
Tribbles Pseudokinases in Colorectal Cancer
by
 and
and
Bibiana I. Ferreira
1,2,3, 
Bruno Santos
1,2,4, 
Wolfgang Link
5,* and
Ana Luísa De Sousa-Coelho
1,2,6,* 1
Centre for Biomedical Research (CBMR), Campus of Gambelas, Universidade do Algarve, 8005-139 Faro, Portugal
2
Algarve Biomedical Center (ABC), Campus de Gambelas, Universidade do Algarve, 8005-139 Faro, Portugal
3
Faculdade de Medicina e Ciências Biomédicas (FMCB), Campus de Gambelas, Universidade do Algarve, 8005-139 Faro, Portugal
4
Serviço de Anatomia Patológica, Centro Hospital Universitário do Algarve (CHUA), 8000-386 Faro, Portugal
5
Instituto de Investigaciones Biomédicas “Alberto Sols” (CSIC-UAM), Arturo Duperier 4, 28029 Madrid, Spain
6
Escola Superior de Saúde (ESS), Campus de Gambelas, Universidade do Algarve, 8005-139 Faro, Portugal
*
Authors to whom correspondence should be addressed.
Cancers 2021, 13(11), 2825; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112825
Submission received: 9 May 2021
/
Revised: 29 May 2021
/
Accepted: 31 May 2021
/
Published: 5 June 2021
(This article belongs to the Special Issue Pseudokinases, Tribbles Proteins and Cancer)
Abstract
:Simple Summary
The Tribbles family of pseudokinases controls a wide number of processes during cancer on-set and progression. However, the exact contribution of each of the three family members is still to be defined. Their functions appear to be context-dependent as they can act as oncogenes or tumor suppressor genes. They act as scaffolds modulating the activity of several signaling pathways involved in different cellular processes. In this review, we discuss the state-of-knowledge for TRIB1, TRIB2 and TRIB3 in the development and progression of colorectal cancer. We take a perspective look at the role of Tribbles proteins as potential biomarkers and therapeutic targets.
Abstract
The Tribbles family of pseudokinases controls a wide number of processes during cancer on-set and progression. However, the exact contribution of each of the three family members is still to be defined. Their function appears to be context-dependent as they can act as oncogenes or tumor suppressor genes. They act as scaffolds modulating the activity of several signaling pathways involved in different cellular processes. In this review, we discuss the state-of-knowledge for TRIB1, TRIB2 and TRIB3 in the development and progression of colorectal cancer. We take a perspective look at the role of Tribbles proteins as potential biomarkers and therapeutic targets. Specifically, we chronologically systematized all available articles since 2003 until 2020, for which Tribbles were associated with colorectal cancer human samples or cell lines. Herein, we discuss: (1) Tribbles amplification and overexpression; (2) the clinical significance of Tribbles overexpression; (3) upstream Tribbles gene and protein expression regulation; (4) Tribbles pharmacological modulation; (5) genetic modulation of Tribbles; and (6) downstream mechanisms regulated by Tribbles; establishing a comprehensive timeline, essential to better consolidate the current knowledge of Tribbles’ role in colorectal cancer.
1. Introduction
Colorectal cancer (CRC) refers to a malignant tumor that arises from the colon or rectum epithelium. They are often referred together due to their many features in common. Worldwide, CRC was the third most common malignancy (1,931,590 cases; 10%) and the second most frequent cause of cancer deaths (935,173 cases; 9.4%), in both genders and all ages, in 2020 [1]. The fact that mortality is approximately half the incidence suggests a relatively good prognosis, which is closely related to improvements in cancer treatment and management. Since survival is highly dependent on the stage of cancer at diagnosis, early detection stands out as one of the best prognosis factors. The majority of cases of this highly heterogeneous disease appear between 50 and 75 years old, being more frequent in men than in women [2]. Risk factors are still unclear, but evidence supports the association of poor diet, lack of physical activity, smoking and an increasing prevalence of overweight, obesity, and type 2 diabetes with CRC [2].
Sporadic cases are the majority of CRC, while germline mutations and hereditary syndromes account for only 6 to 10% of all cases [3]. Two molecular pathways are traditionally associated with sporadic CRC: the adenoma–carcinoma, or chromosomal instability, pathway (70–75%), and the serrated pathway (25–30%) [2]. In the adenoma–carcinoma pathway, CRC evolves from non-malignant precursor lesions called adenomas, for a period of at least 10 years [4]. This process begins with the activation of Wnt signaling due to the loss of the tumor suppressor APC (Adenomatous polyposis coli) through inactivating mutations. Over the years, a series of mutations start to accumulate. KRAS oncogene mutation arises preferentially in early steps, while TP53, SMAD4, PIK3C, and PTEN mutations occur at later phases [5]. For the serrated pathway, the earliest event on sessile serrated adenomas is thought to be the activation of the MAPK pathway through BRAF oncogene mutation leading to a global methylation of CpG islands and consequently silencing of the mismatch repair gene MLH1 or the cyclin-dependent kinase inhibitor p16. In traditional serrated adenomas that comprise less than 1% of all serrated lesions, MAPK pathway activation is more frequently associated with mutation of the KRAS oncogene, although BRAF mutation may also occur [6,7].
Although driver mutations are recognized, over time several of these pathways may overlap or interact. Upon Wnt activation, β-catenin accumulates and translocates into the nucleus where it binds TCF4 to promote the transcription of Jun, c-Myc or CyclinD-1, promoting tumor progression. On the other hand, APC mutation promotes Wnt signal transduction by stabilization of β-catenin [8,9,10]. Overexpression of amplified genes like MYC and MET can also occur through double minute (DM) chromosomes (round-circle, acentric double-strand extra-chromosome DNA that usually exist in pairs). The formation of DM chromosomes is usually regarded as an important sign of genome instability and occurs in 2.6% of CRC [11,12].
As increased EGFR expression has been observed during colorectal carcinogenesis, targeted therapies with monoclonal antibodies such as cetuximab and panitumumab which prevent EGFR activation are commonly used in late-stage CRC. However, mutations in KRAS, NRAS, and BRAF, and amplification of ERBB2 and MET, represent mechanisms of primary and secondary resistance to anti-EGFR therapy. In fact, only patients with wild-type RAS benefit from anti-EGFR therapy. Nonetheless, RAS-BRAF wild type CRC is also associated with resistance to anti-EGFR treatment in the presence of high c-MYC levels [13,14,15,16]. Targeting specific signaling pathways using multiple agents is the best option to achieve a better therapeutic outcome. Clinical trials have shown promising results with a combination of bevacizumab, a monoclonal antibody that inhibits VEGF, and the EGFR inhibitor erlotinib, that blocks these two important pathways in cancer growth [17].
Molecular characterization of CRC can guide therapeutic decisions or define patient prognosis. In fact, a classification based on four consensus molecular subtypes (CMSs) was established: (1) CMS1, associated with hypermutations, microsatellite instability and strong immune activation; (2) CMS2, the canonical subtype, characterized by an epithelial phenotype and increased WNT and MYC activation; (3) CMS3, characterized by metabolic dysregulation; (4) CMS4, which shows prominent transforming growth factor–β (TGFβ) activation, stromal invasion, and increasing angiogenesis [18]. Nonetheless, TNM staging remains the standard for clinical decision-making. In stage I and II, the disease is located in the colon or rectum, in stage III there is ganglionic spread and in stage IV there is already the presence of distant metastasis usually liver and lung [19]. Surgery is the optimal primary treatment for CRC and should be preceded by neoadjuvant radiotherapy. Chemotherapy can also be used in combination with radiotherapy for neoadjuvant treatment. Recurrence and metastatic patterns differ between colon and rectal cancers. Nevertheless, patient’s survival with metastatic CRC varies from 5 to 19 months, depending on disease stage. There is a clear need for new biomarkers predicting CRC behavior and innovative therapeutic targets [20,21].
Tribbles pseudokinase family members (TRIB1 (C8FW or SKIP1), TRIB2 (C5FW) and TRIB3 (NIPK, SKIP3 or LKW)) might be biomarkers of interest in colorectal cancer. TRIB1 lacks, while TRIB2 and TRIB3 have low, affinity for ATP, along with residual phosphotransferase capacity [22]. Instead of phosphorylating target proteins, they function as protein scaffolds that modulate diverse signaling cascades involved in proliferation, survival, differentiation and drug resistance [23]. Tribbles can lead to protein degradation by binding to ubiquitin ligases promoting the ubiquitylation of substrates [24], stabilization by suppressing sumoylation [25], or affecting signal transduction pathways through binding of signaling molecules like MEK and AKT [26]. For example, TRIB2 has been previously identified as an oncoprotein that contributes to the pathogenesis of acute myelogenous leukemia (AML) through the inhibition of C/EBPα function, similarly to slbo inactivation by Drosophila Tribbles [27]. Different reports show that Tribbles proteins exert a role in the development of different types of tumors. However, they might exhibit an oncogenic or tumor suppressive behavior depending on the family member and cellular context [28]. For instance, TRIB2 promoted AKT activation and, consequently, the inactivation of the transcription factor FOXO in melanoma. Expression of FOXO target genes, which can induce apoptosis, are therefore attenuated by TRIB2 overexpression [29,30]. By contrast, TRIB3 was shown to have the opposite effect [31], and its loss was associated with a more aggressive tumor phenotype [32]. Increased levels of TRIB3 inhibited AKT activity and consequently induced cell death via FOXO in glioma cells [33]. These data support a scenario in which the balance between the expression of Tribbles might determine tumor progression, in this particular case through FOXO proteins activity via AKT. Several studies have been published reporting the role of Tribbles proteins in different types of cancer, including TRIB1 in prostate [34], glioma [35], ovarian [36], and thyroid [37] cancers; TRIB2 in melanoma [38], lung [39], liver [40,41,42], acute leukemias [27,43,44], and glioblastoma [45]; and TRIB3 in lung [46], breast [47,48], renal [49], gastric [50], liver [51], retinoblastoma [52], glioblastoma [53], and ovarian [54] cancers; as well as all Tribbles members in colorectal cancer, as herein fully presented. Tribbles pseudokinases may have an immediate impact in cancer treatment being potential therapeutic targets.
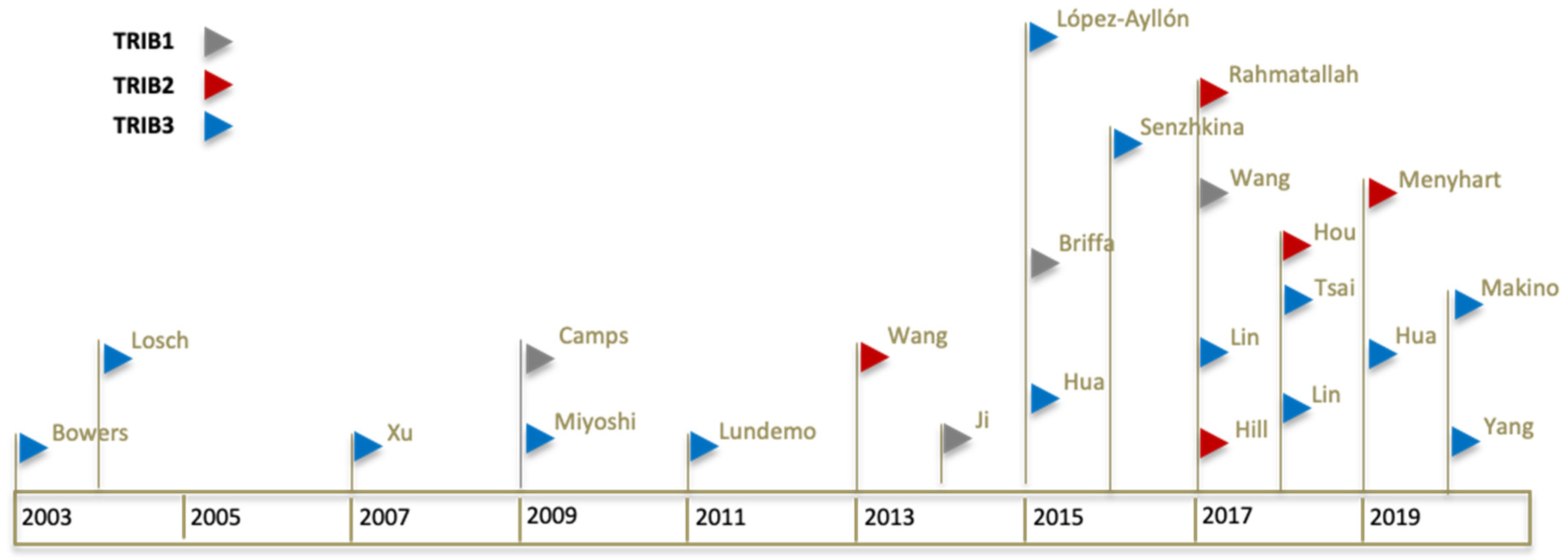
With this review, we established a comprehensive timeline describing what is known about the putative role of each Tribble family member in the development of colorectal cancer (Figure 1). This document should also serve as a guide for future research, describing the available bioinformatic datasets regarding colorectal cancer studies.
