
Clinical and Molecular Heterogeneity in Patients with Innate Resistance to Anti-PD-1 +/− Anti-CTLA-4 Immunotherapy in Metastatic Melanoma Reveals Distinct Therapeutic Targets


 , and
, and
Abstract
:Simple Summary
Abstract
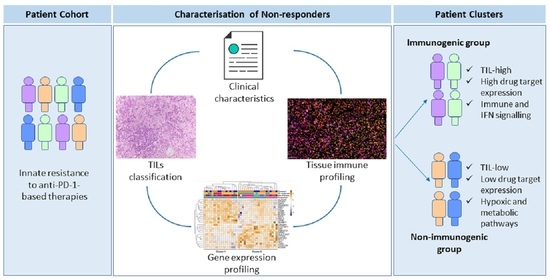
1. Introduction
2. Materials and Methods
2.1. Patient and Sample Selection
2.2. Assessment of TILs Based on Haematoxylin and Eosin (H&E) Staining
2.3. Targeted RNA Sequencing Library Preparation and Sequencing
2.4. Targeted Gene Expression Analysis
2.5. Hierarchical Clustering Analysis of Gene Expression Profiles
2.6. Gene Set Enrichment Analysis (GSEA)
2.7. Multiplex Immunohistochemistry (mIHC)
2.8. Multispectral Image Analysis
2.9. Statistical Analysis
3. Results
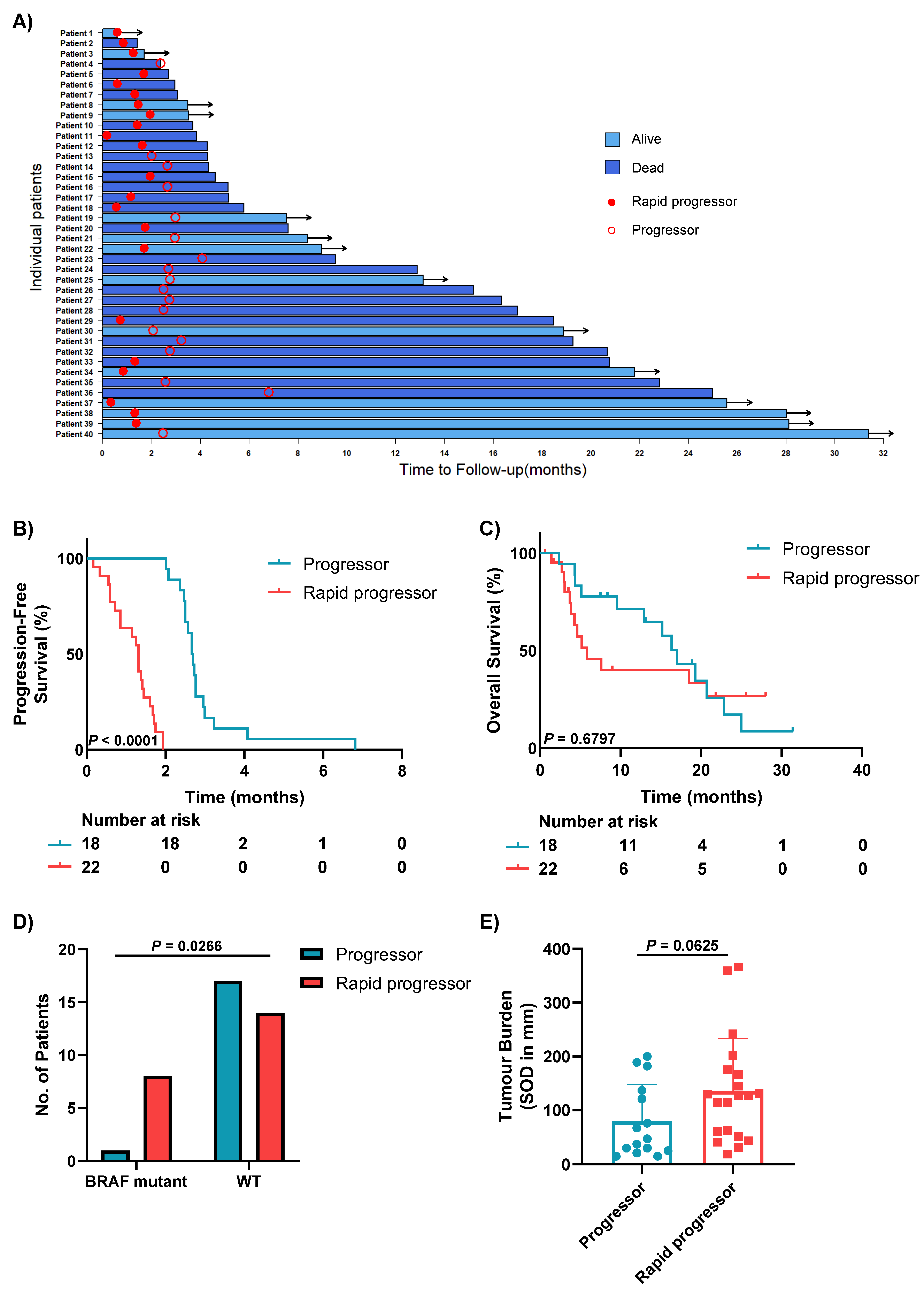
3.1. Patient Characteristics
3.2. Clinical Characteristics of Patients with Different Patterns of Progression
3.3. Rapid Progressors Have Lower Expression of TILs and CD68+ Macrophages Compared to Patients Who Progressed after 2 Months
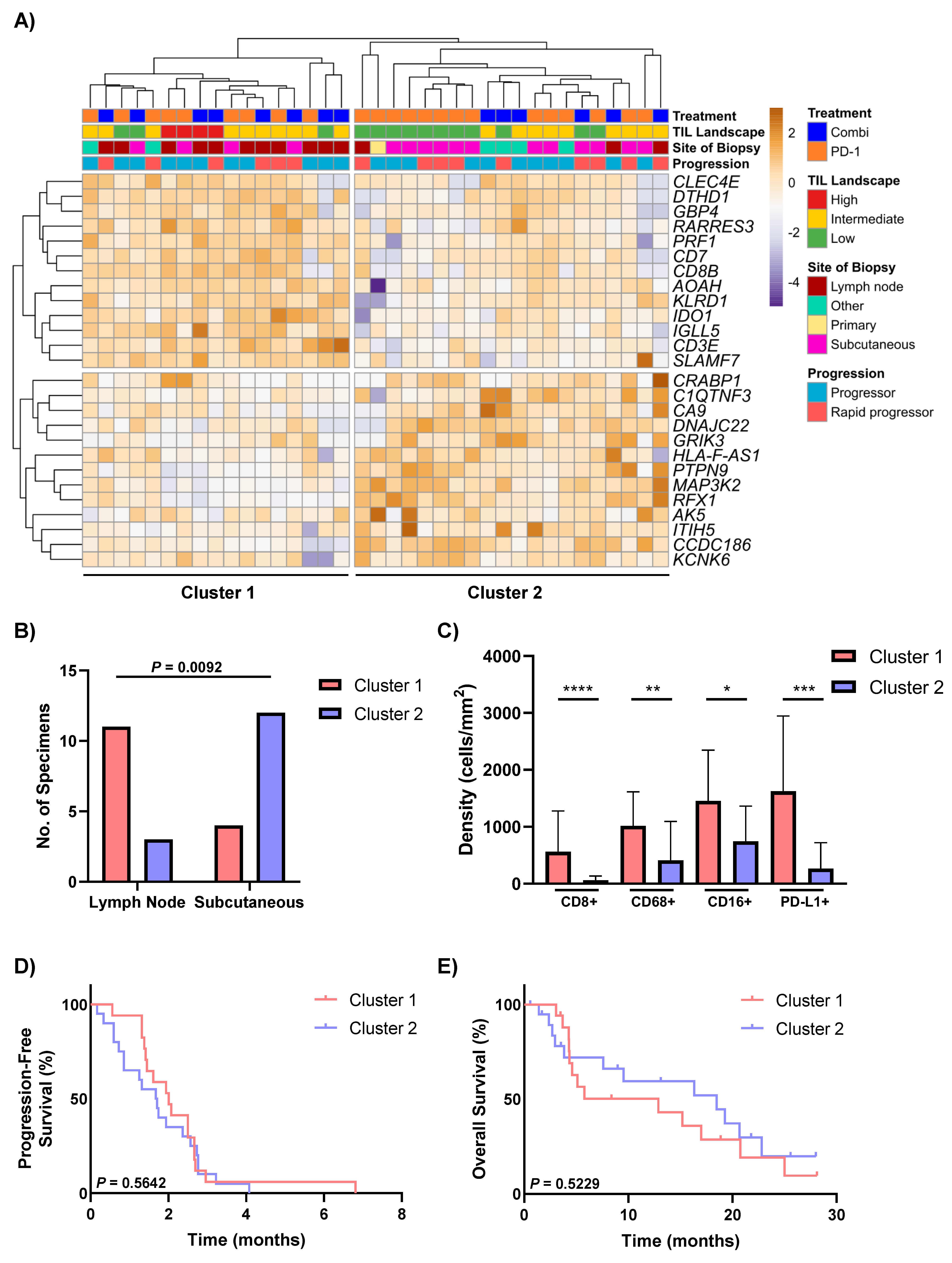
3.4. Differential Expression Analysis Identifies Two Distinct Groups of Non-Responders to Anti-PD-1-Based Therapies
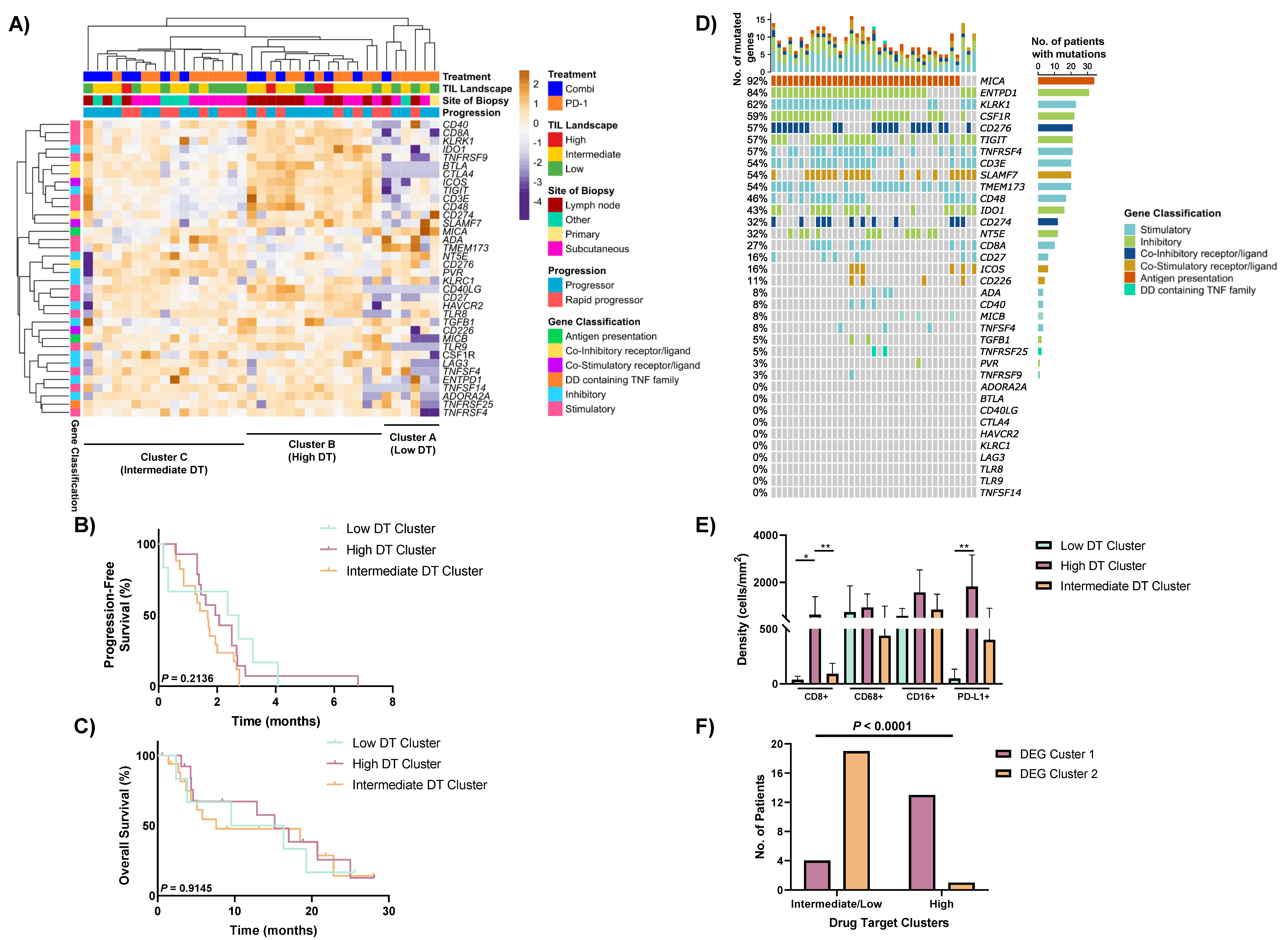
3.5. Heterogeneous Expression of Immune Drug Targets within Non-Responders to Anti-PD-1-Based Therapies
3.6. Drug Target Profiles of Non-Responders Are Associated with TIL Expression and the DEG Clusters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hodi, F.S.; Kluger, H.; Sznol, M.; Carvajal, R.; Lawrence, D.; Atkins, M.; Powderly, J.; Sharfman, W.; Puzanov, I.; Smith, D.; et al. Abstract CT001: Durable, long-term survival in previously treated patients with advanced melanoma (MEL) who received nivolumab (NIVO) monotherapy in a phase I trial. Cancer Res. 2016, 76, CT001-001. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Ferrucci, P.F.; Smylie, M.; et al. Abstract CT075: Overall survival (OS) results from a phase III trial of nivolumab (NIVO) combined with ipilimumab (IPI) in treatment-naïve patients with advanced melanoma (CheckMate 067). Cancer Res. 2017, 77, CT075. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Carlino, M.S.; Long, G.V. Ipilimumab Combined with Nivolumab: A Standard of Care for the Treatment of Advanced Melanoma? Clin. Cancer Res. 2016, 22, 3992–3998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. 2017, 24, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluger, H.M.; Tawbi, H.A.; Ascierto, M.L.; Bowden, M.; Callahan, M.K.; Cha, E.; Chen, H.X.; Drake, C.G.; Feltquate, D.M.; Ferris, R.L.; et al. Defining tumor resistance to PD-1 pathway blockade: Recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J. Immunother. Cancer 2020, 8, e000398. [Google Scholar] [CrossRef] [Green Version]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [Green Version]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade–based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [Green Version]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Holmgaard, R.B.; Zamarin, D.; Munn, D.H.; Wolchok, J.D.; Allison, J.P. Indoleamine 2,3-dioxygenase is a critical resistance mechanism in antitumor T cell immunotherapy targeting CTLA-4. J. Exp. Med. 2013, 210, 1389–1402. [Google Scholar] [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016, 7, 10501. [Google Scholar] [CrossRef]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, J.; Wilmott, J.S.; Madore, J.; Gide, T.N.; Quek, C.; Tasker, A.; Ferguson, A.L.; Chen, J.; Hewavisenti, R.; Hersey, P.; et al. CD103+ tumor-resident CD8+ T cells are associated with improved survival in immunotherapy naive melanoma patients and expand significantly during anti-PD1 treatment. Clin. Cancer Res. 2018, 24, 3036–3045. [Google Scholar] [CrossRef] [Green