
Telomerase and Pluripotency Factors Jointly Regulate Stemness in Pancreatic Cancer Stem Cells

 , , , , , , , , , ,
, , , , , , , , , ,  and add
Show full author list
and add
Show full author list
Abstract
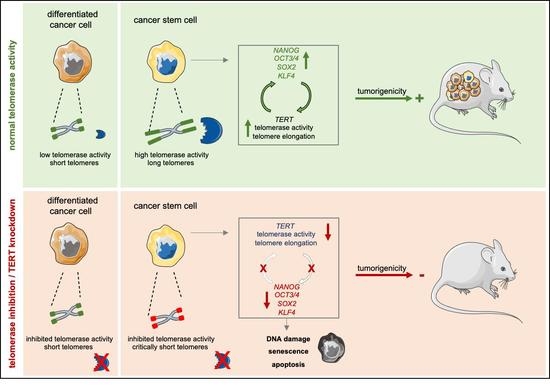
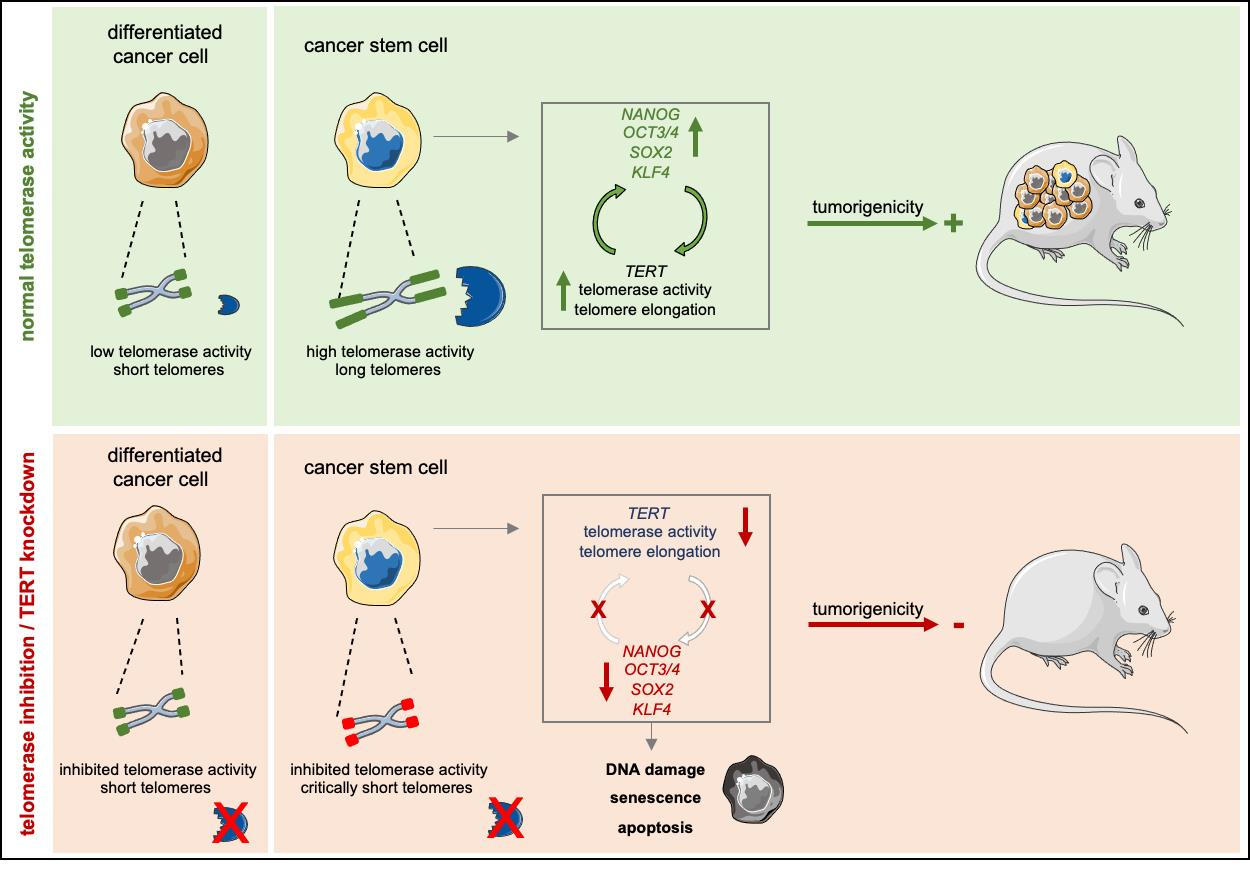
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice, Transplantation and Treatment
2.2. Primary Pancreatic Cancer Cells and Other Cell Lines
2.3. CSC Enrichment
2.3.1. CD133 and ALDH Fluorescence Activated Cell Sorting (FACS)
2.3.2. Sphere Culture
2.4. Pseudorabies Virus Infections
2.5. Telomerase Activity
2.6. Organoid Culture
2.7. Plasmids, Infection and Transfection
2.8. Flow Cytometry
2.9. Q-FISH
2.10. Immunofluorescence
2.11. RNA Isolation and qRT-PCR
2.12. MTT Assay
2.13. Statistical Analysis
3. Results
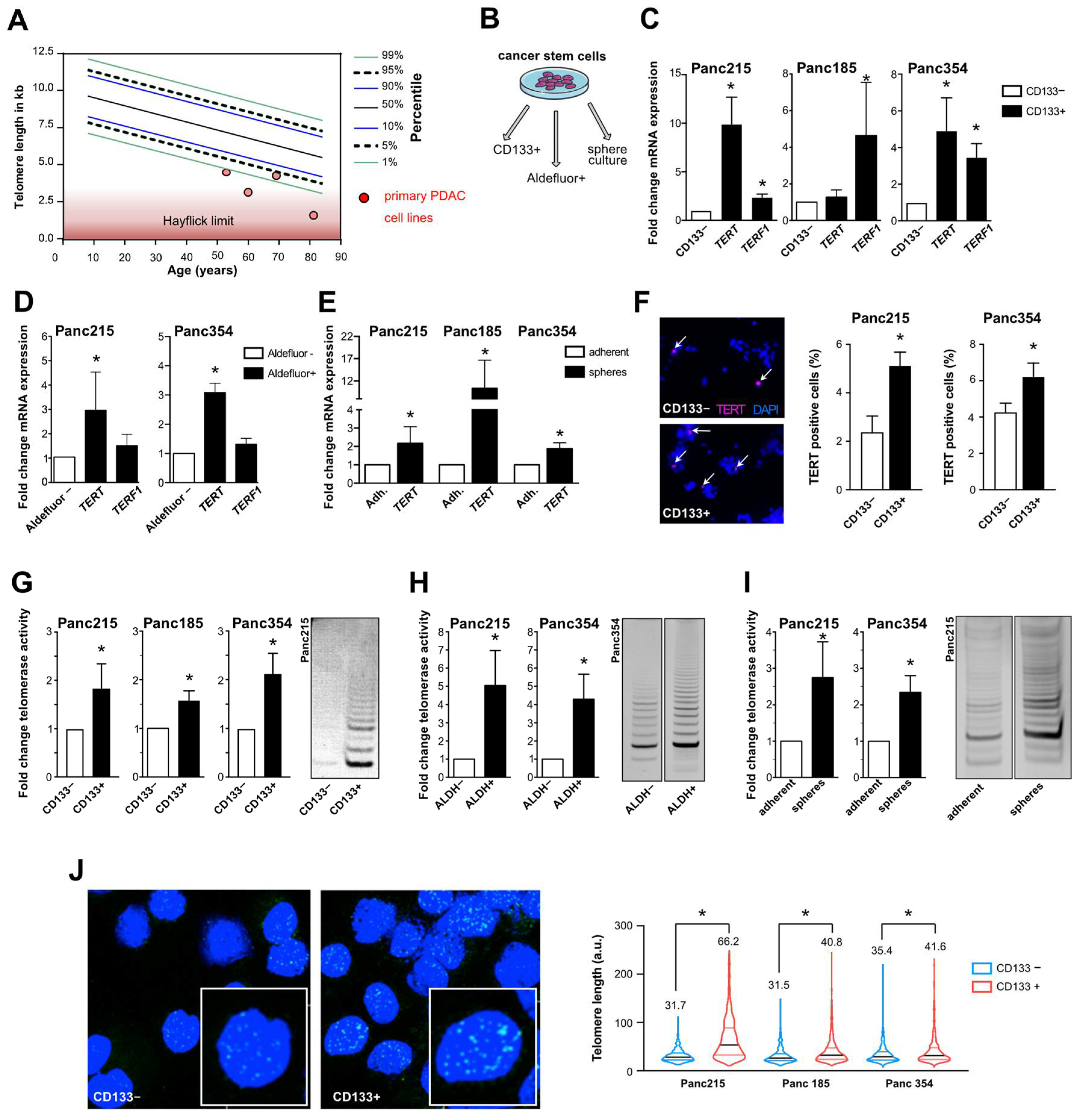
3.1. Telomerase Activity and Telomere Length in Pancreatic CSCs vs. Bulk Tumor Cells
3.2. A Positive Feedback Loop between Stemness Factors and Telomerase Maintains CSC Phenotype
3.3. Telomerase Inhibition Causes DNA Damage and Apoptosis in CSCs
3.4. CSCs Are Depleted upon Telomerase Inhibition
3.5. Telomerase Inhibition as Novel Treatment Strategy for Pancreatic Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2015. CA Cancer J. Clin. 2015, 65, 5–29. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.; Dick, J.; Dirks, P.; Eaves, C.; Jamieson, C.; Jones, D.; Visvader, J.; Weissman, I.; Wahl, G. Cancer Stem Cells—Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.M. Telomerase Catalytic Subunit Homologs from Fission Yeast and Human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef]
- DeLange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [Green Version]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, J.W.; Wright, W.E. Telomeres and telomerase: Three decades of progress. Nat. Rev. Genet. 2019, 20, 299–309. [Google Scholar] [CrossRef]
- Damm, K.; Hemmann, U.; Garin-Chesa, P.; Hauel, N.; Kauffmann, I.; Priepke, H.; Niestroj, C.; Daiber, C.; Enenkel, B.; Guilliard, B.; et al. A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 2001, 20, 6958–6968. [Google Scholar] [CrossRef]
- Hu, Y.; Smyth, G.K. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 2009, 347, 70–78. [Google Scholar] [CrossRef]