2. Tribbles Amplification and Overexpression in Colon Cancer Tissues
The identification of increased genomic copy number (genomic amplification) in chromosome 8, combined with altered gene expression (up-regulation), allowed the first identification of TRIB1 as a putative biologically relevant oncogene, in primary colorectal tumor or established colorectal cancer (CRC) cells lines containing the amplicon [55]. Half a decade later, a study published in 2014, identified TRIB1 as a double minute (DM)-carried gene in a human CRC cell line [56,57]. DMs are small-paired, autonomously replicating, extrachromosomal DNA segments that contain amplified oncogenes, mainly present in solid tumors and identified in many human cancer cell lines [58]. Indeed, along with MYC and FGFR2 (among others), TRIB1 was amplified in NCI-H716 cells compared to normal human peripheral blood DNA. Though the authors also showed elevated TRIB1 mRNA levels in CRC cells, it was compared to non-paired normal colon tissue [56]. In a different study, TRIB1 region was amplified in 7 out of the 15 CRC cell lines tested. TRIB1 was clearly amplified and very highly expressed in NCI-H716 cells. However, there was no correlation between DNA copy number of the TRIB1 region (Chr8: 126,393,571–126,567,050) and TRIB1 protein. The authors did find a weak correlation between TRIB1 copy number and mRNA expression [59]. They also found that TRIB1 copy number was gained in 11% of primary CRC samples (n = 881 patients’ cohort, Oncomine database). From a different cohort, 11 tumors (14.4% out of 76 cases) were TRIB1-amplified; this value was above MYC amplification, for which only 6 tumors were amplified (7.4%) [59]. It was not until 2017 that the TRIB1 gene was shown to be amplified and overexpressed in CRC tissues, when compared with paired surrounding non-tumor tissues from the same patients [60]. By using the TCGA database, the authors described that the copy number of TRIB1 in human CRC tissues (n = 212) was significantly increased, when compared with normal colon tissues (n = 79). Data available from the Gene Expression Omnibus (GEO) [61], revealed that TRIB1 gene expression levels in CRC tissues were elevated, compared with normal colon tissues (microarray expression studies from Oncomine) [60]. The authors experimentally validated the data by western blotting and found that the levels of TRIB1 protein were elevated in 6 out of 8 (75%) CRC tumors when compared to matched adjacent non-tumor tissue. In addition, TRIB1 protein overexpression was detected in 52 out of 75 (69.3%) of CRC cases as assessed by immunohistochemistry (IHC) compared with matched normal tissue [60].
The first hint that TRIB2 expression was related to colon cancer was reported in 2013, when TRIB2 protein was detected in CRC samples using tissue microarray analysis (TMA) by IHC [41]. Later in 2017, TRIB2 mRNA and protein expression were found to be significantly increased in primary colon cancer tissue samples when compared to matched normal tissue samples [30]. However, none of these studies specifically focused on TRIB2 in colon cancer. One year later, it was further confirmed that TRIB2 was overexpressed in colorectal cancer patients, in primary tumor samples obtained from surgical resection, prior to receiving any kind of chemotherapy or radiotherapy [62]. The authors included an initial GEO database analysis, and found that TRIB2 was highly expressed in primary tumor tissues compared with adjacent normal tissues [62]. These data have been experimentally validated by RT-PCR, showing that TRIB2 gene expression was higher in a great proportion of tumors (73.3%), when compared to adjacent tissues. Accordingly, IHC staining showed that the average expression level of TRIB2 was elevated in CRC tissues compared with normal tissues. Further paired analysis also demonstrated that TRIB2 protein level was higher in tumors (76.7%) [62]. All these findings suggest that TRIB2 is highly expressed in a large subset of CRC patients.
Though initially stated as “data not shown”, the very first input on TRIB3 gene overexpression in colon tumor samples, when compared to matched normal colon samples, was published in 2003 [63], much earlier than the other two Tribbles family members (Figure 1). The authors had identified SKIP3, a protein with very high homology to other previously reported mammalian orthologs of Drosophila tribbles [63], later named TRIB3. While in the normal tissues tested (including colon), TRIB3 mRNA levels were preferably elevated in liver, the highest expression was observed in tumor-derived cell lines, which included the colorectal adenocarcinoma SW480 cells [63]. TRIB3 transcript levels were increased in colon adenocarcinoma samples, when compared to normal human tissues. Its tumor-specific localization was further confirmed by in situ hybridization [63]. In human tumor xenografts with HT-29 colorectal adenocarcinoma cells, TRIB3 expression pattern was proximal to a region of cell death, localized to a periapoptotic region. Nonetheless, its localization did not correlate with cell proliferation nor with cells undertaking apoptosis [63]. The following year, another article showed elevated gene expression of TRIB3 in different tumor cell lines [64]. In agreement with the previous report [63], one of the strongest signals was observed in the colorectal adenocarcinoma cell line SW480 [64]. Although not shown, the authors described a significant higher TRIB3 gene expression in colon-carcinoma compared to the corresponding normal tissues. Moreover, they also observed up to 400-fold overexpression of TRIB3 mRNA in human tumor tissues from primary tumors with different TNM stages versus normal colonic mucosa [64]. In 2007, a study on the TRIB3 putative role in tumorigenesis, included the analysis of TRIB3 mRNA expression level in different carcinomas [65]. TRIB3 was highly expressed in colon tumor tissues, moderately in lung and esophageal, very limited in stomach, but not increased in liver or kidney cancers [65]. Interestingly, the authors discovered that TRIB3 was overexpressed in all the colon cancer tissue pairs analyzed [65]. Consolidating the results described in previous studies for SW480 cells [63,64], it was shown that all other gastrointestinal cell lines tested (Caco2, DLD-1, LoVo, HCT116, HT-29, and KM12SM) expressed TRIB3 gene [66]. Moreover, almost 90% of primary CRC samples analyzed had higher levels of TRIB3 mRNA in tumors, compared to the paired normal regions [66]. TRIB3 protein localized both in the nucleus and the cytoplasm of epithelial cells, while very weakly detectable in stromal cells [66]. The expression of TRIB3 protein was positively correlated with its mRNA expression, though not all of the analyzed cases showed higher TRIB3 levels in tumor regions than in normal regions [66]. Consistently with previous results herein described for colon, but not concerning liver [65], an additional report showed high levels of TRIB3 in both colon cancer, hepatocellular carcinoma (HCC), and lung cancer tumor tissues, when compared with adjacent non-tumor tissues [67]. In agreement with the early studies, where TRIB3 transcript levels were evaluated, high TRIB3 protein levels were also found in SW480 cells, and those were associated with TRIB3 mRNA expression [68]. Intriguingly, it was found that two TRIB3 spliced isoforms were overexpressed in colon tumors [69]. The alternatively spliced transcripts uc002wdm/NM_021158 and uc002wdn/AK297546 showed an approximately 10- and 4-fold average increase, respectively, in colorectal cancer specimens, compared to matched morphologically normal tissues [69].
According to these data, it is unquestionable that all members of the Tribbles family are overrepresented in colon cancer and representative colorectal cancer cell lines (Table 1), which might represent useful biomarkers for disease detection. However, it remains to be investigated whether there is a correlation between TRIB1, TRIB2 and TRIB3 expression in these samples. To the best of our knowledge, there are no studies where the authors simultaneously analyzed two or more Tribbles in the same samples.
3. Clinical Significance of Tribbles Overexpression in Colon Cancer
To better understand the clinical impact of Tribbles pseudokinases overexpression in patients with colon cancer, several clinical parameters and clinicopathological features were analyzed and correlated to each member of the Tribbles family level in different samples. With regards to TRIB1 and survival, one initial study did not find statistical differences in survival between patients with or without TRIB1 (nor with MYC) gene amplifications [59]. However, a different study found that high (versus low) TRIB1 gene expression correlated with poor survival in CRC patients [60]. Moreover, TRIB1 protein overexpression was positively associated with distant metastasis and advanced staging [60].
For both melanoma and colon cancer patients, increased levels of TRIB2 gene expression correlated with a significantly worse clinical outcome, which was not observed for pancreatic tumor patients [30]. In agreement, TRIB2 levels in colorectal cancer were inversely correlated with survival rate of CRC patients, and positively correlated with tumor grade [62]. Patients with high expression of TRIB2 experienced not only a worse overall survival, as more recurrence [62].
A recent report analyzed the top ten most frequently mutated genes, which mutations result in a disruptive protein structure [70]. From these, the authors identified the most significantly upregulated genes which correlated with the worst survival outcome. Finally, from these, they selected those genes which were theoretically druggable targets [70]. Using this elegant strategy, it was shown that the mRNA levels of TRIB2 (as well as of DUSP4, dual-specificity MAPK phosphatase (4) were significantly higher in ACVR2A mutant colon cancers compared to wild-type cases [70]. ACVR2A, which encodes activin receptor type IIA, is a component of the TGFβ signaling pathway, shown to function as a tumor suppressor gene in human CRC-derived organoids [71]. Though ACVR2A mutations, reported in several cases of CRC [72], have been previously linked to earlier tumor stages (stages I/II) [73], TRIB2 correlation to tumor staging was not performed within this study [70]. In an independent clinical cohort, high TRIB2 expression was associated with worse recurrence-free survival (RFS), and also linked to mutations in ACVR2A gene [70].
In colonic premalignant polyps, TRIB2 expression was associated with an increased risk for progression to colon cancer [74]. Along with phosphoinositide-3-kinase regulatory subunit 3 (PIK3R3) and poly(ADP-ribose) polymerase-14 (PARP14) (among others), TRIB2 was up-regulated in sessile serrated polyps (SSA/Ps), while expressed at a lower level in both the right colon and hyperplastic polyps (HPs) samples [74]. Improved disease-free survival (DFS), though not OS, were, however, previously described in colorectal patients who expressed higher levels of PIK3R3 [75]. PARP14, by contrast, has been proposed as a novel drug target for different types of cancer, though colon cancer not yet included [76]. Despite sharing histological similarities with the typical hyperplastic polyps (HPs) [77], the SSA/Ps lesions are more prone to develop into cancer [78], which makes this finding clinically relevant and proposes TRIB2 as a useful biomarker to early distinguish colonic lesions [74].
