
Elevated Expression of the RAGE Variant-V in SCLC Mitigates the Effect of Chemotherapeutic Drugs

 , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
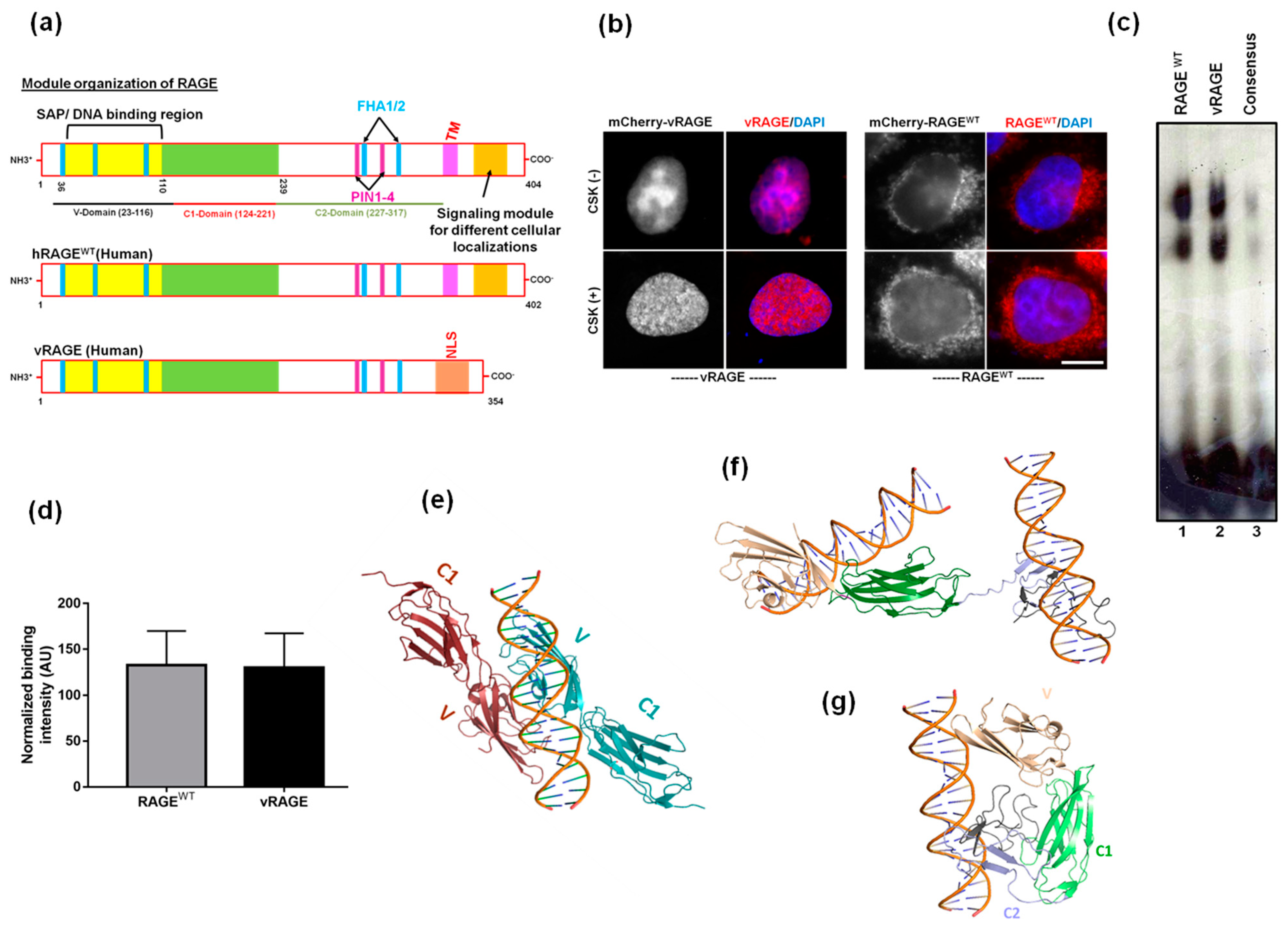
2.1. Both RAGEWT and the Variant RAGE-V Share an Identical Structural Organization of the DNA Binding Modules of RAGE
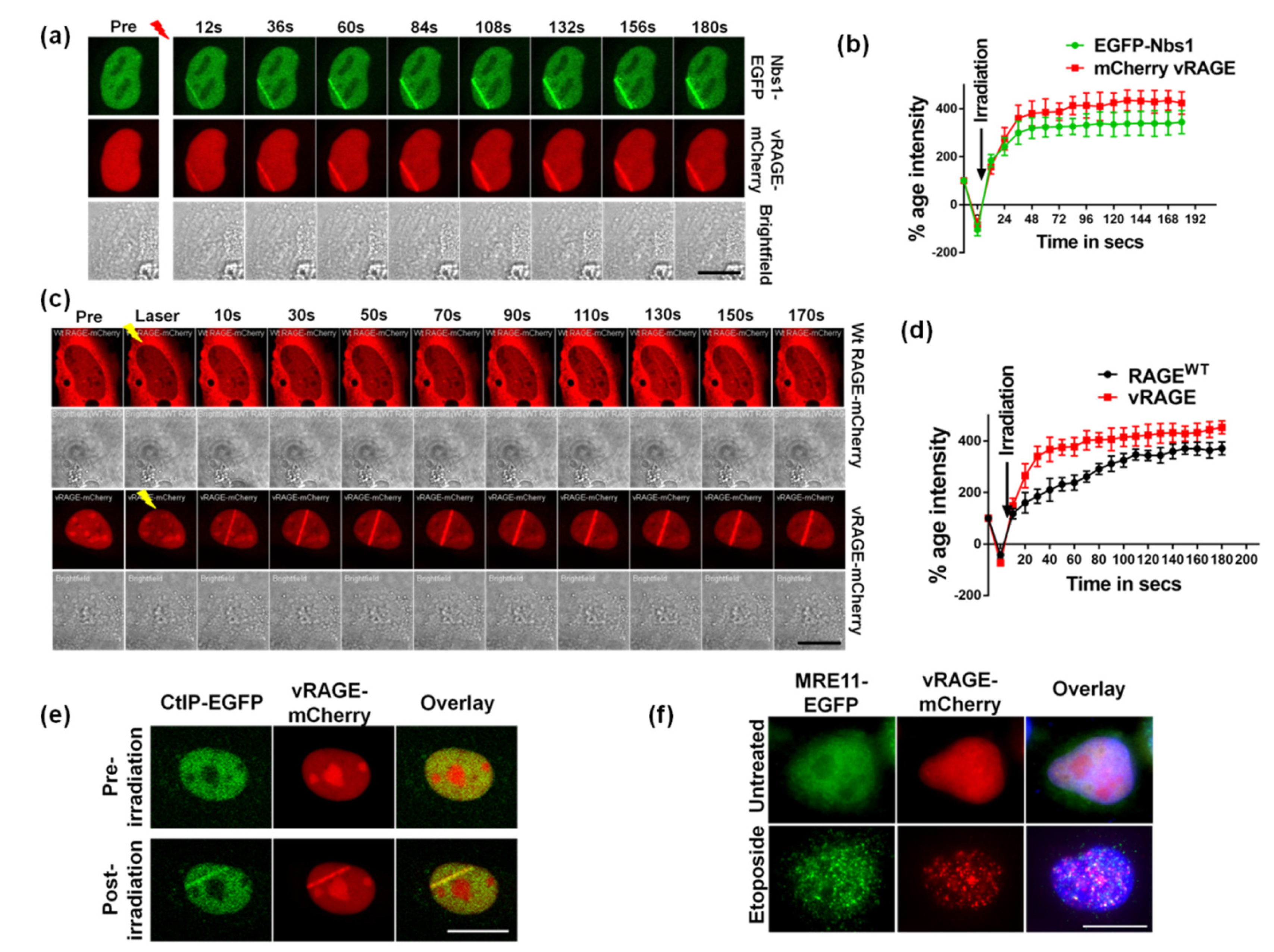
2.2. vRAGE is Involved in the DSB Repair as an Early DDR Factor
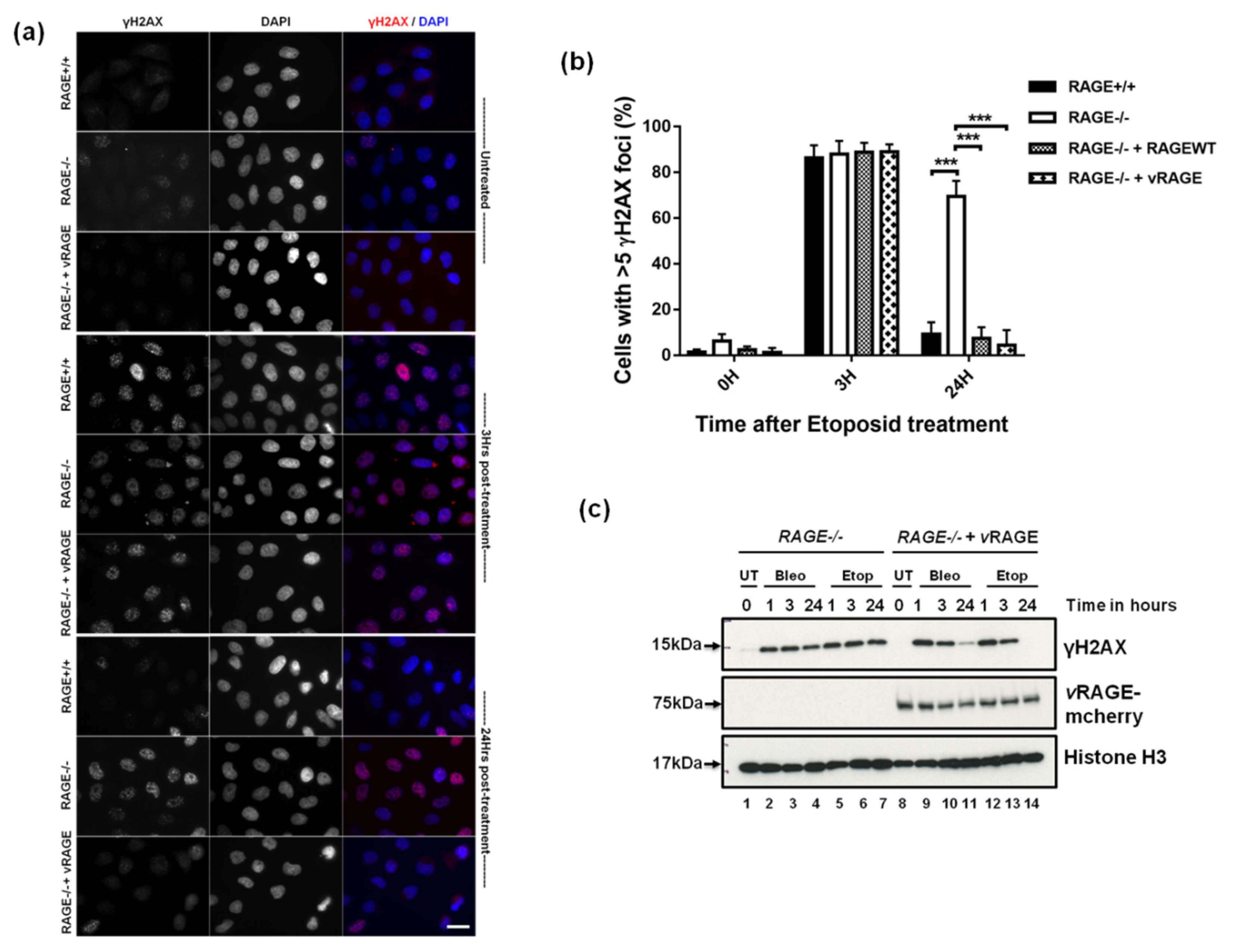
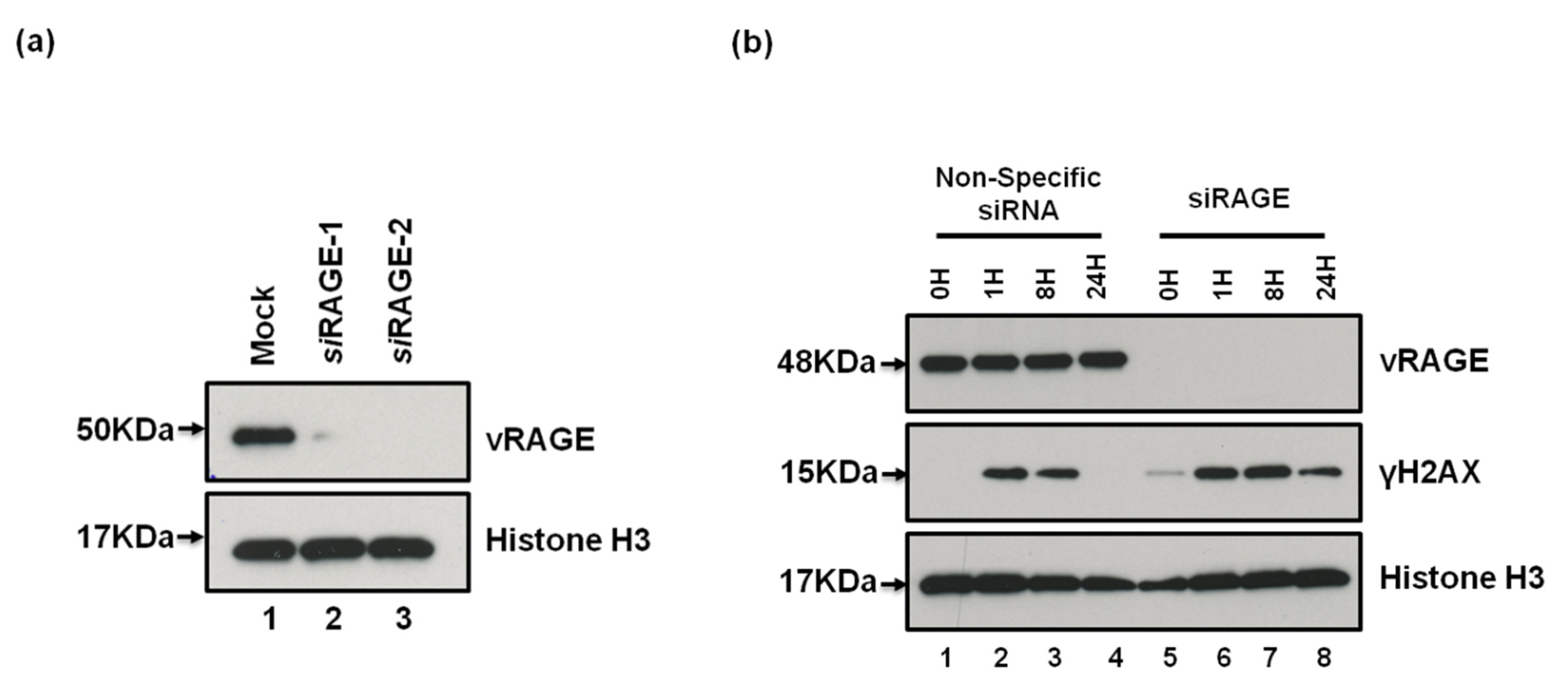
2.3. vRAGE Stimulates the DNA DSBs Repair and Thus, Enhances the Repair Potential of Cells
2.4. vRAGE Is Linked to Aggressively Metastasizing Small Cell Lung Carcinoma
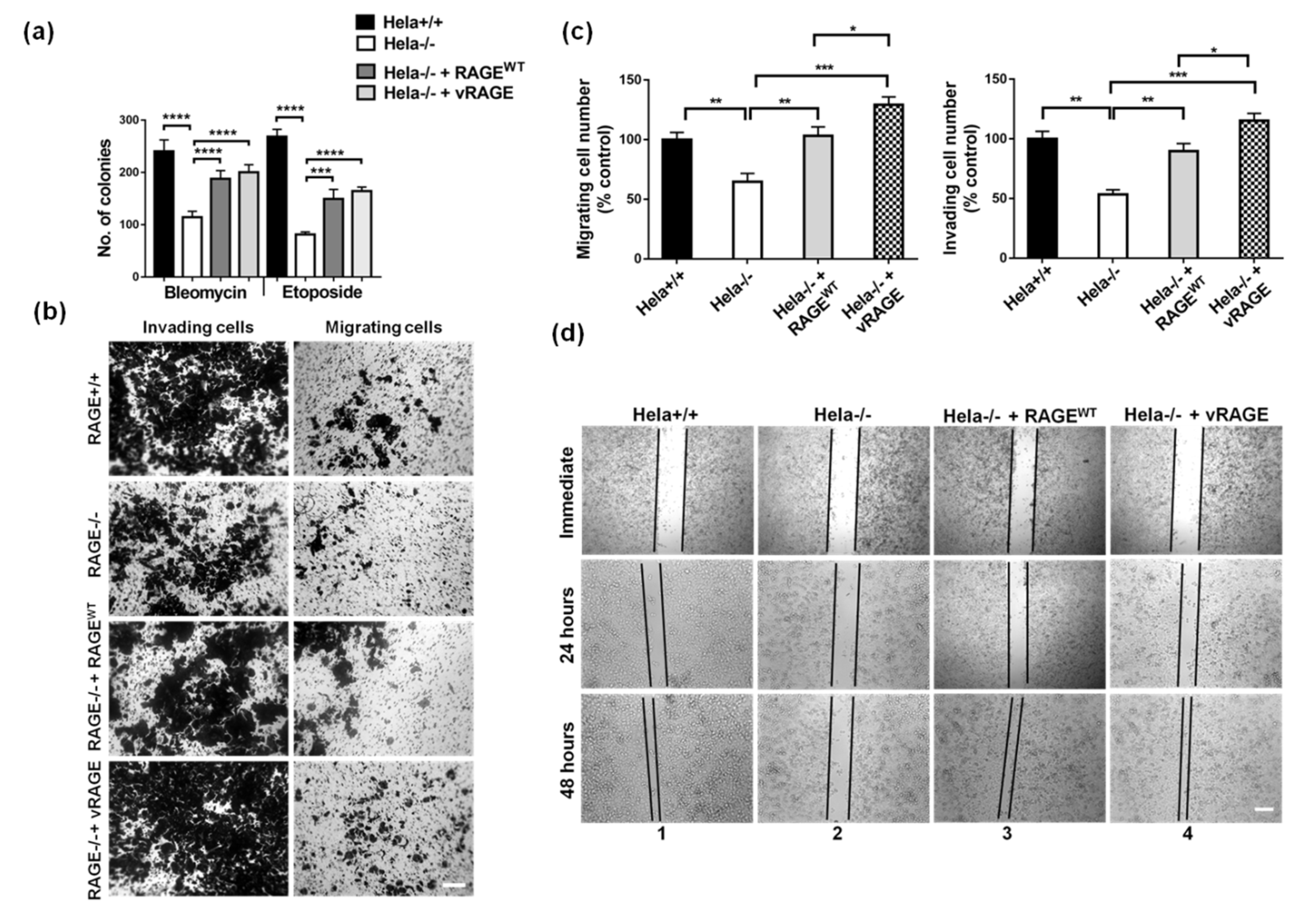
2.5. Radiomimetic Drug Resistance and Metastasis are Cumulatively Associated with vRAGE Over-Expression
3. Discussion
Study Limitations
4. Materials and Methods
4.1. Cell Culture and Transfection
4.2. Cell Lysis and Immunoblotting
4.3. In Vitro Wound Healing
4.4. Matrigel Invasion
4.5. Cellular Immunofluorescence (IF)
4.6. Tissue Sections Immunofluorescence
4.7. H&E Staining
4.8. Monoclonal Antibody Generation
4.9. Laser Micro-Irradiation
4.10. Electrophoretic Mobility Shift Assay (EMSA)
4.11. Molecular Modelling and Docking
4.12. Graph Plotting and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sabari, J.K.; Lok, B.H.; Laird, J.H.; Poirier, J.T.; Rudin, C.M. Unravelling the biology of SCLC: Implications for therapy. Nat. Rev. Clin. Oncol. 2017, 14, 549–561. [Google Scholar] [CrossRef]
- Van Meerbeeck, J.P.; Fennell, D.A.; De Ruysscher, D.K. Small-cell lung cancer. Lancet 2011, 378, 1741–1755. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nat. Cell Biol. 