- Mueller, M.T.; Hermann, P.C.; Witthauer, J.; Rubio-Viqueira, B.; Leicht, S.F.; Huber, S.; Ellwart, J.W.; Mustafa, M.; Bartenstein, P.; D’Haese, J.G.; et al. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology 2009, 137, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Lerma, L.; Alcala, S.; Piñero, C.; Torres, M.; Martin, B.; Lim, F.; Sainz, B.; Tabarés, E. Expression of the immediate early IE180 protein under the control of the hTERT and CEA tumor-specific promoters in recombinant pseudorabies viruses: Effects of IE180 protein on promoter activity and apoptosis induction. Virology 2016, 488, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotta, A.; Cheung, A.Y.; Farra, N.; Vijayaragavan, K.; Séguin, C.A.; Draper, J.S.; Pasceri, P.; Maksakova, I.A.; Mager, D.L.; Rossant, J.; et al. Isolation of human iPS cells using EOS lentiviral vectors to select for pluripotency. Nat. Methods 2009, 6, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Hummel, S.; Ferreira, M.S.V.; Heudobler, D.; Huber, E.; Fahrenkamp, D.; Gremse, F.; Schmid, K.; Muller-Newen, G.; Ziegler, P.; Jost, E.; et al. Telomere shortening in enterocytes of patients with uncontrolled acute intestinal graft-versus-host disease. Blood 2015, 126, 2518–2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, A.; Nakano, M.; Saito, K.; Haruno, R.; Watanabe, T.M.; Ohyanagi, T.; Jin, T.; Okada, Y.; Nagai, T. Expanded palette of Nano-lanterns for real-time multicolor luminescence imaging. Proc. Natl. Acad. Sci. USA 2015, 112, 4352–4356. [Google Scholar] [CrossRef] [Green Version]
- Vinagre, J.; Almeida, A.; Populo, H.; Batista, R.; Lyra, J.; Pinto, V.; Coelho, R.; Celestino, R.; Prazeres, H.; Lima, L.; et al. Frequency of TERT promoter mutations in human cancers. Nat. Commun. 2013, 4, 2185. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.H.; Chen, Y.T.; Lee, Y.H.; Lu, J.; Chien, C.L.; Chen, H.F.; Ho, H.N.; Yu, C.J.; Wang, Z.Q.; Teng, S.C. PARP1 controls KLF4-mediated telomerase expression in stem cells and cancer cells. Nucleic Acids Res. 2017, 45, 10492–10503. [Google Scholar] [CrossRef] [PubMed]
