kaser, A.; Kaneider, N.C. Activation of the GPR35 pathway drives angiogenesis in the tumour microenvironment. Gut 2021. [Google Scholar] [CrossRef]
- Lee, K.; Kwak, J.H.; Pyo, S. Inhibition of LPS-induced inflammatory mediators by 3-hydroxyanthranilic acid in macrophages through suppression of PI3K/NF-κB signaling pathways. Food Funct. 2016, 7, 3073–3082. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.S.; Lee, S.M.; Kim, M.K.; Park, S.G.; Choi, I.W.; Choi, I.; Joo, Y.D.; Park, S.J.; Kang, S.W.; Seo, S.K. The tryptophan metabolite 3-hydroxyanthranilic acid suppresses T cell responses by inhibiting dendritic cell activation. Int. Immunopharmacol. 2013, 17, 721–726. [Google Scholar] [CrossRef]
- Piscianz, E.; Cuzzoni, E.; De Iudicibus, S.; Valencic, E.; Decorti, G.; Tommasini, A. Differential action of 3-hydroxyanthranilic acid on viability and activation of stimulated lymphocytes. Int. Immunopharmacol. 2011, 11, 2242–2245. [Google Scholar] [CrossRef] [PubMed]
- Uzzo, R.G.; Rayman, P.; Kolenko, V.; Clark, P.E.; Bloom, T.; Ward, A.M.; Molto, L.; Tannenbaum, C.; Worford, L.J.; Bukowski, R.; et al. Mechanisms of apoptosis in T cells from patients with renal cell carcinoma. Clin. Cancer Res 1999, 5, 1219–1229. [Google Scholar] [PubMed]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by kynurenines. Adv. Exp. Med. Biol. 2003, 527, 183–190. [Google Scholar] [CrossRef]
- Lee, S.M.; Lee, Y.S.; Choi, J.H.; Park, S.G.; Choi, I.W.; Joo, Y.D.; Lee, W.S.; Lee, J.N.; Choi, I.; Seo, S.K. Tryptophan metabolite 3-hydroxyanthranilic acid selectively induces activated T cell death via intracellular GSH depletion. Immunol. Lett. 2010, 132, 53–60. [Google Scholar] [CrossRef]
- Zaher, S.S.; Germain, C.; Fu, H.; Larkin, D.F.; George, A.J. 3-hydroxykynurenine suppresses CD4+ T-cell proliferation, induces T-regulatory-cell development, and prolongs corneal allograft survival. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2640–2648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walczak, K.; Wnorowski, A.; Turski, W.A.; Plech, T. Kynurenic acid and cancer: Facts and controversies. Cell. Mol. Life Sci. 2020, 77, 1531–1550. [Google Scholar] [CrossRef] [Green Version]
- Walczak, K.; Turski, W.A.; Rzeski, W. Kynurenic acid enhances expression of p21 Waf1/Cip1 in colon cancer HT-29 cells. Pharmacol. Rep. 2012, 64, 745–750. [Google Scholar] [CrossRef] [Green Version]
- Walczak, K.; Zurawska, M.; Kiś, J.; Starownik, R.; Zgrajka, W.; Bar, K.; Turski, W.A.; Rzeski, W. Kynurenic acid in human renal cell carcinoma: Its antiproliferative and antimigrative action on Caki-2 cells. Amino Acids 2012, 43, 1663–1670. [Google Scholar] [CrossRef]
- Walczak, K.; Deneka-Hannemann, S.; Jarosz, B.; Zgrajka, W.; Stoma, F.; Trojanowski, T.; Turski, W.A.; Rzeski, W. Kynurenic acid inhibits proliferation and migration of human glioblastoma T98G cells. Pharmacol. Rep. 2014, 66, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Maes, M. Interactions of Tryptophan and Its Catabolites with Melatonin and the Alpha 7 Nicotinic Receptor in Central Nervous System and Psychiatric Disorders: Role of the Aryl Hydrocarbon Receptor and Direct Mitochondria Regulation. Int. J. Tryptophan Res. 2017, 10. [Google Scholar] [CrossRef]