TRIB3 expression was highest at Duke’s stages B > A (which represents stages with no or infrequent dissemination) > C > D (representing more invasive and metastatic stages), suggesting an inverse correlation between invasion and progression and TRIB3 expression. Nevertheless, although the lowest level was observed in stage D, it was still much higher than in normal colorectal epithelium [64]. Such as for TRIB1 [60], metastasis (M0/M1) was correlated with TRIB3 expression [66]. However, metastatic sites, tumor size, invasion, lymph node metastasis or lymphatic and venous invasion, were not significantly correlated with TRIB3 levels [66]. Higher expression of TRIB3 was inversely correlated with overall survival rate in two independent studies [66,67]. Unquestionable, high TRIB3 expression levels correlated with low survival rates and poor outcomes of patients with CRC [79].
Recently, two independent predictive models included TRIB3 expression levels [80,81]. The analysis of the association between differentially expressed genes and overall survival in a set comprised with 347 colon (but not rectal) cancer patients with a follow-up time greater then 90 days, from TCGA dataset, identified TRIB3 as a high-risk RNA, along with BDNF, KLF4, SESN2 and SMOC1 [80]. Based on the significant correlation of the 5 genes expression with tumor status and tumor stage, the authors developed a predictive multi-mRNA-based model for overall survival, assigning risk scores to each patient and obtaining a prognosis tool, aiming treatment optimization for patients [80]. These results reinforced the usefulness of TRIB3, though combined with other genes, as a potential prognostic biomarker for colon cancer. On the other hand, TRIB3 was associated, together with other immune-related genes such as CHGA, LGALS4, LEP, NOX4, IL17A, HSPD1, and CASP7, with colon cancer prognosis [81]. Based on the gene expression profiles, the authors developed a molecular classifier tool. It was validated in 277 samples from TCGA dataset that had available both gene expression and survival status and time, and in an independent validation dataset (213 colon cancer samples from GEO). Alongside the analysis of the tumor-infiltrating immune cells in colon cancer, a novel predictive tool for colon cancer prognosis, which included TRIB3 levels, was established [81].
These results suggest a significant link between Tribbles expression and patient prognosis (Table 2). Specifically, TRIB1 and TRIB3 might be considered candidates as predictive markers of tumor metastasis development. Additionally, there is abundant data confirming the role of TRIB2 in drug resistance functioning as a predictive biomarker for drug response. This is of utmost importance for the clinicians at the time of deciding the course of treatments.
4. Tribbles Gene and Protein Expression Regulation in Colon Cancer
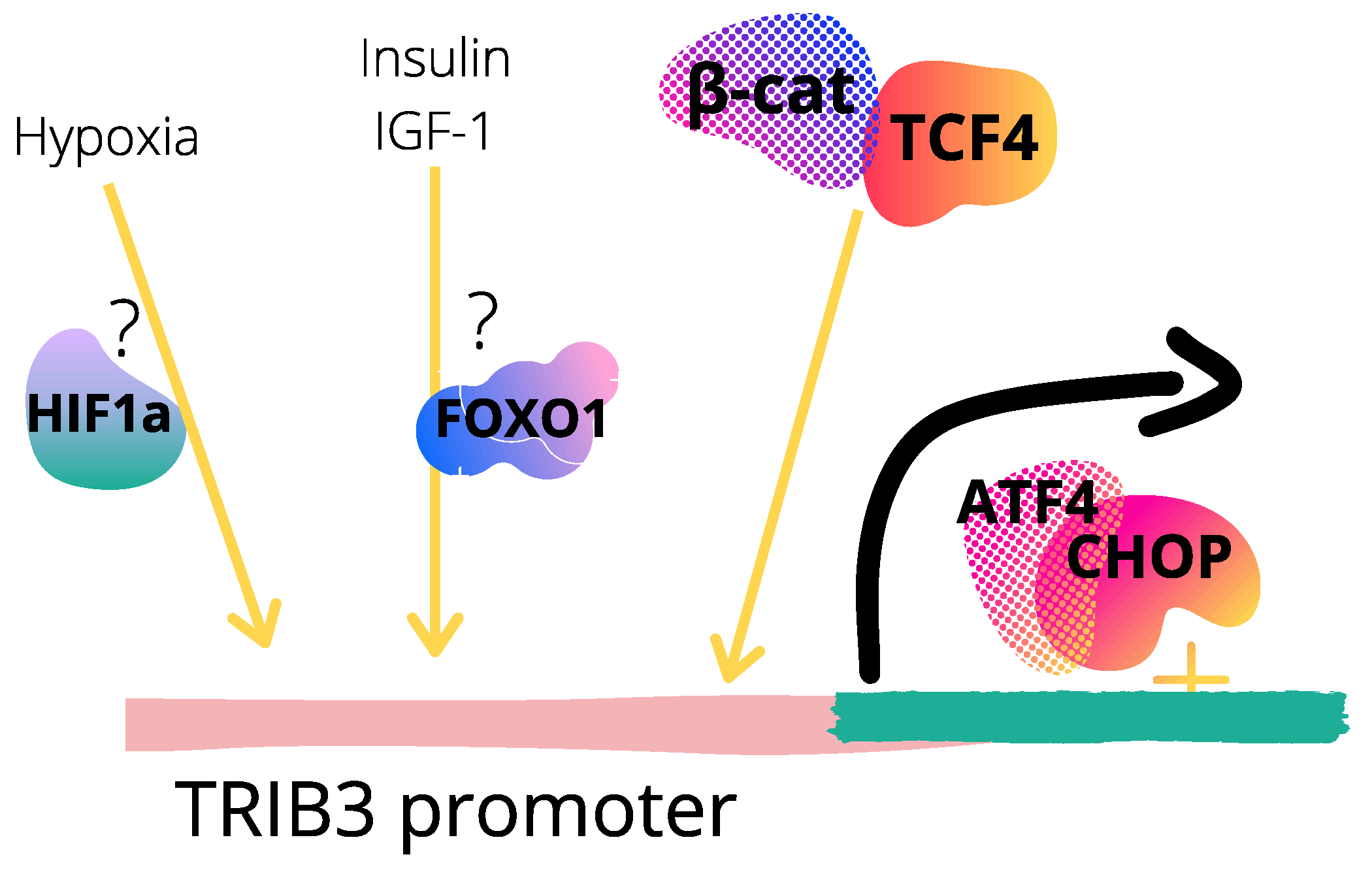
Recognizing how Tribbles are regulated in colon cancer is indispensable to better understand the physiopathology of this type of cancer and Tribbles involvement in the disease development and progression. While for TRIB1, gene amplification might partially support its increased levels not only in colon, but also in other types of cancer [82,83], the mechanism behind TRIB2 overexpression in colon cancer patients is still unclear. In other tumor types, however, different mechanisms for TRIB2 regulation have been described [41,84]. For instance, while Wnt signaling regulated TRIB2 in hepatic cancer HepG2 cells, it had no effect in the LS174T CRC cell line [41]. This might reflect that different transcriptional programs are being differentially activated in different cancer types. By contrast, TRIB3 expression was induced in CRC cells after β-catenin activation by Wnt3a, in a dose- and time-dependent manner. Conversely, decreased TRIB3 expression was observed in response to β-catenin depletion [79]. The authors recognized TRIB3 as a transcriptional target of the β-catenin–TCF4 complex and identified a TCF4-binding site (CACAGCTGCG motif) at the C-terminal domain of the TRIB3 promoter region [79] (Figure 2). Moreover, β-catenin inhibited TRIB3 degradation, contributing to the overall increased levels of TRIB3 in CRCs by inducing its protein stability [79]. Additional mechanisms might be involved in TRIB3 regulation in CRC. In HT-29 colorectal adenocarcinoma cells subjected to hypoxia, TRIB3 was upregulated at the transcript and protein level [63], suggesting TRIB3 could be regulated in CRC by classical hypoxia-related transcription factors, such as HIF1α (Figure 2). More recently, it was shown that TRIB3 was up-regulated concomitantly to the induction of C/EBP-homologous protein (CHOP) in HCT116 cells, while down-regulated following CHOP knockdown [85,86], revealing TRIB3 being downstream of CHOP-activated pathways (Figure 2), as previously proposed [87,88]. In addition, insulin and IGF-1 induced TRIB3 protein expression in human colon cancer HCT-8 cells (Figure 2), as well as in hepatoma HepG2 and lung cancer A549 cell lines [67]. Interestingly, other C/EBP proteins have been linked to the oncogenic role of Tribbles proteins in myeloid cancers [89].
5. Tribbles Pharmacological Modulation in Colon Cancer
Genes that are over-expressed in cancer are more likely to be putative pharmacological targets [90]. Tribbles proteins have emerged as interesting novel therapeutic targets, with their unique pseudokinase domain providing a potential opportunity for drug design approaches [91,92]. To the best of our knowledge, there is lack of studies in colon cancer patients that identified Tribbles differential expression in response to approved cancer treatments.
Recent work demonstrated that afatinib, an approved irreversible electrophilic covalent EGFR/HER2 inhibitor for lung cancer treatment, increased TRIB2 degradation in human acute myeloid leukemia (AML) cancer cells [93], demonstrating for the first time that TRIB2 might be a druggable protein. Early this year, others showed similar results in human hepatoma cell lines, where afatinib treatment (10 µM, 20 h) reduced TRIB2 protein levels and decreased cellular viability over 50% [94]. In SW-48, Colo205 and HT-29 CRC cell lines, though Tribbles levels were not investigated, afatinib treatment (1 or 10 µM, 48 h) also reduced cell viability [95], representing an opportunity for further studies, in order to establish TRIB2 targeting as a potential strategy for treating colon cancer.
Regarding TRIB3, for which more studies have been performed, potential TRIB3 druggability has been suggested, although not yet fully validated in the CRC setting. A study that analyzed different cell lines from the NCI60 database (using CellMiner), representing cancer types that typically receive treatment with erlotinib, such as breast cancer, colon cancer and NSCLC, identified TRIB3 at the top three genes that showed the strongest correlation with EGFR inhibition [96]. The average expression of the seven genes identified (LCN2, MET, MMP7, PTPRZ1, TRIB3, UGT1A6 and COL17A1 was lower in more sensitive cells [96]. These results suggest that TRIB3 might be a useful clinical predictive biomarker for erlotinib action in the absence of EGFR-activating mutations for colon cancer patients. Conversely, high expression of TRIB3 could be related to CRC resistance to anti-EGFR therapies, and future research should clarify this potential relationship. Interestingly, it was recently shown that increased levels of TRIB3 were associated with elevated EGFR stability and signaling activity in lung cancer, suggesting that the disruption of the TRIB3-EGFR interaction could be a novel therapeutic target [97]. Nevertheless, whether TRIB3 might be in fact a downstream erlotinib therapeutic target still remains to be elucidated.
Another study showed that transcript and protein levels of TRIB3 were upregulated in SW620 cells treated with the bioactive chemical modified fatty acid (FA) analog saturated 3-thia FA tetradecylthioacetic acid (TTA), compared to non-treated cells [98]. TTA not only inhibited the growth of colon cancer cells, but also induced the expression of several other genes involved in endoplasmic reticulum (ER) stress and unfolded protein response (UPR), such as CHOP and C/EBPβ, whereas Cyclin D1 was down-regulated [98]. After treating HCT116 cells with the photosensitizer hypericin and photodynamic therapy (HY-PDT) [99], at the higher concentration range, TRIB3 protein levels were induced, along with induction of CHOP, activation of the autophagy signal and cell death [85]. Nevertheless, TRIB3 up-regulation was CHOP-dependent, reflecting a most likely indirect action of HY-PDT in TRIB3 modulation [85]. Importantly, TRIB3 has been previously located downstream of CHOP-mediated pathways [87]. Similarly, after treatment of HCT116 cells with a specific extract from Antrodia cinnamomea, a native Taiwanese rare mushroom, TRIB3 transcript levels were induced in a dose-dependent manner [86]. Though CHOP itself was also up-regulated, none of the other downstream genes of the apoptosis and autophagy CHOP-mediated pathways were affected. Interestingly, this extract (ACF2) inhibited cellular viability in different human colorectal cancer cell lines such as HCT116, HT29, SW480, Caco-2 and Colo205 [86]. Though the authors validated that ACF2 inhibited the growth of CRC cells subcutaneously inoculated into athymic nude mice, the levels of TRIB3 were not evaluated in the tumors [86]. In both primary CRCs and HCT-8 ileocecal adenocarcinoma cells, a fusion peptide named P2-T3A6, which inhibited cell viability and migration, demonstrated having binding affinity with TRIB3. As a result, TRIB3 protein degradation was accelerated, together with inhibition of TRIB3 gene transcription [79]. Interestingly, P2-T3A6 peptide caused disruption of the β-catenin–TRIB3 interaction, though it did not affect other TRIB3 known interactions, such as with SMAD3 [100], p62 [67] or AKT [31], which might reflect an indirect peptide action on TRIB3 regulation by β-catenin [79], as previously described above. Though there is few evidence regarding pharmacological modulation of TRIB3 in CRC, all cases most likely represent an indirect effect through β-catenin or CHOP pathways (Figure 3).