- Celeghin, A.; Giunco, S.; Freguja, R.; Zangrossi, M.; Nalio, S.; Dolcetti, R.; Rossi, A.D. Short-term inhibition of TERT induces telomere length-independent cell cycle arrest and apoptotic response in EBV-immortalized and transformed B cells. Cell Death Dis. 2016, 7, e2562. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, E.; Hermann, P.C.; Mueller, M.-T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Frappart, P.O.; Walter, K.; Gout, J.; Beutel, A.K.; Morawe, M.; Arnold, F.; Breunig, M.; Barth, T.F.; Marienfeld, R.; Schulte, L.; et al. Pancreatic cancer-derived organoids—A disease modeling tool to predict drug response. United Eur. Gastroenterol. J. 2020, 8, 594–606. [Google Scholar] [CrossRef] [Green Version]
- Beier, F.; Beier, C.P.; Aschenbrenner, I.; Hildebrandt, G.C.; Brummendorf, T.H.; Beier, D. Identification of CD133(-)/telomerase(low) progenitor cells in glioblastoma-derived cancer stem cell lines. Cell Mol. Neurobiol. 2011, 31, 337–343. [Google Scholar] [CrossRef]
- Marian, C.O.; Wright, W.E.; Shay, J.W. The effects of telomerase inhibition on prostate tumor-initiating cells. Int. J. Cancer 2010, 127, 321–331. [Google Scholar] [CrossRef]
- Joseph, I.; Tressler, R.; Bassett, E.; Harley, C.; Buseman, C.M.; Pattamatta, P.; Wright, W.E.; Shay, J.W.; Go, N.F. The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res. 2010, 70, 9494–9504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Daly, H.; Kull, M.; Zimmermann, S.; Pantic, M.; Waller, C.F.; Martens, U.M. Selective cytotoxicity and telomere damage in leukemia cells using the telomerase inhibitor BIBR1532. Blood 2005, 105, 1742–1749. [Google Scholar] [CrossRef]
- Sexton, A.N.; Regalado, S.G.; Lai, C.S.; Cost, G.J.; O’Neil, C.M.; Urnov, F.D.; Gregory, P.D.; Jaenisch, R.; Collins, K.; Hockemeyer, D. Genetic and molecular identification of three human TPP1 functions in telomerase action: Recruitment, activation, and homeostasis set point regulation. Genes Dev. 2014, 28, 1885–1899. [Google Scholar] [CrossRef] [Green Version]
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA Damage Foci at Dysfunctional Telomeres. Curr. Biol. 2003, 13, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Brassat, U.; Balabanov, S.; Bali, D.; Dierlamm, J.; Braig, M.; Hartmann, U.; Sirma, H.; Gunes, C.; Wege, H.; Fehse, B.; et al. Functional p53 is required for effective execution of telomerase inhibition in BCR-ABL-positive CML cells. Exp. Hematol. 2011, 39, 66–76. [Google Scholar] [CrossRef]