- Zhang, C.; Ding, X.P.; Zhao, Q.N.; Yang, X.J.; An, S.M.; Wang, H.; Xu, L.; Zhu, L.; Chen, H.Z. Role of α7-nicotinic acetylcholine receptor in nicotine-induced invasion and epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. Oncotarget 2016, 7, 59199–59208. [Google Scholar] [CrossRef] [Green Version]
- Bu, X.; Zhang, A.; Chen, Z.; Zhang, X.; Zhang, R.; Yin, C.; Zhang, J.; Zhang, Y.; Yan, Y. Migration of gastric cancer is suppressed by recombinant Newcastle disease virus (rL-RVG) via regulating α7-nicotinic acetylcholine receptors/ERK-EMT. BMC Cancer 2019, 19, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Kang, X.; Liu, G.; Zhang, B.; Hu, X.; Feng, Y. α7-Nicotinic Acetylcholine Receptor Promotes Cholangiocarcinoma Progression and Epithelial-Mesenchymal Transition Process. Dig. Dis. Sci. 2019, 64, 2843–2853. [Google Scholar] [CrossRef]
- Aali, N.; Motalleb, G. The effect of nicotine on the expressions of the α7 nicotinic receptor gene and Bax and Bcl-2 proteins in the mammary gland epithelial-7 breast cancer cell line and its relationship to drug resistance. Cell. Mol. Biol. Lett. 2015, 20, 948–964. [Google Scholar] [CrossRef] [PubMed]
- Di Serio, C.; Cozzi, A.; Angeli, I.; Doria, L.; Micucci, I.; Pellerito, S.; Mirone, P.; Masotti, G.; Moroni, F.; Tarantini, F. Kynurenic acid inhibits the release of the neurotrophic fibroblast growth factor (FGF)-1 and enhances proliferation of glia cells, in vitro. Cell. Mol. Neurobiol. 2005, 25, 981–993. [Google Scholar] [CrossRef]
- Zhao, D.; Lu, Y.; Yang, C.; Zhou, X.; Xu, Z. Activation of FGF receptor signaling promotes invasion of non-small-cell lung cancer. Tumour Biol. 2015, 36, 3637–3642. [Google Scholar] [CrossRef]
- Winterhoff, B.; Konecny, G.E. Targeting fibroblast growth factor pathways in endometrial cancer. Curr. Probl. Cancer 2017, 41, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.P.; Shang, K.; Chen, H.; Ding, F.; Wang, Z.; Liang, C.; Xu, Y.; Sun, M.H.; Li, Y.Y. FGF-1/-3/FGFR4 signaling in cancer-associated fibroblasts promotes tumor progression in colon cancer through Erk and MMP-7. Cancer Sci. 2015, 106, 1278–1287. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, R.; Yu, W.; Ye, Y.; Cheng, Y.; Han, L.; Dong, L.; Chen, Y.; Wei, X.; Yu, J. FGF1 and IGF1-conditioned 3D culture system promoted the amplification and cancer stemness of lung cancer cells. Biomaterials 2017, 149, 63–76. [Google Scholar] [CrossRef]
- Li, J.; Wei, Z.; Li, H.; Dang, Q.; Zhang, Z.; Wang, L.; Gao, W.; Zhang, P.; Yang, D.; Liu, J.; et al. Clinicopathological significance of fibroblast growth factor 1 in non-small cell lung cancer. Hum. Pathol. 2015, 46, 1821–1828. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Yang, B.; Chen, J.; Xiong, H.; Li, Y.; Pan, Z.; Cao, Y.; Chen, J.; Li, T.; Zhou, S.; et al. Upregulation of long non-coding RNA RAB1A-2 induces FGF1 expression worsening lung cancer prognosis. Cancer Lett. 2018, 438, 116–125. [Google Scholar] [CrossRef]
- Shi, H.; Fu, C.; Wang, W.; Li, Y.; Du, S.; Cao, R.; Chen, J.; Sun, D.; Zhang, Z.; Wang, X.; et al. The FGF-1-specific single-chain antibody scFv1C9 effectively inhibits breast cancer tumour growth and metastasis. J. Cell. Mol. Med. 2014, 18, 2061–2070. [Google Scholar] [CrossRef]
- Lipok, M.; Szlachcic, A.; Kindela, K.; Czyrek, A.; Otlewski, J. Identification of a peptide antagonist of the FGF1-FGFR1 signaling axis by phage display selection. FEBS Open Biol. 2019, 9, 914–924. [Google Scholar] [CrossRef] [PubMed]