Version]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255e236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenhauer, E.A.; Therasse, P.; Bogaerts, J.; Schwartz, L.H.; Sargent, D.; Ford, R.; Dancey, J.; Arbuck, S.; Gwyther, S.; Mooney, M.; et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 2009, 45, 228–247. [Google Scholar] [CrossRef]
- Azimi, F.; Scolyer, R.A.; Rumcheva, P.; Moncrieff, M.; Murali, R.; McCarthy, S.W.; Saw, R.P.; Thompson, J.F. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J. Clin. Oncol. 2012, 30, 2678–2683. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gershenwald, J.E.; Scolyer, R.A. Melanoma Staging: American Joint Committee on Cancer (AJCC) 8th Edition and Beyond. Ann. Surg. Oncol. 2018, 25, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.-C.; et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clin. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, R.; Mezquita, L.; Texier, M.; Lahmar, J.; Audigier-Valette, C.; Tessonnier, L.; Mazieres, J.; Zalcman, G.; Brosseau, S.; Le Moulec, S.; et al. Hyperprogressive Disease in Patients With Advanced Non-Small Cell Lung Cancer Treated With PD-1/PD-L1 Inhibitors or With Single-Agent Chemotherapy. JAMA Oncol. 2018, 4, 1543–1552. [Google Scholar] [CrossRef]
- Kanjanapan, Y.; Day, D.; Wang, L.; Al-Sawaihey, H.; Abbas, E.; Namini, A.; Siu, L.L.; Hansen, A.; Razak, A.A.; Spreafico, A.; et al. Hyperprogressive disease in early-phase immunotherapy trials: Clinical predictors and association with immune-related toxicities. Cancer 2019, 125, 1341–1349. [Google Scholar] [CrossRef]
- Saâda-Bouzid, E.; Defaucheux, C.; Karabajakian, A.; Coloma, V.P.; Servois, V.; Paoletti, X.; Even, C.; Fayette, J.; Guigay, J.; Loirat, D.; et al. Hyperprogression during anti-PD-1/PD-L1 therapy in patients with recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2017, 28, 1605–1611. [Google Scholar] [CrossRef]