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Bunn, P.A.; Minna, J.D.; Augustyn, A.; Gazdar, A.F.; Ouadah, Y.; Krasnow, M.A.; Berns, A.; Brambilla, E.; Rekhtman, N.; Massion, P.P.; et al. Small Cell Lung Cancer: Can Recent Advances in Biology and Molecular Biology Be Translated into Improved Outcomes? J. Thorac. Oncol. 2016, 11, 453–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Ferguson, T.B.; Bennett, D.E.; Burford, T.H. Oat cell carcinoma of the lung: A review of 138 cases. Cancer 1969, 23, 517–524. [Google Scholar] [CrossRef]
- Foy, V.; Schenk, M.W.; Baker, K.; Gomes, F.; Lallo, A.; Frese, K.K.; Forster, M.; Dive, C.; Blackhall, F. Targeting DNA damage in SCLC. Lung Cancer 2017, 114, 12–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, T.; Gay, C.M.; Byers, L.A. Targeting DNA damage repair in small cell lung cancer and the biomarker landscape. Transl. Lung Cancer Res. 2018, 7, 50–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Kumar, V.; Fleming, T.; Terjung, S.; Gorzelanny, C.; Gebhardt, C.; Agrawal, R.; Mall, M.A.; Ranzinger, J.; Zeier, M.; Madhusudhan, T.; et al. Homeostatic nuclear RAGE–ATM interaction is essential for efficient DNA repair. Nucleic Acids Res. 2017, 45, 10595–10613. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D.; Lotze, M.T.; Zeh, I.H.J. RAGE regulates autophagy and apoptosis following oxidative injury. Autophagy 2011, 7, 442–444. [Google Scholar] [CrossRef] [Green Version]
- Bucciarelli, L.G.; Kaneko, M.; Ananthakrishnan, R.; Harja, E.; Lee, L.K.; Hwang, Y.; Lerner, S.; Bakr, S.; Li, Q.; Lu, Y.; et al. Receptor for advanced-glycation end products: Key modulator of myocardial ischemic injury. Circulation 2006, 113, 1226–1234. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Agrawal, R.; Pandey, A.; Kopf, S.; Hoeffgen, M.; Kaymak, S.; Bandapalli, O.R.; Gorbunova, V.; Seluanov, A.; Mall, M.A.; et al. Compromised DNA repair is responsible for diabetes-associated fibrosis. EMBO J. 2020, 39, e103477. [Google