- Pae, H.O.; Oh, G.S.; Lee, B.S.; Rim, J.S.; Kim, Y.M.; Chung, H.T. 3-Hydroxyanthranilic acid, one of L-tryptophan metabolites, inhibits monocyte chemoattractant protein-1 secretion and vascular cell adhesion molecule-1 expression via heme oxygenase-1 induction in human umbilical vein endothelial cells. Atherosclerosis 2006, 187, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Sharma, R.; Khaket, T.P.; Dutta, C.; Chakraborty, B.; Mukherjee, T.K. Breast cancer metastasis: Putative therapeutic role of vascular cell adhesion molecule-1. Cell. Oncol. 2017, 40, 199–208. [Google Scholar] [CrossRef]
- Zhang, D.; Bi, J.; Liang, Q.; Wang, S.; Zhang, L.; Han, F.; Li, S.; Qiu, B.; Fan, X.; Chen, W.; et al. VCAM1 Promotes Tumor Cell Invasion and Metastasis by Inducing EMT and Transendothelial Migration in Colorectal Cancer. Front. Oncol. 2020, 10, 1066. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Keep, R.F.; Mostarica-Stojkovic, M.; Andjelkovic, A.V. CCL2 regulates angiogenesis via activation of Ets-1 transcription factor. J. Immunol. 2006, 177, 2651–2661. [Google Scholar] [CrossRef] [Green Version]
- Gálvez, B.G.; Genís, L.; Matías-Román, S.; Oblander, S.A.; Tryggvason, K.; Apte, S.S.; Arroyo, A.G. Membrane type 1-matrix metalloproteinase is regulated by chemokines monocyte-chemoattractant protein-1/ccl2 and interleukin-8/CXCL8 in endothelial cells during angiogenesis. J. Biol. Chem. 2005, 280, 1292–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, M.; Bendas, G. Vascular cell adhesion molecule-1 (VCAM-1)—an increasing insight into its role in tumorigenicity and metastasis. Int. J. Cancer 2015, 136, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Leuthauser, S.W.; Oberley, L.W.; Oberley, T.D. Antitumor activity of picolinic acid in CBA/J mice. J. Natl. Cancer Inst. 1982, 68, 123–126. [Google Scholar]
- Ruffmann, R.; Schlick, R.; Chirigos, M.A.; Budzynsky, W.; Varesio, L. Antiproliferative activity of picolinic acid due to macrophage activation. Drugs Exp. Clin. Res. 1987, 13, 607–614. [Google Scholar] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients with Advanced Solid Tumors: Phase I Results from a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Kristeleit, R.; Davidenko, I.; Shirinkin, V.; El-Khouly, F.; Bondarenko, I.; Goodheart, M.J.; Gorbunova, V.; Penning, C.A.; Shi, J.G.; Liu, X.; et al. A randomised, open-label, phase 2 study of the IDO1 inhibitor epacadostat (INCB024360) versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)-only epithelial ovarian cancer, primary peritoneal carcinoma, or fallopian tube cancer. Gynecol. Oncol. 2017, 146, 484–490. [Google Scholar] [CrossRef]
- Komiya, T.; Huang, C.H. Updates in the Clinical Development of Epacadostat and Other Indoleamine 2,3-Dioxygenase 1 Inhibitors (IDO1) for Human Cancers. Front. Oncol. 2018, 8, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, C.M.; Chi, P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Qin, L.; Adamson, T.; Condy, M.M.; Biniakewitz, M.; Phelan, H.; et al. A phase II study of epacadostat and pembrolizumab in patients with advanced sarcoma. J. Clin. Oncol. 2019, 37, 11049. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Gettinger, S.; Chow, L.; Gordon, M.; Awad, M.M.; Cha, E.; Gong, X.; Zhou, G.; Walker, C.; Leopold, L.; et al. Phase 1 study of epacadostat in combination with atezolizumab for patients with previously treated advanced nonsmall cell lung cancer. Int. J. Cancer 2020, 147, 1963–1969. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04106414?term=BMS-986205&draw=2&rank=5 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03695250?term=BMS-986205&draw=2&rank=6 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03661320?term=BMS-986205&draw=2&rank=11 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02658890?term=BMS-986205&draw=2&rank=14 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03854032?term=BMS-986205&draw=2&rank=19 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03459222?term=BMS-986205&draw=2&rank=21 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02996110?term=BMS-986205&draw=2&rank=22 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02935634?term=BMS-986205&draw=2&rank=24 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03329846?term=BMS-986205&draw=3&rank=7 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03915405?term=KHK2455&draw=2&rank=2 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03343613?term=LY3381916&draw=2&rank=1 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://0-faseb-onlinelibrary-wiley-com.brum.beds.ac.uk/doi/abs/10.1096/fasebj.31.1_supplement.754.9 (accessed on 15 April 2021).
- Wu, J.S.; Lin, S.Y.; Liao, F.Y.; Hsiao, W.C.; Lee, L.C.; Peng, Y.H.; Hsieh, C.L.; Wu, M.H.; Song, J.S.; Yueh, A.; et al. Identification of Substituted Naphthotriazolediones as Novel Tryptophan 2,3-Dioxygenase (TDO) Inhibitors through Structure-Based Virtual Screening. J. Med. Chem. 2015, 58, 7807–7819. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. Targeting the Inhibition of Tryptophan 2,3-Dioxygenase (TDO-2) for Cancer Treatment. ACS Med. Chem. Lett. 2016, 8, 11–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazemi, M.H.; Najafi, A.; Karami, J.; Ghazizadeh, F.; Yousefi, H.; Falak, R.; Safari, E. Immune and metabolic checkpoints blockade: Dual wielding against tumors. Int. Immunopharmacol. 2021, 94, 107461. [Google Scholar] [CrossRef]
- Kumar, S.; Jaipuri, F.A.; Waldo, J.P.; Potturi, H.; Marcinowicz, A.; Adams, J.; Van Allen, C.; Zhuang, H.; Vahanian, N.; Link, C., Jr.; et al. Discovery of indoximod prodrugs and characterization of clinical candidate NLG802. Eur. J. Med. Chem. 2020, 198, 112373. [Google Scholar] [CrossRef] [PubMed]
- Le Naour, J.; Galluzzi, L.; Zitvogel, L.; Kroemer, G.; Vacchelli, E. Trial watch: IDO inhibitors in cancer therapy. Oncoimmunology 2020, 9, 1777625. [Google Scholar] [CrossRef] [PubMed]
- Soliman, H.H.; Minton, S.E.; Han, H.S.; Ismail-Khan, R.; Neuger, A.; Khambati, F.; Noyes, D.; Lush, R.; Chiappori, A.A.; Roberts, J.D.; et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget 2016, 7, 22928–22938. [Google Scholar] [CrossRef] [Green Version]
- Emadi, A.; Holtzman, N.; Imran, M. Indoximod in Combination with Idarubicin and Cytarabine for Upfront Treatment of Patients with Newly Diagnosed Acute Myeloid Leukemia (AML): Phase 1 Report. In Proceedings of the 22nd Congress of the European Hematology Association (EHA), Madrid, Spain, 22–25 June 2017. [Google Scholar]
- Fox, E.; Oliver, T.; Rowe, M.; Thomas, S.; Zakharia, Y.; Gilman, P.B.; Muller, A.J.; Prendergast, G.C. Indoximod: An Immunometabolic Adjuvant That Empowers T Cell Activity in Cancer. Front. Oncol. 2018, 8, 370. [Google Scholar] [CrossRef] [Green Version]