- Joshi, K.; Atwal, D.; Ravilla, R.; Pandey, Y.; Yarlagadda, N.; Kakadia, S.; Makhoul, I.; Hutchins, L.; Mahmoud, F. Immunotherapy Outcomes in Advanced Melanoma in Relation to Age. Perm. J. 2020, 24, 19.093. [Google Scholar] [CrossRef]
- Pires da Silva, I.; Wang, K.Y.X.; Wilmott, J.S.; Holst, J.; Carlino, M.S.; Park, J.J.; Quek, C.; Wongchenko, M.; Yan, Y.; Mann, G.; et al. Distinct Molecular Profiles and Immunotherapy Treatment Outcomes of V600E and V600K BRAF-Mutant Melanoma. Clin Cancer Res. 2019, 25, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Zuazo-Ibarra, M.; Arasanz, H.; Fernández-Hinojal, G.; María, G.-C.; Hernández-Marín, B.; Martínez-Aguillo, M.; Jose Lecumberri, M.; Fernández, A.; Teijeira, L.; Vera, R.; et al. Highly differentiated CD4 T cells Unequivocally Identify Primary Resistance and Risk of Hyperprogression to PD-L1/PD-1 Immune Checkpoint Blockade in Lung Cancer. bioRxiv 2018, 320176. [Google Scholar] [CrossRef]
- Lo Russo, G.; Moro, M.; Sommariva, M.; Cancila, V.; Boeri, M.; Centonze, G.; Ferro, S.; Ganzinelli, M.; Gasparini, P.; Huber, V.; et al. Antibody-Fc/FcR Interaction on Macrophages as a Mechanism for Hyperprogressive Disease in Non-small Cell Lung Cancer Subsequent to PD-1/PD-L1 Blockade. Clin. Cancer Res. 2019, 25, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Daud, A.I.; Loo, K.; Pauli, M.L.; Sanchez-Rodriguez, R.; Sandoval, P.M.; Taravati, K.; Tsai, K.; Nosrati, A.; Nardo, L.; Alvarado, M.D.; et al. Tumor immune profiling predicts response to anti–PD-1 therapy in human melanoma. J. Clin. Investig. 2016, 126, 3447–3452. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Furness, A.J.S.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017, 171, 934.e915–949.e915. [Google Scholar] [CrossRef] [Green Version]
- Rizos, H.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Fung, C.; Hyman, J.; Haydu, L.E.; Mijatov, B.; Becker, T.M.; Boyd, S.C.; et al. BRAF inhibitor resistance mechanisms in metastatic melanoma: Spectrum and clinical impact. Clin. Cancer Res. 2014, 20, 1965–1977. [Google Scholar] [CrossRef] [Green Version]