6. Genetic Modulation of Tribbles in Colon Cancer
It is challenging to distinguish between driver and passenger mutations. Reports on genetic alterations of Tribbles genes in human cancer are rare. A recent study described a novel fusion transcript derived from a chromosomal translocation between the TRIB2 and the PRKCE genes, in pulmonary carcinoid tumors [101]. Nevertheless, to the best of our knowledge, no Tribbles mutations have been associated with CRC so far.
The first report on the effect of TRIB1 downregulation in colorectal cancer cells was published in 2014, and showed that interfering with TRIB1 (cells stably transfected with shRNA vectors, specifically knocking down TRIB1 gene expression) led to the decrease of the number of double-minute chromosomes (DMs) formed, along with genomic instability and cytotoxic DNA damage in the NCI-H716 tumor cells [56]. Although not specifically tested for TRIB1, the authors showed that downregulation of other DM-carried oncogenes amplified along the same chromosome, such as MYC and FGFR2, led to impairments of cellular proliferation and invasion, suggesting a parallelism with TRIB1 [56]. TRIB1 stable or transient overexpression in SW480 and LoVo cells, respectively, showed enhanced migratory and invasion ability of the cells, compared with controls [60]. Moreover, SW480-TRIB1 cells exhibited increased metastasis capacity, evaluated in vitro by cell adhesion to extracellular matrix (ECM) assays [60]. Conversely, silencing TRIB1 expression by shRNA against TRIB1, in both SW480-TRIB1 and COLO320HSR cells, reduced the level of cell migration and invasion [60], establishing a role for TRIB1 in colon cancer cells motility.
Similar to the findings previously described for TRIB1, siRNA down-regulation of TRIB2 in SW48 and LoVo CRC cells suppressed proliferation, induced cell cycle arrest (increased G0/G1-phase ratios along with reduced the S-phase ratios) and promoted cellular senescence [62]. Although cell growth was affected, TRIB2 knock-down did not induce apoptosis. The role of TRIB2 in the cancer progression phenotype was further confirmed in these cells, since both proliferation and cell cycle progression were accelerated by TRIB2 overexpression, along with decreased rates of cellular senescence [62].
In 5 different CRC cells (DLD-1, LoVo, HCT116, KM12SM and SW480), TRIB3 knock-down was obtained by siRNA [66]. For all cell lines, silencing of TRIB3 correlated with decreased cellular proliferation. A very robust effect was observed at day 4 after plating, mainly in DLD-1, LoVo, and HCT116 TRIB3-depleted cells [66]. The first in vivo experiment regarding Tribbles cellular genetic manipulation was reported in 2015, where the authors aimed to evaluate the role of TRIB3 in CRC tumorigenesis [67]. Genetically diabetic KK-Ay mouse, a model of human T2D, showed not only higher TRIB3 expression in the liver and lungs, but also the xenografted tumors from the diabetic model displayed elevated TRIB3 expression, when compared with C57BL/6 mice [67]. After being inoculated in BALB/c nude mice, TRIB3 knock-down in HCT-8 CRC cells led to decreased metastasis and growth, revealing an antitumor role of TRIB3 silencing. A coincident TRIB3-dependent phenotype was observed for human hepatoma HepG2 cells [67]. These results might be particularly important for T2D patients, who are at greater risk to develop liver or colorectal cancers [102]. The first in vivo modulation of TRIB3 was reported in 2019. Few years later from previous report, the same research group down-regulated TRIB3 expression in C57BL/6J-ApcMin/J mice fed a high-fat diet (Trib3-KD), aiming to specifically explore the role of TRIB3 in intestinal tumorigenesis [79]. Despite no differences in weight changes between groups, Trib3-KD mice revealed higher survival rate and no colon or small intestinal tumors at the 10 weeks of age time point. By contrast, 75% and 50% of control mice developed tumors within their colons and small intestines, respectively [79]. Interestingly, mice overexpressing TRIB3 (TRIB3-OE) showed a slower increase in the high-fat diet-induced body weight gain, despite the shorter colon length. As anticipated, TRIB3-OE mice showed a heavier tumor burden, reflected by a decrease in respective survival rates [79].
In colorectal cancer, the epithelial to mesenchymal transition (EMT) is associated with a more invasive or metastatic phenotype, as cells show increased motility (mesenchymal properties), while losing their epithelial characteristics [103]. A very recent report showed that down-regulating TRIB3 in different colon carcinoma cell lines, such as SW480, HCT116, CaCO2, SW48 and SKCO1, led to a mesenchymal-epithelial transition (MET) phenotype [104].
It is evident that there is still much more to be learned about the role of these proteins in the process of cancer progression. What we do know so far, is that the majority of the reports pinpoint Tribbles as oncogenes in several cancer types, including colon cancer (Table 3). This suggests that modulation of Tribbles protein levels might have a positive impact delaying the oncogenic process. It is still to be determine if down-regulation of each Tribbles protein individually would trigger a positive feedback on any of the other Tribbles.
7. Downstream Mechanisms Regulated by Tribbles in Colon Cancer
A better understanding of the downstream pathways, potentially regulated by Tribbles proteins, is essential to better direct the therapeutic options available, based on Tribbles expression and the mechanism of action involved. Though an association does not always reflects causation, analyses from tissue microarray consisting of 118 Dukes’ A and B CRC patients revealed a significant correlation of TRIB1 protein expression with ERK signaling pathway activation, and AKT and MYC protein abundance [59]. In agreement, TRIB1 overexpression (TRIB1-OE) in SW480 cells also increased ERK, as well as Src and FAK (focal adhesion kinase), protein phosphorylation [60]. In addition, TRIB1 overexpression led to up-regulation of MMP-2 and MMP-9 proteolytic enzymes protein levels. Conversely, RNA interference TRIB1 silencing in TRIB1-OE cells fully reversed the previous phenotype [60]. Moreover, specific ERK, Scr and FAK inhibitors independently down-regulated MMP-2 (but not MMP-9) expression in TRIB1-OE cells [60].
In TRIB2 knocked-down SW48 and LoVo cells, increased p21 protein and mRNA was observed, while p53 (in contrast with previous report in a different cancer type, ie, melanoma [30]), cyclin D1 or p16 levels remained unchanged [62]. Conversely, overexpression of TRIB2 showed the inverse phenotype (i.e., decreased p21), in a p53-independent manner. The authors demonstrated TRIB2 negatively regulated p21 at the promoter level, through TRIB2 kinase-like domain binding and cooperation with AP4, which was elevated in CRC tumor compared with the corresponding normal tissues [62]. Though TRIB2 did not directly influence AP4 expression, upon TRIB2 over-expression, AP4 protein was further enriched at the p21 promoter [62]. The observed cellular events were validated in a CRC xenograft nude mice in vivo model, subcutaneously injected with stable TRIB2-knockdown SW48 cells, where p21 expression levels were significantly increased, compared to controls [62].
TRIB3 has been previously described, in other cellular types, to negatively regulate Akt-mTOR pathway by direct binding to AKT and consequent dephosphorylation [31]. In agreement, depletion of TRIB3 in HCT116 cells reversed the reduction of phospho-Akt and phospho-mTOR in response to A. cinnamomea-derived extract ACF2 treatment [86]. Intricately, TRIB3 silencing also decreased ACF2-induced LC3-II levels [86]. Making use of bioinformatic analysis, Hua et al. identified an enrichment of the Wnt signaling gene set in TRIB3-overexpressing cells in patients with CRC [79]. Experimentally, the authors verified that β-catenin transcriptional activation correlated with TRIB3 levels in different CRC cell lines. Moreover, when comparing tumor with adjacent normal tissues, higher TRIB3 and β-catenin expression, along with proteins co-localization, was observed. By contrast, colon tissues from TRIB3-KD mice showed much lower expression of genes regulated by Wnt signaling to β-catenin, compared to control mice, such as Axin2, c-Myc, Cyclin D1, and TRIB3 itself. TRIB3 and TCF4 were shown to interact, while depletion of TRIB3 decreased the formation of the β-catenin-TCF4 complex. Finally, it was demonstrated that the up-regulation of these genes regulated by Wnt, in response to TRIB3 overexpression, was β-catenin dependent, as cellular β-catenin depletion reversed the phenotype [79]. In the context of colon cancer, all three Tribbles seem to induce signaling pathways that are commonly associated with tumor progression.
8. Conclusions and Future Perspectives
There has been accumulating evidence that all members of the Tribbles family play a role in tumorigenesis. Whether they contribute to cancer progression or impairment is still an open question. This review highlights the existing research on the contribution of TRIB1, TRIB2 and TRIB3 to colorectal cancer as well its potential as biomarker for disease progression and/or prognosis. There is now accumulating data that TRIB2 acts as an oncogene in several tumor types, including colon cancer. This is not as clear for TRIB3, as it was found that TRIB3 overexpression negatively regulates the mTOR pathway, which is hyperactivated in several tumor types. On the contrary, different studies show that TRIB3 overexpression is positively correlated with poor prognosis and survival of colon cancer patients, highlighting the potential of using TRIB3 as a predictive biomarker. These data suggests that Tribbles family members role might be cell context dependent. Moving forward, it will be important to thoroughly study the interaction of all family members as they might have redundant roles in specific contexts. The generation of in vivo knock-out models using CRISPR technology would certainly contribute to decipher the role of each Tribble isoform in CRC development. On the other hand, colon cancer stem cells (CCSC) have been associated with patient relapse and chemoresistance, with diverse molecular mechanisms involved [105]. Importantly, high Tribbles levels have been linked with quiescent stem cell population, at least in hematopoietic diseases [106]. Thus, it is of great interest to understand the role of this family of proteins in the maintenance of CCSC and how they contribute to drug resistance. Furthermore, as new data emerge suggesting that this family of pseudokinases might be therapeutically modulated, it is of outmost importance to thoroughly understand the mechanisms by which these proteins are governed. This will potentiate the development of novel pharmacological approaches or the repurposing of existing drugs that specifically target the Tribbles proteins and improve patient care.
Author Contributions
Conceptualization, A.L.D.S.-C.; Methodology, A.L.D.S.-C.; Validation, A.L.D.S.-C., B.I.F. and W.L.; Formal Analysis, A.L.D.S.-C.; Investigation, A.L.D.S.-C.; Data Curation, A.L.D.S.-C.; Writing–Original Draft Preparation, A.L.D.S.-C., B.S. and B.I.F.; Writing–Review & Editing, A.L.D.S.-C., B.S., W.L. and B.I.F.; Supervision, A.L.D.S.-C., W.L. and B.I.F.; Project Administration, B.I.F.; Funding Acquisition, W.L. and B.I.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Spanish Ministry of Science, Innovation and Universities through Grant RTI2018-094629-B-I00 to W.L. This work was supported by the European Commission project TRIBBLES–748585 and Fundação para a Ciência e a Tecnologia (FCT) (grant PTDC/MED-ONC/4167/2020 “ENDURING”) to B.I.F.