Scholar] [CrossRef]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T.; et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: A novel pathway for in-flammatory cell recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Malherbe, P.; Richards, J.G.; Gaillard, H.; Thompson, A.; Diener, C.; Schuler, A.; Huber, G. cDNA cloning of a novel secreted isoform of the human receptor for advanced glycation end products and charac-terization of cells co-expressing cell-surface scavenger receptors and Swedish mutant amyloid precursor protein. Brain Res. Mol. Brain Res. 1999, 71, 159–170. [Google Scholar] [CrossRef]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Ding, Q.; Keller, J.N. Splice variants of the receptor for advanced glycosylation end products (RAGE) in human brain. Neurosci. Lett. 2004, 373, 67–72. [Google Scholar] [CrossRef]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2007, 22, 1572–1580. [Google Scholar] [CrossRef]
- Kalea, A.Z.; Reiniger, N.; Yang, H.; Arriero, M.; Schmidt, A.M.; Hudson, B.I. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J. 2009, 23, 1766–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterenczak, K.A.; Nolte, I.; Escobar, H.M. RAGE Splicing Variants in Mammals. Adv. Struct. Saf. Stud. 2012, 963, 265–276. [Google Scholar] [CrossRef]
- Katsuoka, F.; Kawakami, Y.; Arai, T.; Imuta, H.; Fujiwara, M.; Kanma, H.; Yamashita, K. Type II Alveolar Epithelial Cells in Lung Express Receptor for Advanced Glycation End Products (RAGE) Gene. Biochem. Biophys. Res. Commun. 1997, 238, 512–516. [Google Scholar] [CrossRef]
- Brett, J.; Schmidt, A.M.; Du Yan, S.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the Distribution of a Newly Characterized Receptor for Advanced Glycation End Products in Tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar] [PubMed]