- Wilmott, J.S.; Tembe, V.; Howle, J.R.; Sharma, R.; Thompson, J.F.; Rizos, H.; Lo, R.S.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Intratumoral molecular heterogeneity in a BRAF-mutant, BRAF inhibitor-resistant melanoma: A case illustrating the challenges for personalized medicine. Mol. Cancer Ther. 2012, 11, 2704–2708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, K.; Krijgsman, O.; Cornelissen-Steijger, P.; Shahrabi, A.; Weeber, F.; Song, J.Y.; Kuilman, T.; Vis, D.J.; Wessels, L.F.; Voest, E.E.; et al. Intra- and inter-tumor heterogeneity in a vemurafenib-resistant melanoma patient and derived xenografts. EMBO Mol. Med. 2015, 7, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Reuben, A.; Spencer, C.N.; Prieto, P.A.; Gopalakrishnan, V.; Reddy, S.M.; Miller, J.P.; Mao, X.; De Macedo, M.P.; Chen, J.; Song, X.; et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med. 2017, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Haqq, C.; Nosrati, M.; Sudilovsky, D.; Crothers, J.; Khodabakhsh, D.; Pulliam, B.L.; Federman, S.; Miller, J.R.; Allen, R.E.; Singer, M.I.; et al. The gene expression signatures of melanoma progression. Proc. Natl. Acad. Sci. USA 2005, 102, 6092. [Google Scholar] [CrossRef] [Green Version]
- Gide, T.N.; Allanson, B.M.; Menzies, A.M.; Ferguson, P.M.; Madore, J.; Saw, R.P.M.; Thompson, J.F.; Long, G.V.; Wilmott, J.S.; Scolyer, R.A. Inter- and intra-patient heterogeneity of indoleamine 2,3-dioxygenase expression in primary and metastatic melanoma cells and the tumor microenvironment. Histopathology 2018, 74, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Akhtar, R.; Birney, E.; Bower, L.; Cerdeno-Ta´rraga, A.; Cheng, Y.; Cleland, I.; Faruque, N.; Goodgame, N.; Gibson, R.; et al. The European Nucleotide Archive. Nucleic Acids Res. 2011, 39, D28–D31. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Patients, n (%) | 40 (100) |