Conflicts of Interest
The authors A.L.D.S.-C., B.S. and B.I.F. declare no conflict of interest. W.L. is the scientific co-founder of Refoxy Pharmaceuticals GmbH, Berlin and is required by his institution to state so in his publications. The funders had no role in the design and writing of the manuscript.
References
- International Agency for Research on Cancer (IARC). Age Standardized (World) Incidence Rates, Colorectal Cancer, Males, All Ages [Internet]. Globocan 2020: Colon Fact Sheet. 2020. Available online: https://gco.iarc.fr/today (accessed on 21 April 2021).
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. World Cancer Report: Cancer Research for Cancer Prevention; International Agency for Research on Cancer (IARC): Lyon, France, 2020; p. 630. [Google Scholar]
- Valle, L.; Vilar, E.; Tavtigian, S.V.; Stoffel, E.M. Genetic predisposition to colorectal cancer: Syndromes, genes, classification of genetic variants and implications for precision medicine. J. Pathol. 2019, 247, 574–588. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Zoratto, F.; Rossi, L.; Verrico, M.; Papa, A.; Basso, E.; Zullo, A.; Tomao, L.; Romiti, A.; Russo, G.L.; Tomao, S. Focus on genetic and epigenetic events of colorectal cancer pathogenesis: Implications for molecular diagnosis. Tumor Biol. 2014, 35, 6195–6206. [Google Scholar] [CrossRef]
- Haque, T.; Greene, K.G.; Crockett, S.D. Serrated neoplasia of the colon: What do we really know? Curr. Gastroenterol. Rep. 2014, 16, 380. [Google Scholar] [CrossRef]
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386. [Google Scholar] [CrossRef]
- Shang, S.; Hua, F.; Hu, Z.-W. The regulation of β-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [Green Version]
- Kendziorra, E.; Ahlborn, K.; Spitzner, M.; Rave-Fränk, M.; Emons, G.; Gaedcke, J.; Kramer, F.; Wolff, H.A.; Becker, H.; Beissbarth, T.; et al. Silencing of the Wnt transcription factor TCF4 sensitizes colorectal cancer cells to (chemo-) radiotherapy. Carcinogenesis 2011, 32, 1824–1831. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, I.; Barman, N.; Maiti, M.; Sarkar, R. Assessment of beta-catenin expression by immunohistochemistry in colorectal neoplasms and its role as an additional prognostic marker in colorectal adenocarcinoma. Med. Pharm. Rep. 2019, 92, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Liu, J.; You, J.; Wu, D.; Yu, Y.; Liu, C.; Wang, L.; Wang, F.; Xu, L.; Wang, L.; et al. Met promotes the formation of double minute chromosomes induced by Sei-1 in NIH-3T3 murine fibroblasts. Oncotarget 2016, 7, 56664–56675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Mao, R.; Lv, H.; Xu, J.; Yan, L.; Liu, Y.; Shi, M.; Ji, G.; Yu, Y.; Bai, J.; et al. Frequency of double minute chromosomes and combined cytogenetic abnormalities and their characteristics. J. Appl. Genet. 2010, 52, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Williet, N.; Petcu, C.A.; Rinaldi, L.; Cottier, M.; Del Tedesco, E.; Clavel, L.; Dumas, O.; Jarlot, C.; Bouarioua, N.; Roblin, X.; et al. The level of epidermal growth factor receptors expression is correlated with the advancement of colorectal adenoma: Validation of a surface biomarker. Oncotarget 2017, 8, 16507–16517. [Google Scholar] [CrossRef] [Green Version]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to Anti-EGFR Therapy in Colorectal Cancer: From Heterogeneity to Convergent Evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef] [Green Version]
- Kearney, M.R.; Chen, E.Y.; Vaccaro, G.M.; Strother, J.; Burt, A.; Todd, K.; Donovan, J.; Kampa-Schittenhelm, K.; Lopez, C.D. A Phase II Study Alternating Erlotinib With Second-line mFOLFOX6 or FOLFIRI for Metastatic Colorectal Cancer. Anticancer Res. 2018, 39, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strippoli, A.; Cocomazzi, A.; Basso, M.; Cenci, T.; Ricci, R.; Pierconti, F.; Cassano, A.; Fiorentino, V.; Barone, C.; Bria, E.; et al. c-MYC Expression Is a Possible Keystone in the Colorectal Cancer Resistance to EGFR Inhibitors. Cancers 2020, 12, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaveh, S.; Ebrahimi, P.; Rezapour, A.; Mozafari, M.; Sayehmiri, K. Bevacizumab and erlotinib versus bevacizumab for colorectal cancer treatment: Systematic review and meta-analysis. Int. J. Clin. Pharm. 2019, 41, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350. [Google Scholar] [CrossRef]
- Amin, M.B.; Edge, S.; Greene, F.; Byrd, D.R.; Brookland, R.K.; Washington, M.K.; Gershenwald, J.E.; Compton, C.C.; Hess, K.R.; Sullivan, D.C.; et al. AJCC Cancer Staging Manual. Available online: https://0-www-springer-com.brum.beds.ac.uk/gp/book/9783319406176 (accessed on 21 April 2021).
- Riihimäki, M.; Hemminki, A.; Sundquist, J.; Hemminki, K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 2016, 6, 29765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; He, Y.; Wang, Y.; Li, X.; Young, J.; Ioannidis, J.P.A.; Dunlop, M.G.; Theodoratou, E. Risk factors and risk prediction models for colorectal cancer metastasis and recurrence: An umbrella review of systematic reviews and meta-analyses of observational studies. BMC Med. 2020, 18, 172. [Google Scholar] [CrossRef]
- Byrne, D.P.; Foulkes, D.M.; A Eyers, P. Pseudokinases: Update on their functions and evaluation as new drug targets. Future Med. Chem. 2017, 9, 245–265. [Google Scholar] [CrossRef] [Green Version]
- Eyers, P.A.; Keeshan, K.; Kannan, N. Tribbles in the 21st Century: The Evolving Roles of Tribbles Pseudokinases in Biology and Disease. Trends Cell Biol. 2017, 27, 284–298. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.M.; Nakatani, Y.; Jamieson, S.A.; Dai, W.; Lucet, I.S.; Mace, P.D. Molecular Mechanism of CCAAT-Enhancer Binding Protein Recruitment by the TRIB1 Pseudokinase. Structure 2015, 23, 2111–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.; Wang, F.; Cao, W.-B.; Lv, X.-X.; Hua, F.; Cui, B.; Yu, J.-J.; Zhang, X.-W.; Shang, S.; Liu, S.-S.; et al. TRIB3 Promotes APL Progression through Stabilization of the Oncoprotein PML-RARα and Inhibition of p53-Mediated Senescence. Cancer Cell 2017, 31, 697–710.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, T.; Kanno, Y.; Yamazaki, Y.; Takahara, T.; Miyata, S.; Nakamura, T. Trib1 links the MEK1/ERK pathway in myeloid leukemogenesis. Blood 2010, 116, 2768–2775. [Google Scholar] [CrossRef] [PubMed]
- Richmond, L.; Keeshan, K. Pseudokinases: A tribble-edged sword. FEBS J. 2020, 287, 4170–4182. [Google Scholar] [CrossRef] [Green Version]
- Zanella, F.; Renner, O.; García, B.; Callejas, S.; Dopazo, A.; Peregrina, S.; Carnero, A.; Link, W. Human TRIB2 is a repressor of FOXO that contributes to the malignant phenotype of melanoma cells. Oncogene 2010, 29, 2973–2982. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.; Madureira, P.A.; Ferreira, B.; Baptista, I.; Machado, S.; Colaço, L.; Dos Santos, M.; Liu, N.; Dopazo, A.; Ugurel, S.; et al. TRIB2 confers resistance to anti-cancer therapy by activating the serine/threonine protein kinase AKT. Nat. Commun. 2017, 8, 14687. [Google Scholar] [CrossRef]
- Du, K.; Herzig, S.; Kulkarni, R.N.; Montminy, M. TRB3: A tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 2003, 300, 1574–1577. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Lorente, M.; García-Taboada, E.; Gómez, E.P.; Dávila, D.; García, P.Z.; Flores, J.M.; Rodríguez, A.; Hegedus, Z.; Mosén-Ansorena, D.; et al. Loss of Tribbles pseudokinase-3 promotes Akt-driven tumorigenesis via FOXO inactivation. Cell Death Differ. 2014, 22, 131–144. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Carracedo, A.; Salanueva, Í.J.; Hernández-Tiedra, S.; Lorente, M.; Egia, A.; Vázquez, P.; Blázquez, C.; Torres, S.; García, S.; et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J. Clin. Investig. 2009, 119, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.-Y.; Huang, Y.-Q.; Zhang, Y.-Q.; Han, Z.-D.; He, H.-C.; Ling, X.-H.; Fu, X.; Dai, Q.-S.; Cai, C.; Chen, J.-H.; et al. MicroRNA-224 inhibits progression of human prostate cancer by downregulating TRIB1. Int. J. Cancer 2014, 135, 541–550. [Google Scholar] [CrossRef]
- Tang, B.; Wu, W.; Zhang, Q.; Sun, Y.; Cui, Y.; Wu, F.; Wei, X.; Qi, G.; Liang, X.; Tang, F.; et al. Inhibition of tribbles protein-1 attenuates radioresistance in human glioma cells. Sci. Rep. 2015, 5, 15961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puiffe, M.-L.; Le Page, C.; Filali-Mouhim, A.; Zietarska, M.; Ouellet, V.; Tonin, P.N.; Chevrette, M.; Provencher, D.M.; Mes-Masson, A.-M. Characterization of Ovarian Cancer Ascites on Cell Invasion, Proliferation, Spheroid Formation, Gene Expression in an In Vitro Model of Epithelial Ovarian Cancer. Neoplasia 2007, 9, 820-IN8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puskas, L.G.; Juhasz, F.; Zarva, A.; Hackler, L.; Farid, N.R. Gene profiling identifies genes specific for well-differentiated epithelial thyroid tumors. Cell. Mol. Biol. 2005, 51, 177–186. [Google Scholar]
- Hill, R.; Kalathur, R.K.R.; Colaço, L.; Brandão, R.; Ugurel, S.; Futschik, M.; Link, W. TRIB2 as a biomarker for diagnosis and progression of melanoma. Carcinogenesis 2015, 36, 469–477. [Google Scholar] [CrossRef]
- Grandinetti, K.B.; Stevens, T.A.; Ha, S.; Salamone, R.J.; Walker, J.R.; Zhang, J.; Agarwalla, S.; Tenen, D.G.; Peters, E.C.; Reddy, V.A. Overexpression of TRIB2 in human lung cancers contributes to tumorigenesis through downregulation of C/EBPα. Oncogene 2011, 30, 3328–3335. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Tong, M.; Huang, J.; Zhang, Y.; Qiao, Y.; Weng, W.; Liu, W.; Wang, J.; Sun, F. TRIB2 inhibits Wnt/β-Catenin/TCF4 signaling through its associated ubiquitin E3 ligases, β-TrCP, COP1 and Smurf1, in liver cancer cells. FEBS Lett. 2014, 588, 4334–4341. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Park, J.-S.; Wei, Y.; Rajurkar, M.; Cotton, J.L.; Fan, Q.; Lewis, B.C.; Ji, H.; Mao, J. TRIB2 Acts Downstream of Wnt/TCF in Liver Cancer Cells to Regulate YAP and C/EBPα Function. Mol. Cell 2013, 51, 211–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, Y.; Zhang, Y.; Wang, J. Ubiquitin E3 ligase SCF(β-TRCP) regulates TRIB2 stability in liver cancer cells. Biochem. Biophys. Res. Commun. 2013, 441, 555–559. [Google Scholar] [CrossRef]