- Bahary, N.; Garrido-Laguna, I.; Cinnar, P. Phase 2 trial of the indoleamine2,3-dioxygenase pathway (IDO) inhibitor indoximod plus gemcitabine/nab-paclitaxel for the treatment of metastatic pancreas cancer: Interim analysis. In Proceedings of the 2016 Annual Meeting of the American Society of Clinical Oncology, Chicago, IL, USA, 3–7 June 2016. [Google Scholar]
- Zakharia, J.; Johnson, T. A Phase I/II study of the combination of indoximod and temozolomide for adult patients with temozolomide-refractory primary malignant brain tumors. J. Clin. Oncol. 2014, 32, TPS2107. [Google Scholar] [CrossRef]
- Mariotti, V.; Han, H.; Ismail-Khan, R.; Tang, S.C.; Dillon, P.; Montero, A.J.; Poklepovic, A.; Melin, S.; Ibrahim, N.K.; Kennedy, E.; et al. Effect of Taxane Chemotherapy with or without Indoximod in Metastatic Breast Cancer: A Randomized Clinical Trial. JAMA Oncol. 2021, 7, 61–69. [Google Scholar] [CrossRef]
- Soliman, H.H.; Jackson, E.; Neuger, T.; Dees, E.C.; Harvey, R.D.; Han, H.; Ismail-Khan, R.; Minton, S.; Vahanian, N.N.; Link, C.; et al. A first in man phase I trial of the oral immunomodulator, indoximod, combined with docetaxel in patients with metastatic solid tumors. Oncotarget 2014, 5, 8136–8146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Bolduc, A.R.; Hoda, M.N.; Gamble, D.N.; Dolisca, S.B.; Bolduc, A.K.; Hoang, K.; Ashley, C.; McCall, D.; Rojiani, A.M.; et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J. Immunother. Cancer 2014, 2, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.H.; LoRusso, P.; Burris, H.; Gordon, M.; Bang, Y.J.; Hellmann, M.D.; Cervantes, A.; de Olza, M.O.; Marabelle, A.; Hodi, F.S.; et al. Phase I Study of the Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitor Navoximod (GDC-0919) Administered with PD-L1 Inhibitor (Atezolizumab) in Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3220–3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak-Kapoor, A.; Hao, Z.; Sadek, R.; Dobbins, R.; Marshall, L.; Vahanian, N.N.; Jay Ramsey, W.; Kennedy, E.; Mautino, M.R.; Link, C.J.; et al. Phase Ia study of the indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor navoximod (GDC-0919) in patients with recurrent advanced solid tumors. J. Immunother. Cancer 2018, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03208959 (accessed on 15 April 2021).
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03641794?term=DN1406131&draw=2&rank=1 (accessed on 15 April 2021).
- Gyulveszi, G.; Fischer Ch Mirolo, M. A novel, highly potent dual IDO1/TDO inhibitor to reverse metabolic suppression of immune cells in the tumor micro-environment. Cancer Res. 2016, 76. [Google Scholar] [CrossRef]
- Gullapalli, S.; Roychowdhury, A.; Khaladkar, T. EPL-1410, a novel fused heterocycle based orally active dual inhibitor of ido1/tdo2 as a potential immuno-oncology therapeutic. In Proceedings of the Annual Meeting of American Association for Cancer Research, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Kim, C.; Kim, J.; Kim, J.S.; Chon, H.; Kim, J.H. A novel dual inhibitor of IDO and TDO, CMG017, potently suppresses the kynurenine pathway and overcomes resistance to immune checkpoint inhibitors. J. Clin. Oncol. 2019, 37, e14228. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03364049 (accessed on 15 April 2021).