|---|---|
| Age median (range) | 67 (34–96) |
| Sex, n (%) Male | 26 (65) |
| ECOG, n (%) >=1 | 19 (48) |
| Mutational Status, n (%) BRAF-Mutant V600E Other NRAS-Mutant WT 1 | 5 (13) 4 (10) 13 (33) 18 (45) |
| LDH, n (%) >ULN | 15 (38) |
| AJCC v8 2, n (%) IIIc/M1a/M1b M1c M1d | 16 (40) 12 (30) 12 (30) |
| Liver Metastases, n (%) | 11 (28) |
| Treatment, n (%) Anti-PD-1 Anti-CTLA-4 + Anti-PD-1 | 26 (65) 14 (35) |
| Toxicity, n (%) Any ≥G3 | 8 (20) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gide, T.N.; Pires da Silva, I.; Quek, C.; Ferguson, P.M.; Batten, M.; Shang, P.; Ahmed, T.; Menzies, A.M.; Carlino, M.S.; Saw, R.P.M.; et al. Clinical and Molecular Heterogeneity in Patients with Innate Resistance to Anti-PD-1 +/− Anti-CTLA-4 Immunotherapy in Metastatic Melanoma Reveals Distinct Therapeutic Targets. Cancers 2021, 13, 3186. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133186
Gide TN, Pires da Silva I, Quek C, Ferguson PM, Batten M, Shang P, Ahmed T, Menzies AM, Carlino MS, Saw RPM, et al. Clinical and Molecular Heterogeneity in Patients with Innate Resistance to Anti-PD-1 +/− Anti-CTLA-4 Immunotherapy in Metastatic Melanoma Reveals Distinct Therapeutic Targets. Cancers. 2021; 13(13):3186. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133186
Chicago/Turabian StyleGide, Tuba N., Inês Pires da Silva, Camelia Quek, Peter M. Ferguson, Marcel Batten, Ping Shang, Tasnia Ahmed, Alexander M. Menzies, Matteo S. Carlino, Robyn P. M. Saw, and et al. 2021. "Clinical and Molecular Heterogeneity in Patients with Innate Resistance to Anti-PD-1 +/− Anti-CTLA-4 Immunotherapy in Metastatic Melanoma Reveals Distinct Therapeutic Targets" Cancers 13, no. 13: 3186. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13133186