- Keeshan, K.; He, Y.; Wouters, B.J.; Shestova, O.; Xu, L.; Sai, H.; Rodriguez, C.G.; Maillard, I.; Tobias, J.W.; Valk, P.; et al. Tribbles homolog 2 inactivates C/EBPα and causes acute myelogenous leukemia. Cancer Cell 2006, 10, 401–411. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C.; Lohan, F.; Campos, J.; Ohlsson, E.; Salomè, M.; Forde, C.; Artschwager, R.; Liskamp, R.M.; Cahill, M.R.; A Kiely, P.; et al. The presence of C/EBPα and its degradation are both required for TRIB2-mediated leukaemia. Oncogene 2016, 35, 5272–5281. [Google Scholar] [CrossRef] [Green Version]
- O’Connor, C.; Yalla, K.; Salomé, M.; Moka, H.A.; Castañeda, E.G.; Eyers, P.A.; Keeshan, K. Trib2 expression in granulocyte-monocyte progenitors drives a highly drug resistant acute myeloid leukaemia linked to elevated Bcl2. Oncotarget 2018, 9, 14977–14992. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zuo, J.; Wahafu, A.; Wang, M.; Li, R.; Xie, W. Combined elevation of TRIB2 and MAP3K1 indicates poor prognosis and chemoresistance to temozolomide in glioblastoma. CNS Neurosci. Ther. 2020, 26, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Luo, Y.; Chen, J.-H.; Hu, J.; Luo, Y.-Z.; Wang, W.; Zeng, Y.; Xiao, L. Knockdown of TRB3 induces apoptosis in human lung adenocarcinoma cells through regulation of Notch 1 expression. Mol. Med. Rep. 2013, 8, 47–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wennemers, M.; Bussink, J.; Scheijen, B.; Nagtegaal, I.D.; Van Laarhoven, H.W.; A Raleigh, J.; A Varia, M.; Heuvel, J.J.; Rouschop, K.M.; Sweep, F.C.; et al. Tribbles homolog 3 denotes a poor prognosis in breast cancer and is involved in hypoxia response. Breast Cancer Res. 2011, 13, R82. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.-M.; Sun, W.; Wang, Z.-H.; Liang, X.; Hua, F.; Li, K.; Lv, X.-X.; Zhang, X.-W.; Liu, Y.-Y.; Yu, J.-J.; et al. TRIB3 supports breast cancer stemness by suppressing FOXO1 degradation and enhancing SOX2 transcription. Nat. Commun. 2019, 10, 5720. [Google Scholar] [CrossRef] [Green Version]
- Hong, B.; Zhou, J.; Ma, K.; Zhang, J.; Xie, H.; Zhang, K.; Li, L.; Cai, L.; Zhang, N.; Zhang, Z.; et al. TRIB3 Promotes the Proliferation and Invasion of Renal Cell Carcinoma Cells via Activating MAPK Signaling Pathway. Int. J. Biol. Sci. 2019, 15, 587–597. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Xia, J.; Bin, J.; Liao, Y.; Li, N.; Wu, Z.; Wang, H.; Sun, L.; Liao, W. Overexpression of TRIB3 promotes angiogenesis in human gastric cancer. Oncol. Rep. 2016, 36, 2339–2348. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-J.; Li, F.-F.; Zhang, Y.-J.; Jiang, M.; Ren, W.-H. TRIB3 promotes hepatocellular carcinoma growth and predicts poor prognosis. Cancer Biomark. 2020, 29, 307–315. [Google Scholar] [CrossRef]
- Bao, X.-Y.; Sun, M.; Peng, T.-T.; Han, D.-M. TRIB3 promotes proliferation, migration, and invasion of retinoblastoma cells by activating the AKT/mTOR signaling pathway. Cancer Biomark. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Chen, H.; Zhong, D.; Wei, W.; Liu, L.; Duan, Q.; Han, B.; Li, G. TRIB3 facilitates glioblastoma progression via restraining autophagy. Aging 2020, 12, 25020–25034. [Google Scholar] [CrossRef]
- Wang, S.; Wang, C.; Li, X.; Hu, Y.; Gou, R.; Guo, Q.; Nie, X.; Liu, J.; Zhu, L.; Lin, B. Down-regulation of TRIB3 inhibits the progression of ovarian cancer via MEK/ERK signaling pathway. Cancer Cell Int. 2020, 20, 418. [Google Scholar] [CrossRef] [PubMed]
- Camps, J.; Nguyen, Q.T.; Padilla-Nash, H.M.; Knutsen, T.; McNeil, N.E.; Wangsa, D.; Hummon, A.B.; Grade, M.; Ried, T.; Difilippantonio, M.J. Integrative genomics reveals mechanisms of copy number alterations responsible for transcriptional deregulation in colorectal cancer. Genes Chromosom. Cancer 2009, 48, 1002–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, W.; Bian, Z.; Yu, Y.; Yuan, C.; Liu, Y.; Yu, L.; Li, C.; Zhu, J.; Jia, X.; Guan, R.; et al. Expulsion of micronuclei containing amplified genes contributes to a decrease in double minute chromosomes from malignant tumor cells. Int. J. Cancer 2014, 134, 1279–1288. [Google Scholar] [CrossRef]
- Tanaka, H.; Watanabe, T. Mechanisms Underlying Recurrent Genomic Amplification in Human Cancers. Trends Cancer 2020, 6, 462–477. [Google Scholar] [CrossRef]
- Kuttler, F.; Mai, S. Formation of non-random extrachromosomal elements during development, differentiation and oncogenesis. Semin. Cancer Biol. 2007, 17, 56–64. [Google Scholar] [CrossRef]
- Briffa, R.; Um, I.; Faratian, D.; Zhou, Y.; Turnbull, A.K.; Langdon, S.P.; Harrison, D.J. Multi-Scale Genomic, Transcriptomic and Proteomic Analysis of Colorectal Cancer Cell Lines to Identify Novel Biomarkers. PLoS ONE 2015, 10, e0144708. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, N.; Pang, B.; Tong, D.; Sun, D.; Sun, H.; Zhang, C.; Sun, W.; Meng, X.; Bai, J.; et al. TRIB1 promotes colorectal cancer cell migration and invasion through activation MMP-2 via FAK/Src and ERK pathways. Oncotarget 2017, 8, 47931–47942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.; Guo, K.; Sun, X.; Hu, F.; Chen, Q.; Luo, X.; Wang, G.; Hu, J.; Sun, L. TRIB2 functions as novel oncogene in colorectal cancer by blocking cellular senescence through AP4/p21 signaling. Mol. Cancer 2018, 17, 172. [Google Scholar] [CrossRef]
- Bowers, A.J.; Scully, S.; Boylan, J.F. SKIP3, a novel Drosophila tribbles ortholog, is overexpressed in human tumors and is regulated by hypoxia. Oncogene 2003, 22, 2823–2835. [Google Scholar] [CrossRef] [Green Version]
- Lösch, S.; Buchholz, M.; Gress, T.M.; Weidle, U.H. LKW, a Putative Dual-specificity Kinase which is Down-regulated in Several Invasive Systems. Cancer Genom. Proteom. 2004, 1, 177–188. [Google Scholar]
- Xu, J.; Lv, S.; Qin, Y.; Shu, F.; Xu, Y.; Chen, J.; Xu, B.-E.; Sun, X.; Wu, J. TRB3 interacts with CtIP and is overexpressed in certain cancers. Biochim. Biophys. Acta (BBA) Gen. Subj. 2007, 1770, 273–278. [Google Scholar] [CrossRef]
- Miyoshi, N.; Ishii, H.; Mimori, K.; Takatsuno, Y.; Kim, H.; Hirose, H.; Sekimoto, M.; Doki, Y.; Mori, M. Abnormal expression of TRIB3 in colorectal cancer: A novel marker for prognosis. Br. J. Cancer 2009, 101, 1664–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, F.; Li, K.; Yu, J.-J.; Lv, X.; Yan, J.; Zhang, X.-W.; Sun, W.; Lin, H.; Shang, S.; Wang, F.; et al. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat. Commun. 2015, 6, 7951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.-J.; Wu, I.-J.; Hong, J.-Y.; Liu, B.-H.; Liang, R.-Y.; Yuan, T.-M.; Chuang, S.-M. Capsaicin-induced TRIB3 upregulation promotes apoptosis in cancer cells. Cancer Manag. Res. 2018, 10, 4237–4248. [Google Scholar] [CrossRef] [Green Version]
- Snezhkina, A.V.; Krasnov, G.S.; Zaretsky, A.R.; Zhavoronkov, A.; Nyushko, K.M.; Moskalev, A.A.; Karpova, I.Y.; Afremova, A.I.; Lipatova, A.V.; Kochetkov, D.V.; et al. Differential expression of alternatively spliced transcripts related to energy metabolism in colorectal cancer. BMC Genom. 2016, 17, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Menyhart, O.; Kakisaka, T.; Pongor, L.S.; Uetake, H.; Goel, A.; Győrffy, B. Uncovering Potential Therapeutic Targets in Colorectal Cancer by Deciphering Mutational Status and Expression of Druggable Oncogenes. Cancers 2019, 11, 983. [Google Scholar] [CrossRef] [Green Version]
- Takeda, H.; Kataoka, S.; Nakayama, M.; Ali, M.A.E.; Oshima, H.; Yamamoto, D.; Park, J.-W.; Takegami, Y.; An, T.; Jenkins, N.A.; et al. CRISPR-Cas9–mediated gene knockout in intestinal tumor organoids provides functional validation for colorectal cancer driver genes. Proc. Natl. Acad. Sci. USA 2019, 116, 15635–15644. [Google Scholar] [CrossRef] [Green Version]
- Jung, B.; Staudacher, J.J.; Beauchamp, D. Transforming Growth Factor β Superfamily Signaling in Development of Colorectal Cancer. Gastroenterology 2017, 152, 36–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahmatallah, Y.; Khaidakov, M.; Lai, K.K.; Goyne, H.E.; Lamps, L.W.; Hagedorn, C.H.; Glazko, G. Platform-independent gene expression signature differentiates sessile serrated adenomas/polyps and hyperplastic polyps of the colon. BMC Med. Genom. 2017, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.; Li, G.; Hu, F.; Hou, Z.; Chen, Q.; Li, G.; Luo, X.; Hu, J.; Feng, Y. PIK3R3 promotes chemotherapeutic sensitivity of colorectal cancer through PIK3R3/NF-kB/TP pathway. Cancer Biol. Ther. 2018, 19, 222–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, W.; Wu, H.-J.; Cao, L.-Q.; Li, H.-J.; He, C.-X.; Zhao, D.; Xing, L.; Li, P.-Q.; Jin, X.; Cao, H.-L. Research Progress on PARP14 as a Drug Target. Front. Pharmacol. 2019, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Farris, A.B.; Misdraji, J.; Srivastava, A.; Muzikansky, A.; Deshpande, V.; Lauwers, G.Y.; Mino-Kenudson, M. Sessile serrated adenoma: Challenging discrimination from other serrated colonic polyps. Am. J. Surg. Pathol. 2008, 32, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Sweetser, S.; Smyrk, T.C.; Sugumar, A. Serrated polyps: Critical precursors to colorectal cancer. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 627–635. [Google Scholar] [CrossRef]