- Sawasdichai, A.; Chen, H.-T.; Hamid, N.A.; Jayaraman, P.-S.; Gaston, K. In situ Subcellular Fractionation of Adherent and Non-adherent Mammalian Cells. J. Vis. Exp. 2010, 2010, e1958. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Sulaj, A.; Fleming, T.; Nawroth, P.P. Purification and Characterization of the Soluble form of the Receptor for Advanced Glycation End-Products (sRAGE): A Novel Fast, Economical and Convenient Method. Exp. Clin. Endocrinol. Diabetes 2018, 126, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Sirois, C.M.; Jin, T.; Miller, A.L.; Bertheloot, D.; Nakamura, H.; Horvath, G.L.; Mian, A.; Jiang, J.; Schrum, J.; Bossaller, L.; et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J. Exp. Med. 2013, 210, 2447–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Mohanty, S.K.; Stephens, J.; Heale, J.T.; Gomez-Godinez, V.; Shi, L.Z.; Kim, J.-S.; Yokomori, K.; Berns, M.W. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res. 2009, 37, e68. [Google Scholar] [CrossRef]
- Polo, S.E.; Blackford, A.N.; Chapman, J.R.; Baskcomb, L.; Gravel, S.; Rusch, A.; Thomas, A.; Blundred, R.; Smith, P.; Kzhyshkowska, J.; et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell 2012, 45, 505–516. [Google Scholar] [CrossRef] [Green Version]
- Seluanov, A.; Vaidya, A.; Gorbunova, V. Establishing Primary Adult Fibroblast Cultures From Rodents. J. Vis. Exp. 2010, 2010, e2033. [Google Scholar] [CrossRef] [Green Version]
- Bartling, B.; Hofmann, H.-S.; Weigle, B.; Silber, R.-E.; Simm, A. Down-regulation of the receptor for advanced glycation end-products (RAGE) supports non-small cell lung carcinoma. Carcinogenesis 2004, 26, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Abel, M.; Ritthaler, U.; Zhang, Y.; Deng, Y.; Schmidt, A.M.; Greten, J.; Sernau, T.; Wahl, P.; Andrassy, K.; Ritz, E. Expression of receptors for advanced glycosylated end-products in renal disease. Nephrol. Dial. Transplant. 1995, 10, 1662–1667. [Google Scholar] [PubMed]