- Gallmeier, E.; Hermann, P.C.; Mueller, M.T.; Machado, J.G.; Ziesch, A.; De Toni, E.N.; Palagyi, A.; Eisen, C.; Ellwart, J.W.; Rivera, J.; et al. Inhibition of ataxia telangiectasia- and Rad3-related function abrogates the in vitro and in vivo tumorigenicity of human colon cancer cells through depletion of the CD133(+) tumor-initiating cell fraction. Stem Cells 2011, 29, 418–429. [Google Scholar] [CrossRef]
- Liu, C.C.; Ma, D.L.; Yan, T.D.; Fan, X.; Poon, Z.; Poon, L.F.; Goh, S.A.; Rozen, S.G.; Hwang, W.Y.; Tergaonkar, V.; et al. Distinct Responses of Stem Cells to Telomere Uncapping-A Potential Strategy to Improve the Safety of Cell Therapy. Stem Cells 2016, 34, 2471–2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, L.G.; Swartz, M.A. Capturing complex 3D tissue physiology in vitro. Nat. Rev. Mol. Cell. Biol. 2006, 7, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, L. Linking Telomere Regulation to Stem Cell Pluripotency. Trends Genet. 2017, 33, 16–33. [Google Scholar] [CrossRef]
- Yang, C.; Przyborski, S.; Cooke, M.J.; Zhang, X.; Stewart, R.; Anyfantis, G.; Atkinson, S.P.; Saretzki, G.; Armstrong, L.; Lako, M. A key role for telomerase reverse transcriptase unit in modulating human embryonic stem cell proliferation, cell cycle dynamics, and in vitro differentiation. Stem Cells 2008, 26, 850–863. [Google Scholar] [CrossRef]
- Hoffmeyer, K.; Raggioli, A.; Rudloff, S.; Anton, R.; Hierholzer, A.; Del Valle, I.; Hein, K.; Vogt, R.; Kemler, R. Wnt/beta-catenin signaling regulates telomerase in stem cells and cancer cells. Science 2012, 336, 1549–1554. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.W.; Hou, P.S.; Tseng, S.F.; Chien, C.L.; Wu, K.J.; Chen, H.F.; Ho, H.N.; Kyo, S.; Teng, S.C. Kruppel-like transcription factor 4 contributes to maintenance of telomerase activity in stem cells. Stem Cells 2010, 28, 1510–1517. [Google Scholar] [CrossRef]
- Chan, K.K.; Zhang, J.; Chia, N.Y.; Chan, Y.S.; Sim, H.S.; Tan, K.S.; Oh, S.K.; Ng, H.H.; Choo, A.B. KLF4 and PBX1 directly regulate NANOG expression in human embryonic stem cells. Stem Cells 2009, 27, 2114–2125. [Google Scholar] [CrossRef]
- Nakatake, Y.; Fukui, N.; Iwamatsu, Y.; Masui, S.; Takahashi, K.; Yagi, R.; Yagi, K.; Miyazaki, J.; Matoba, R.; Ko, M.S.; et al. Klf4 cooperates with Oct3/4 and Sox2 to activate the Lefty1 core promoter in embryonic stem cells. Mol. Cell. Biol. 2006, 26, 7772–7782. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.H.; Li, S.Z.; Zhang, Z.Y.; Hu, X.M.; Hou, P.N.; Gao, L.; Du, R.L.; Zhang, X.D. Nemo-like kinase is critical for p53 stabilization and function in response to DNA damage. Cell Death Differ. 2014, 21, 1656–1663. [Google Scholar] [CrossRef] [Green Version]