- Hua, F.; Shang, S.; Yang, Y.-W.; Zhang, H.-Z.; Xu, T.-L.; Yu, J.-J.; Zhou, D.-D.; Cui, B.; Li, K.; Lv, X.-X.; et al. TRIB3 Interacts With β-Catenin and TCF4 to Increase Stem Cell Features of Colorectal Cancer Stem Cells and Tumorigenesis. Gastroenterology 2019, 156, 708–721.e15. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.-R.; Pan, X.-B. Prognostic lncRNAs, miRNAs, and mRNAs Form a Competing Endogenous RNA Network in Colon Cancer. Front. Oncol. 2019, 9, 712. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Jin, W.; Liu, H.; Gan, D.; Cui, C.; Han, C.; Wang, Z. Immune-Related Prognostic Model in Colon Cancer: A Gene Expression-Based Study. Front. Genet. 2020, 11. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yakushijin, K.; Kawamori, Y.; Minagawa, K.; Katayama, Y.; Matsui, T. Translocation (7;9)(q22;q34) in therapy-related myelodysplastic syndrome after allogeneic bone marrow transplantation for acute myeloblastic leukemia. Cancer Genet. Cytogenet. 2007, 176, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, W.; Arai, Y.; Kasai, F.; Fujiwara, Y.; Haruta, M.; Hosaka, R.; Nishida, K.; Kurosumi, M.; Kobayashi, Y.; Akagi, K.; et al. Association of germline or somatic TP53 missense mutation with oncogene amplification in tumors developed in patients with Li-Fraumeni or Li-Fraumeni-like syndrome. Genes Chromosom. Cancer 2011, 50, 535–545. [Google Scholar] [CrossRef]
- Rishi, L.; Hannon, M.; Salomè, M.; Hasemann, M.; Frank, A.-K.; Campos, J.; Timoney, J.; O’Connor, C.; Cahill, M.R.; Porse, B.; et al. Regulation of Trib2 by an E2F1-C/EBPα feedback loop in AML cell proliferation. Blood 2014, 123, 2389–2400. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Yang, L.; Shi, H.; Du, W.; Qi, Y.; Qiu, C.; Liang, X.; Shi, W.; Liu, J. Endoplasmic reticulum-targeting photosensitizer Hypericin confers chemo-sensitization towards oxaliplatin through inducing pro-death autophagy. Int. J. Biochem. Cell Biol. 2017, 87, 54–68. [Google Scholar] [CrossRef]
- Tsai, D.-H.; Chung, C.-H.; Lee, K.-T. Antrodia cinnamomea induces autophagic cell death via the CHOP/TRB3/Akt/mTOR pathway in colorectal cancer cells. Sci. Rep. 2018, 8, 17424. [Google Scholar] [CrossRef]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4–CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef]
- Su, N.; Kilberg, M.S. C/EBP Homology Protein (CHOP) Interacts with Activating Transcription Factor 4 (ATF4) and Negatively Regulates the Stress-dependent Induction of the Asparagine Synthetase Gene. J. Biol. Chem. 2008, 283, 35106–35117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, S.J.; Mack, E.A.; Rome, K.S.; Pear, W.S. Tribbles in normal and malignant haematopoiesis. Biochem. Soc. Trans. 2015, 43, 1112–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.; Nim, S.; Teyra, J.; Datti, A.; Wrana, J.L.; Sidhu, S.S.; Moffat, J.; Kim, P.M. A systematic approach to identify novel cancer drug targets using machine learning, inhibitor design and high-throughput screening. Genome Med. 2014, 6, 57. [Google Scholar] [CrossRef]
- Foulkes, D.M.; Byrne, M.P.; Bailey, F.P.; Eyers, P.A. Tribbles pseudokinases: Novel targets for chemical biology and drug discovery? Biochem. Soc. Trans. 2015, 43, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Byrne, D.P.; Harris, J.A.; Kannan, N.; Eyers, P.A. Cataloguing the dead: Breathing new life into pseudokinase research. FEBS J. 2020, 287, 4150–4169. [Google Scholar] [CrossRef]
- Foulkes, D.M.; Byrne, D.P.; Yeung, W.; Shrestha, S.; Bailey, F.P.; Ferries, S.; Eyers, C.E.; Keeshan, K.; Wells, C.; Drewry, D.H.; et al. Covalent inhibitors of EGFR family protein kinases induce degradation of human Tribbles 2 (TRIB2) pseudokinase in cancer cells. Sci. Signal. 2018, 11, eaat7951. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Chen, Y.; Yang, Y.; Zhang, X.; Ma, L.; Xue, X.; Qiao, Y.; Wang, J. TRIB2 modulates proteasome function to reduce ubiquitin stability and protect liver cancer cells against oxidative stress. Cell Death Dis. 2021, 12, 42. [Google Scholar] [CrossRef] [PubMed]
- Miele, E.; Abballe, L.; Spinelli, G.P.; Besharat, Z.M.; Catanzaro, G.; Chiacchiarini, M.; Vacca, A.; Po, A.; Capalbo, C.; Ferretti, E. BRAF mutant colorectal cancer: ErbB2 expression levels as predictive factor for the response to combined BRAF/ErbB inhibitors. BMC Cancer 2020, 20, 129. [Google Scholar] [CrossRef] [Green Version]
- López-Ayllón, B.D.; De Castro-Carpeño, J.; Rodríguez-Antolín, C.; Pernía, O.; De Cáceres, I.I.; Belda-Iniesta, C.; Perona, R.; Sastre, L. Biomarkers of erlotinib response in non-small cell lung cancer tumors that do not harbor the more common epidermal growth factor receptor mutations. Int. J. Clin. Exp. Pathol. 2015, 8, 2888–2898. [Google Scholar] [PubMed]
- Yu, J.-J.; Zhou, D.-D.; Yang, X.-X.; Cui, B.; Tan, F.-W.; Wang, J.; Li, K.; Shang, S.; Zhang, C.; Lv, X.-X.; et al. TRIB3-EGFR interaction promotes lung cancer progression and defines a therapeutic target. Nat. Commun. 2020, 11, 3660. [Google Scholar] [CrossRef]
- Lundemo, A.G.; Pettersen, C.H.; Berge, K.; Berge, R.K.; A Schønberg, S. Tetradecylthioacetic acid inhibits proliferation of human SW620 colon cancer cells—gene expression profiling implies endoplasmic reticulum stress. Lipids Health Dis. 2011, 10, 190. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Zeng, Y.; Zhang, Z.; Fu, J.; You, L.; He, Y.; Hao, Y.; Gu, Z.; Yu, Z.; Qu, C.; et al. Hypericin-mediated photodynamic therapy for the treatment of cancer: A review. J. Pharm. Pharmacol. 2021, 73, 425–436. [Google Scholar] [CrossRef]
- Hua, F.; Mu, R.; Liu, J.; Xue, J.; Wang, Z.; Lin, H.; Yang, H.; Chen, X.; Hu, Z. TRB3 interacts with SMAD3 promoting tumor cell migration and invasion. J. Cell Sci. 2011, 124, 3235–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyanaga, A.; Masuda, M.; Motoi, N.; Tsuta, K.; Nakamura, Y.; Nishijima, N.; Watanabe, S.-I.; Asamura, H.; Tsuchida, A.; Seike, M.; et al. Whole-exome and RNA sequencing of pulmonary carcinoid reveals chromosomal rearrangements associated with recurrence. Lung Cancer 2020, 145, 85–94. [Google Scholar] [CrossRef]
- Peeters, P.J.; Bazelier, M.T.; Leufkens, H.G.; de Vries, F.; De Bruin, M.L. The Risk of Colorectal Cancer in Patients With Type 2 Diabetes: Associations With Treatment Stage and Obesity. Diabetes Care 2015, 38, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.; Datta, P.K. Regulation of EMT in Colorectal Cancer: A Culprit in Metastasis. Cancers 2017, 9, 171. [Google Scholar] [CrossRef] [Green Version]
- Makino, S.; Takahashi, H.; Okuzaki, D.; Miyoshi, N.; Haraguchi, N.; Hata, T.; Matsuda, C.; Yamamoto, H.; Mizushima, T.; Mori, M.; et al. DCLK1 integrates induction of TRIB3, EMT, drug resistance and poor prognosis in colorectal cancer. Carcinogenesis 2020, 41, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Hervieu, C.; Christou, N.; Battu, S.; Mathonnet, M. The Role of Cancer Stem Cells in Colorectal Cancer: From the Basics to Novel Clinical Trials. Cancers 2021, 13, 1092. [Google Scholar] [CrossRef] [PubMed]
- Salomé, M.; Hopcroft, L.; Keeshan, K. Inverse and correlative relationships between TRIBBLES genes indicate non-redundant functions during normal and malignant hemopoiesis. Exp. Hematol. 2018, 66, 63–78.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Timeline of Tribbles publications in colorectal cancer. Chronological representation of each significant publication. Each publication is represented by a coloured flag symbol (▷). Flag colours are associated with a different Tribble family member (TRIB1 in grey flag, TRIB2 in red flag, and TRIB3 in blue flag). First author last name is shown for each flag. A total of four articles studying or including TRIB1, five for TRIB2, and 14 for TRIB3, in colon cancer, were identified from 2003 to 2020 and are included in the review.
Figure 1.
Timeline of Tribbles publications in colorectal cancer. Chronological representation of each significant publication. Each publication is represented by a coloured flag symbol (▷). Flag colours are associated with a different Tribble family member (TRIB1 in grey flag, TRIB2 in red flag, and TRIB3 in blue flag). First author last name is shown for each flag. A total of four articles studying or including TRIB1, five for TRIB2, and 14 for TRIB3, in colon cancer, were identified from 2003 to 2020 and are included in the review.

Figure 2.
TRIB3 upstream regulation in colorectal cancer. Proposed mechanisms regarding TRIB3 regulation at the promoter level based on references [63,67,79,85,86] are shown. TRIB3 is a transcriptional target of the ATF4–CHOP [87] and β-catenin–TCF4 [79] complexes through direct binding at specific sites at TRIB3 genomic sequence, localized at +201 to +312 and at −12 to −3, respectively. TRIB3 is proposed to be positively or negatively regulated by HIF1α or FOXO1 at the transcriptional level, respectively, in response to specific stimuli that modulate transcription factors merged (TF) activity such as hypoxia or feeding signals. TF are depicted as random elements from Canva design.
Figure 2.
TRIB3 upstream regulation in colorectal cancer. Proposed mechanisms regarding TRIB3 regulation at the promoter level based on references [63,67,79,85,86] are shown. TRIB3 is a transcriptional target of the ATF4–CHOP [87] and β-catenin–TCF4 [79] complexes through direct binding at specific sites at TRIB3 genomic sequence, localized at +201 to +312 and at −12 to −3, respectively. TRIB3 is proposed to be positively or negatively regulated by HIF1α or FOXO1 at the transcriptional level, respectively, in response to specific stimuli that modulate transcription factors merged (TF) activity such as hypoxia or feeding signals. TF are depicted as random elements from Canva design.

Figure 3.
Proposed mechanisms for TRIB3 pharmacological modulation in colorectal cancer. Compounds that alter TRIB3 expression based in references [79,85,86,98], are depicted on figure. TRIB3 was upregulated in response to FA-TTA and ACF2 extract treatments, along with CHOP induction. TRIB3 levels were elevated after HY-PDT, in a CHOP-dependent fashion. P2-T3A6 treatment caused disruption of the β-catenin–TRIB3 interaction, leading to decreased TRIB3 levels. Symbols: ▲ represents up-regulation (in blue color) and ▼ represents down-regulation (in pink color) of TRIB3. Transcription factors are depicted as random elements from Canva design. FA-TTA, bioactive chemical modified fatty acid analog saturated 3-thiatetradecylthioacetic acid; HY-PDT, photosensitizer hypericin and photodynamic therapy; ACF2 is a specific extract from Antrodia cinnamomea, a native Taiwanese rare mushroom; P2-T3A6, fusion peptide.
Figure 3.