- Hamilton, G.; Rath, B. Circulating tumor cell interactions with macrophages: Implications for biology and treatment. Transl. Lung Cancer Res. 2017, 6, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Carstens, R.P. Alternative splicing regulates distinct subcellular localization of Epithelial splicing regulatory protein 1 (Esrp1) isoforms. Sci. Rep. 2017, 7, 3848. [Google Scholar] [CrossRef] [PubMed]
- Constien, R.; Forde, A.; Liliensiek, B.; Gröne, H.-J.; Nawroth, P.; Hämmerling, G.; Arnold, B. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis 2001, 30, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Satar, N.A.; Fakiruddin, K.S.; Lim, M.N.; Mok, P.L.; Zakaria, N.; Fakharuzi, N.A.; Rahman, A.Z.A.; Zakaria, Z.; Yahaya, B.H.; Baharuddin, P. Novel triple-positive markers identified in human non-small cell lung cancer cell line with chemotherapy-resistant and putative cancer stem cell characteristics. Oncol. Rep. 2018, 40, 669–681. [Google Scholar] [CrossRef] [Green Version]
- Barrandon, Y.; Green, H. Three clonal types of keratinocyte with different capacities for multiplication. Proc. Natl. Acad. Sci. USA 1987, 84, 2302–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trotter, K.W.; Fraser, I.D.C.; Scott, G.K.; Stutts, M.J.; Scott, J.D.; Milgram, S.L. Alternative Splicing Regulates the Subcellular Localization of a-Kinase Anchoring Protein 18 Isoforms. J. Cell Biol. 1999, 147, 1481–1492. [Google Scholar] [CrossRef]
- Peng, Y.; Horwitz, N.; Lakatta, E.; Lin, L. Mouse RAGE Variant 4 Is a Dominant Membrane Receptor that Does Not Shed to Generate Soluble RAGE. PLoS ONE 2016, 11, e0153657. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Smith, E.A. Diaphanous-1 affects the nanoscale clustering and lateral diffusion of receptor for advanced glycation end-products (RAGE). Biochim. Biophys. Acta Biomembr. 