- Kjeldsen, J.W.; Iversen, T.Z.; Engell-Noerregaard, L.; Mellemgaard, A.; Andersen, M.H.; Svane, I.M. Durable Clinical Responses and Long-Term Follow-Up of Stage III-IV Non-Small-Cell Lung Cancer (NSCLC) Patients Treated with IDO Peptide Vaccine in a Phase I Study-A Brief Research Report. Front. Immunol. 2018, 9, 2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, R.B.; Køllgaard, T.; Andersen, R.S.; van den Berg, J.H.; Svane, I.M.; thor Straten, P.; Andersen, M.H. Spontaneous cytotoxic T-Cell reactivity against indoleamine 2,3-dioxygenase-2. Cancer Res. 2011, 71, 2038–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hjortsø, M.D.; Larsen, S.K.; Kongsted, P.; Met, Ö.; Frøsig, T.M.; Andersen, G.H.; Ahmad, S.M.; Svane, I.M.; Becker, J.C.; Straten, P.T.; et al. Tryptophan 2,3-dioxygenase (TDO)-reactive T cells differ in their functional characteristics in health and cancer. Oncoimmunology 2015, 4, e968480. [Google Scholar] [CrossRef]
- Nitschke, N.J.; Bjoern, J.; Iversen, T.Z.; Andersen, M.H.; Svane, I.M. Indoleamine 2,3-dioxygenase and survivin peptide vaccine combined with temozolomide in metastatic melanoma. Stem Cell Investig. 2017, 4, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjoern, J.; Iversen, T.Z.; Nitschke, N.J.; Andersen, M.H.; Svane, I.M. Safety, immune and clinical responses in metastatic melanoma patients vaccinated with a long peptide derived from indoleamine 2,3-dioxygenase in combination with ipilimumab. Cytotherapy 2016, 18, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/study/NCT01961115?term=IDO+vaccine&draw=3&rank=6 (accessed on 15 April 2021).
- Sherr, D.; Kenison-Whte, J.; Wang, Z. The aryl hydrocarbon receptor as driver of cancer immunity. In Proceedings of the AACR Annual Meeting, Chicago, IL, USA, 14–18 April 2018. [Google Scholar]
- Gutcher, I.; Kober, C.; Roese, L. Blocking tumor-associated immune suppression with BAY-218, a novel, selective aryl hydrocarbon receptor (AhR) inhibitor. In Proceedings of the AACR Annual Meeting, Atlanta, GA, USA, 29 March–3 April 2019. [Google Scholar]
- Garcia, C.; Lemar, H.; Galang, C.; Joseph, J.; Gonzalez-Lopez, M.; Hager, J.; Dillon, M.P.; Aswad, J.F. Abstract 3255: A novel small molecule inhibitor of AhR suppresses the polarization and activity of M2 macrophages. Cancer Res. 2019, 79, 3255. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04069026 (accessed on 15 April 2021).