Proposed mechanisms for TRIB3 pharmacological modulation in colorectal cancer. Compounds that alter TRIB3 expression based in references [79,85,86,98], are depicted on figure. TRIB3 was upregulated in response to FA-TTA and ACF2 extract treatments, along with CHOP induction. TRIB3 levels were elevated after HY-PDT, in a CHOP-dependent fashion. P2-T3A6 treatment caused disruption of the β-catenin–TRIB3 interaction, leading to decreased TRIB3 levels. Symbols: ▲ represents up-regulation (in blue color) and ▼ represents down-regulation (in pink color) of TRIB3. Transcription factors are depicted as random elements from Canva design. FA-TTA, bioactive chemical modified fatty acid analog saturated 3-thiatetradecylthioacetic acid; HY-PDT, photosensitizer hypericin and photodynamic therapy; ACF2 is a specific extract from Antrodia cinnamomea, a native Taiwanese rare mushroom; P2-T3A6, fusion peptide.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Tribbles amplification and overexpression in colon cancer tissues. Main result obtained for each study included in the section is shown for each Tribble member. Details of the samples, databases and methodology used by the authors is detailed under respective column. Whenever available, the number of samples analyzed is specified under n. nd, not defined.
Table 1.
Tribbles amplification and overexpression in colon cancer tissues. Main result obtained for each study included in the section is shown for each Tribble member. Details of the samples, databases and methodology used by the authors is detailed under respective column. Whenever available, the number of samples analyzed is specified under n. nd, not defined.
| Tribbles | Main Result | Samples | Databases/Experimental Methods | Author, Year (Reference) | |
|---|---|---|---|---|---|
| Disease | Control | ||||
| TRIB1 | Genomic amplification; Increased copy number. | Primary colorectal tumors (n = 31); CRC cell lines: DLD-1, HCT116, p53HCT116, SW48, LoVo, SW480, SW837, HT-29, T84, Colo 201, Colo 320DM, LS411N, SK-CO-1, NCI-H508, NCI-H716 (from ATCC). | Normal colon RNA isolated postmortem from different donors without a history of colorectal cancer (n = 5) (from Ambion). | Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis; Oligonucleotide-based Human Genome Microarrays (Agilent Technologies). | Camps, 2009 [55] |
| Genomic amplification; DM-carried gene; 5 × mRNA increase. | NCI-H716 cell line. | Normal human peripheral blood; Non-paired normal colon tissue. | Immunoblotting analysis with anti-TRIB1 antibody (Abnova); Affymetrix GeneChip Human Mapping SNP6.0 array (analysis of DNA copy number changes); qRT-PCR. | Ji, 2014 [56] | |
| Genomic amplification in 7 out of the 15 cell lines tested; 11–14.4% gain in CRC samples. | DLD-1, HCT116, HCT116p53-/-, SW48, LoVo, SW480, SW837, HT29, T84, Colo 201, Colo 320DM, LS411N, SK-CO-1, NCI H508 and NCI H716 cells (ECACC or ATCC); primary CRC samples (n = 881); CRC tumors (n = 76). | nd | Oncomine database; Comparative genomic hybridization (CGH) NimbleGen microarray (Roche): GSE72296; Illumina Whole Genome Gene Expression Profiling: GSE72544; Fluorescence in situ hybridisation (FISH): TRIB1/CEN8p probe (Abnova); Tissue microarray (TMA), automated quantitative analysis (AQUA) with anti-TRIB1 rabbit polyclonal antibody. | Briffa, 2015 [59] | |
| Genomic amplification; Increase in mRNA and protein levels. | (1) Human CRC tissues (n = 212); (2) CRC (n = 70); (3) Colon adenocarcinoma (n = 4(1); (4) CRC tumor (n = 8). | Normal colon tissues: (1) n = 79; (2) n = 12; (3) n = 5; (4) paired surrounding non-tumor tissues (n = 8). | Oncomine: (1) TCGA; GEO: (2) GSE9348; (3) GSE5206 (GEO); (4) Western blot & IHC (protein levels). | Wang, 2017 [60] | |
| TRIB2 | Detection in CRC samples. | Human CRC samples. | nd | Human CRC tissue microarray slides (Biomax, Genvelop or UMass Cancer Center Tissue Bank). | Wang, 2013 [41] |
| Increase in mRNA and protein levels. | Primary colon cancer tissue (n = 14). | Adjacent paired non-tumor colon samples (n = 14). | Immunoblot analysis. | Hill, 2017 [30] | |
| (1) Higher protein levels; (2) overexpressed in 73.3% of tumors. | (1) CRC patients samples; (2) Primary tumor (n = 186); (3) Adenocarcinoma (n = 45) | (1) Normal colorectal tissues (15 pairs); Normal colon: (2) n = 54; (3) n = 34. | (1) RT-PCR; IHC staining; GEO: (2) GSE41258; (3) GSE20916. | Hou, 2018 [62] | |
| TRIB3 | Gene and protein overexpression. | (1) Colon tumor; (2) SW480 cells; (3) Xenografts with HT-29 cells. | (1) Matched normal colon samples. | Northern blot analysis; Real-time PCR; In situ hybridization. | Bowers, 2003 [63] |
| Gene overexpression. | (1) SW480 cells; (2) Tumor (n = 241); (3) human tumor tissues from primary cancers with different TNM stages (n = 24). | (1) nd; (2) Corresponding normal tissues from individual patients; (3) Normal colonic mucosa (n = 24) | Cancer profiling array; LightCycler PCR technology. | Lösch, 2004 [64] | |
| Overexpression. | Colon tumor tissues. | Colon pairs (n = 11) | qRT-PCR; data not shown. | Xu, 2007 [65] | |
| Gene and protein overexpression. | (1) Caco2, DLD-1, LoVo, HCT116, HT-29, KM12 SM, SW480 cells; (2) Primary CRC specimens (n = 202) | (1) nd; (2) Adjacent normal colorectal mucosa (n = 202) | IHC staining; Gel RT-PCR; Real-time RT-PCR. | Miyoshi, 2009 [66] | |
| Protein overexpression. | Colon tumors (n = 76) | Adjacent non-tumour tissues of the human colon (n = 72) | Tissue microarray (TMA); Immunohistologic staining. | Hua, 2015 [67] | |
| Gene and protein overexpression. | SW480 cells | - | Western blot; gel RT-PCR. | Lin, 2018 [68] | |
| Alternative transcripts overexpression | Colorectal cancer specimens (n = 40) | Normal tissues matched morphologically (n = 40) | TCGA RNA-Seq datasets; Gene Ontology database. | Snezhkina, 2016 [69] | |
Table 2.
Clinical significance of Tribbles overexpression in colon cancer. Main results regarding the correlation between the expression of each Tribbles family member and its clinical impact. Sample details, databases and methodology used by the authors is detailed under the respective column.
Table 2.
Clinical significance of Tribbles overexpression in colon cancer. Main results regarding the correlation between the expression of each Tribbles family member and its clinical impact. Sample details, databases and methodology used by the authors is detailed under the respective column.
| Tribbles | Main Result | Samples/Databases/Experimental Methods | Author, Year (Reference) |
|---|---|---|---|
| TRIB1 | No association with survival. | Dukes’ A and B stages CRC patients (n = 118); Tissue microarray. | Briffa, 2015 [59] |
| Lower Disease Free or Specific Survival. | CRC patients; PrognoScan tool and gene expression databases (GSE17536: n = 145, GSE14333: n = 226 and GSE17537: n = 49); GEO database-GSE17537; anti-TRIB1 ICH correlation with clinicopathological features. | Wang, 2017 [60] | |
| TRIB2 | Worse clinical outcome. | Gene Expression Omnibus (GEO) database: GSE17536 | Hill, 2017 [30] |
| Worse overall survival (OS); more recurrence; higher tumor grade. | Gene Expression Omnibus (GEO) databases: GSE21510; GSE25071; GSE17536. The Cancer Genome Atlas (TCGA). | Hou, 2018 [62] | |
| Worse survival outcome in ACVR2A mutant colon cancers; Worse recurrence-free survival. | Independent clinical cohort (formalin-fixed paraffin-embedded (FFPE) cancer tissues from 171 CRC patients; GEO datasets with published survival times (GSE17538, GSE12945, GSE31595, GSE14333, GSE37892, GSE33114, GSE41258, GSE39582, GSE30540, GSE18088, GSE26682, and GSE1329(4); Drug–Gene Interaction database DGIdb 3.0; TCGA repository. | Menyhart, 2019 [70] | |
| Up-regulation in cancer-prone sessile serrated polyps. | Human colonic premalignant polyps: sessile serrated polyps (SSA/Ps), right colon and hyperplastic polyps (HPs) samples (GEO datasets: GSE10714, GSE45270, GSE76987, GSE4384(1). | Rahmatallah, 2017 [74] | |
| TRIB3 | Lower expression in more invasive/metastatic stages (stage D). | Dukes’ A, B, C and D stages CRC patients. | Lösch, 2004 [64] |
| Metastasis (M0/M(1) correlation; inverse correlation with OS. | Primary CRC specimens and adjacent normal colorectal mucosa from 202 patients who underwent surgery for CRC (Kyusyu University, 1992–2002; Osaka University, 2002–2006). | Miyoshi, 2009 [66] | |
| Inverse correlation with OS. | Surgically removed human primary colon carcinoma and normal colon tissue specimens (Alenabio, Xian, China). | Hua, 2015 [67] | |
| Low survival rate; poor outcomes. | CRC dataset from The Cancer Genome Atlas (TCGA). GEO dataset: GSE41258. | Hua, 2019 [79] | |
| Significant correlation with tumor status & stage. | Colon cancer patients with a follow-up time greater then 90 days (n = 347); TCGA dataset. | Huang, 2019 [80] | |
| Association with prognosis. | 277 samples (GSE17538); Gene expression profiles (TCGA). | Yang, 2020 [81] |
Table 3.
Tribbles genetic modulation response in colon cancer. Main results obtained for cellular genetic manipulation of Tribbles is described, separately for either up- or down-regulation, and respective reference (author, year). Not applicable (na) is shown when the data was not available.
Table 3.
Tribbles genetic modulation response in colon cancer. Main results obtained for cellular genetic manipulation of Tribbles is described, separately for either up- or down-regulation, and respective reference (author, year). Not applicable (na) is shown when the data was not available.
| Tribbles | Genetic Manipulation | Author, Year (Reference) | |
|---|---|---|---|
| Up-Regulation | Down-Regulation | ||
| TRIB1 | na | Decrease in DM formation; genomic instability; cytotoxicity (NCI-H716 cells). | Ji, 2014 [56] |
| Increased migration, invasion and metastatic capacity (SW480 and LoVo cells). | Reduced migration and invasion (SW480-TRIB1 and COLO320HSR cells). | Wang, 2017 [60] | |
| TRIB2 | Decreased cellular senescence, increased proliferation and cell cycle progression. | Decreased proliferation, induced cell cycle arrest and cellular senescence (SW48 and LoVo cells) | Hou, 2018 [62] |
| TRIB3 | na | Decreased proliferation (DLD-1, LoVo, HCT116, KM12SM and SW480 cells). | Miyoshi, 2009 [66] |
| na | Decreased metastasis and growth (HCT-8 cells in nude mice). | Hua, 2015 [67] | |
| Decreased survival rate and tumor burden (TRIB3-OE mice). | Increased survival rate, no colon tumor development (Trib3-KD mice). | Hua, 2019 [79] | |
| na | Mesenchymal-epithelial transition phenotype (SW480, HCT116, CaCO2, SW48 and SKCO1 cells). | Makino, 2020 [104] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ferreira, B.I.; Santos, B.; Link, W.; De Sousa-Coelho, A.L. Tribbles Pseudokinases in Colorectal Cancer. Cancers 2021, 13, 2825. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112825
AMA Style
Ferreira BI, Santos B, Link W, De Sousa-Coelho AL. Tribbles Pseudokinases in Colorectal Cancer. Cancers. 2021; 13(11):2825. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112825
Chicago/Turabian StyleFerreira, Bibiana I., Bruno Santos, Wolfgang Link, and Ana Luísa De Sousa-Coelho. 2021. "Tribbles Pseudokinases in Colorectal Cancer" Cancers 13, no. 11: 2825. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112825
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.