2019, 1861, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and their role in Cancer and Inflammation. J. Transl. Med. 2009, 7, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierhaus, A.; Haslbeck, K.-M.; Humpert, P.M.; Liliensiek, B.; Dehmer, T.; Morcos, M.; Sayed, A.A.; Andrassy, M.; Schiekofer, S.; Schneider, J.G.; et al. Loss of pain perception in diabetes is dependent on a receptor of the immunoglobulin superfamily. J. Clin. Investig. 2004, 114, 1741–1751. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, T.; Bierhaus, A.; Nawroth, P.P. RAGE (receptor for advanced glycation end products): A central player in the in-flammatory response. Microbes Infect. 2004, 6, 1219–1225. [Google Scholar] [CrossRef]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Gröne, H.-J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Rao, X.; Wang, Y.; Feng, W.; Liang, H.; Liu, Y. Roles of alternative splicing in modulating transcriptional regulation. BMC Syst. Biol. 2017, 11, 89. [Google Scholar] [CrossRef] [Green Version]
- Eblen, S.T. Regulation of chemoresistance via alternative messenger RNA splicing. Biochem. Pharmacol. 2012, 83, 1063–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollig, A.; Miksicek, R.J. An estrogen receptor-alpha splicing variant mediates both positive and negative effects on gene tran-scription. Mol. Endocrinol. 2000, 14, 634–649. [Google Scholar]
- Berge, E.O.; Staalesen, V.; Straume, A.H.; Lillehaug, J.R.; Lønning, P.E. Chk2 splice variants express a dominant-negative effect on the wild-type Chk2 kinase activity. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1803, 386–395. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, Y.; Yu, W.; Ma, L.; Ji, X.; Xiao, W. Expression of the receptor for advanced glycation end-products and frequency of polymorphism in lung cancer. Oncol. Lett. 2015, 10, 51–60. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef]