- Chin, L.; Artandi, S.E.; Shen, Q.; Tam, A.; Lee, S.L.; Gottlieb, G.J.; Greider, C.W.; DePinho, R.A. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999, 97, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.; Ludlow, A.T.; Min, J.; Robin, J.D.; Stadler, G.; Mender, I.; Lai, T.P.; Zhang, N.; Wright, W.E.; Shay, J.W. Regulation of the Human Telomerase Gene TERT by Telomere Position Effect-Over Long Distances (TPE-OLD): Implications for Aging and Cancer. PLoS Biol. 2016, 14, e2000016. [Google Scholar] [CrossRef] [Green Version]
- Bechter, O.E.; Eisterer, W.; Dlaska, M.; Kuhr, T.; Thaler, J. CpG island methylation of the hTERT promoter is associated with lower telomerase activity in B-cell lymphocytic leukemia. Exp. Hematol. 2002, 30, 26–33. [Google Scholar] [CrossRef]
- Guilleret, I.; Yan, P.; Grange, F.; Braunschweig, R.; Bosman, F.T.; Benhattar, J. Hypermethylation of the human telomerase catalytic subunit (hTERT) gene correlates with telomerase activity. Int. J. Cancer 2002, 101, 335–341. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walter, K.; Rodriguez-Aznar, E.; Ferreira, M.S.V.; Frappart, P.-O.; Dittrich, T.; Tiwary, K.; Meessen, S.; Lerma, L.; Daiss, N.; Schulte, L.-A.; et al. Telomerase and Pluripotency Factors Jointly Regulate Stemness in Pancreatic Cancer Stem Cells. Cancers 2021, 13, 3145. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133145
Walter K, Rodriguez-Aznar E, Ferreira MSV, Frappart P-O, Dittrich T, Tiwary K, Meessen S, Lerma L, Daiss N, Schulte L-A, et al. Telomerase and Pluripotency Factors Jointly Regulate Stemness in Pancreatic Cancer Stem Cells. Cancers. 2021; 13(13):3145. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133145
Chicago/Turabian StyleWalter, Karolin, Eva Rodriguez-Aznar, Monica S. Ventura Ferreira, Pierre-Olivier Frappart, Tabea Dittrich, Kanishka Tiwary, Sabine Meessen, Laura Lerma, Nora Daiss, Lucas-Alexander Schulte, and et al. 2021. "Telomerase and Pluripotency Factors Jointly Regulate Stemness in Pancreatic Cancer Stem Cells" Cancers 13, no. 13: 3145. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133145