- Lucarelli, G.; Rutigliano, M.; Ferro, M.; Giglio, A.; Intini, A.; Triggiano, F.; Palazzo, S.; Gigante, M.; Castellano, G.; Ranieri, E.; et al. Activation of the kynurenine pathway predicts poor outcome in patients with clear cell renal cell carcinoma. Urol. Oncol. 2017; 35, 461.e15–461.e27. [Google Scholar] [CrossRef]
- Pasikanti, K.K.; Esuvaranathan, K.; Hong, Y.; Ho, P.C.; Mahendran, R.; Raman Nee Mani, L.; Chiong, E.; Chan, E.C.Y. Urinary metabotyping of bladder cancer using two-dimensional gas chromatography time-of-flight mass spectrometry. J. Proteome Res. 2013, 12, 3865–3873. [Google Scholar] [CrossRef] [PubMed]
- Dinges, S.S.; Hohm, A.; Vandergrift, L.A.; Nowak, J.; Habbel, P.; Kaltashov, I.A.; Cheng, L.L. Cancer metabolomic markers in urine: Evidence, techniques and recommendations. Nat. Rev. Urol. 2019, 16, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Cavia-Saiz, M.; Rodríguez, P.M.; Ayala, B.L.; García-González, M.; Coma-Del Corral, M.J.; Girón, C.G. The role of plasma IDO activity as a diagnostic marker of patients with colorectal cancer. Mol. Biol. Rep. 2014, 41, 2275–2279. [Google Scholar] [CrossRef]
- Schenk, A.; Esser, T.; Knoop, A.; Thevis, M.; Herden, J.; Heidenreich, A.; Bloch, W.; Joisten, N.; Zimmer, P. Effect of a Single Bout of Aerobic Exercise on Kynurenine Pathway Metabolites and Inflammatory Markers in Prostate Cancer Patients-A Pilot Randomized Controlled Trial. Metabolites 2020, 11, 4. [Google Scholar] [CrossRef]
- Loeser, H.; Kraemer, M.; Gebauer, F.; Bruns, C.; Schröder, W.; Zander, T.; Alakus, H.; Hoelscher, A.; Buettner, R.; Lohneis, P.; et al. Indoleamine 2,3-Dioxygenase (IDO) Expression Is an Independent Prognostic Marker in Esophageal Adenocarcinoma. J. Immunol. Res. 2020, 2020, 2862647. [Google Scholar] [CrossRef] [PubMed]
- Balakrishna, P.; George, S.; Hatoum, H.; Mukherjee, S. Serotonin Pathway in Cancer. Int. J. Mol. Sci. 2021, 22, 1268. [Google Scholar] [CrossRef] [PubMed]
- Sarrouilhe, D.; Mesnil, M. Serotonin and human cancer: A critical view. Biochimie 2019, 161, 46–50. [Google Scholar] [CrossRef]
- Zamani, A.; Qu, Z. Serotonin activates angiogenic phosphorylation signaling in human endothelial cells. FEBS Lett. 2012, 586, 2360–2365. [Google Scholar] [CrossRef]
- Peters, M.A.; Walenkamp, A.M.; Kema, I.P.; Meijer, C.; de Vries, E.G.; Oosting, S.F. Dopamine and serotonin regulate tumor behavior by affecting angiogenesis. Drug Resist. Updates 2014, 17, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Herr, N.; Bode, C.; Duerschmied, D. The Effects of Serotonin in Immune Cells. Front. Cardiovasc. Med. 2017, 4, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iken, K.; Chheng, S.; Fargin, A.; Goulet, A.C.; Kouassi, E. Serotonin upregulates mitogen-stimulated B lymphocyte proliferation through 5-HT1A receptors. Cell. Immunol. 1995, 163, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwiatkowska, I.; Hermanowicz, J.M.; Przybyszewska-Podstawka, A.; Pawlak, D. Not Only Immune Escape—The Confusing Role of the TRP Metabolic Pathway in Carcinogenesis. Cancers 2021, 13, 2667. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112667
Kwiatkowska I, Hermanowicz JM, Przybyszewska-Podstawka A, Pawlak D. Not Only Immune Escape—The Confusing Role of the TRP Metabolic Pathway in Carcinogenesis. Cancers. 2021; 13(11):2667. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112667
Chicago/Turabian StyleKwiatkowska, Iwona, Justyna Magdalena Hermanowicz, Alicja Przybyszewska-Podstawka, and Dariusz Pawlak. 2021. "Not Only Immune Escape—The Confusing Role of the TRP Metabolic Pathway in Carcinogenesis" Cancers 13, no. 11: 2667. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112667