- Akisik, E.; Yazici, H.; Dalay, N. ARLTS1, MDM2 and RAD51 gene variations are associated with familial breast cancer. Mol. Biol. Rep. 2010, 38, 343–348. [Google Scholar] [CrossRef]
- Nogueira, A.; Assis, J.; Catarino, R.; Medeiros, R. DNA repair and cytotoxic drugs: The potential role of RAD51 in clinical outcome of non-small-cell lung cancer patients. Pharmacogenomics 2013, 14, 689–700. [Google Scholar] [CrossRef]
- Thakur, S.; Zhang, H.B.; Peng, Y.; Le, H.; Carroll, B.T.; Ward, T.; Yao, J.; Farid, L.M.; Couch, F.J.; Wilson, R.B.; et al. Localization of BRCA1 and a splice variant identifies the nuclear localization signal. Mol. Cell. Biol. 1997, 17, 444–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieche, I.; Lidereau, R. Increased level of exon 12 alternatively spliced BRCA2 transcripts in tumor breast tissue compared with normal tissue. Cancer Res. 1999, 59, 2546–2550. [Google Scholar]
- Deb, S.P. Function and dysfunction of the human oncoprotein MDM2. Front Biosci. 2002, 7, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Stevens, M.; Oltean, S. Modulation of the Apoptosis Gene Bcl-x Function Through Alternative Splicing. Front. Genet. 2019, 10, 804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Y.; Tang, H.; Havlioglu, N. Alternative pre-mRNA splicing and regulation of programmed cell death. Prog. Mol. Subcell. Biol. 2003, 31, 153–185. [Google Scholar]
- Syken, J.; De-Medina, T.; Munger, K. TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mito-chondrial modulators of apoptosis with opposing functions. Proc. Natl. Acad. Sci. USA 1999, 96, 8499–8504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuszynska, I.; Magnus, M.; Jonak, K.; Dawson, W.; Bujnicki, J.M. NPDock: A web server for protein–nucleic acid docking. Nucleic Acids Res. 2015, 43, W425–W430. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Zhang, D.; Zhou, P.; Li, B.; Huang, S.-Y. HDOCK: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017, 45, W365–W373. [Google Scholar] [CrossRef] [PubMed]
- Heo, L.; Feig, M. PREFMD: A web server for protein structure refinement via molecular dynamics simulations. Bioinformatics 2018, 34, 1063–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anthropometric Variables | Control (n = 20) | SCLC (n = 80) | ||
|---|---|---|---|---|
| Male | 12 | 63 | ||
| Female | 8 | 17 | ||
| Control | SCLC Stage-I | SCLC Stage-II | SCLC Stage-III | |
| Total | 20 | 22 | 40 | 18 |
| Male | 12 | 16 | 29 | 18 |
| Female | 8 | 6 | 11 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madhavan, B.K.; Han, Z.; Singh, B.; Bordt, N.; Kaymak, S.; Bandapalli, O.R.; Kihm, L.; Shahzad, K.; Isermann, B.; Herzig, S.; et al. Elevated Expression of the RAGE Variant-V in SCLC Mitigates the Effect of Chemotherapeutic Drugs. Cancers 2021, 13, 2843. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112843
Madhavan BK, Han Z, Singh B, Bordt N, Kaymak S, Bandapalli OR, Kihm L, Shahzad K, Isermann B, Herzig S, et al. Elevated Expression of the RAGE Variant-V in SCLC Mitigates the Effect of Chemotherapeutic Drugs. Cancers. 2021; 13(11):2843. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112843
Chicago/Turabian StyleMadhavan, Bindhu K., Zhe Han, Bishal Singh, Nico Bordt, Serap Kaymak, Obul Reddy Bandapalli, Lars Kihm, Khurrum Shahzad, Berend Isermann, Stephan Herzig, and et al. 2021. "Elevated Expression of the RAGE Variant-V in SCLC Mitigates the Effect of Chemotherapeutic Drugs" Cancers 13, no. 11: 2843. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13112843