
Genomic Analysis of Response to Neoadjuvant Chemotherapy in Esophageal Adenocarcinoma

 ,
,  , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Overview of Patients and Sequencing Strategy
2.2. Whole-Genome Sequencing Analysis
2.3. Copy Number and Clonality Analysis
2.4. Genomic Instability Analysis
2.5. Mutational Signature and Neoantigen Analysis
2.6. Expression Profiling by Bulk RNA Sequencing (RNA-seq) and Gene Set Enrichment Analysis (GSEA)
2.7. Survival Analysis
2.8. DDR Pathway Deregulation Analysis
2.9. Classification of Genes Relevant for Genomics-Driven Therapy
2.10. Identification of Specific Mutations with Therapeutic Relevance
2.11. Statistics
3. Results
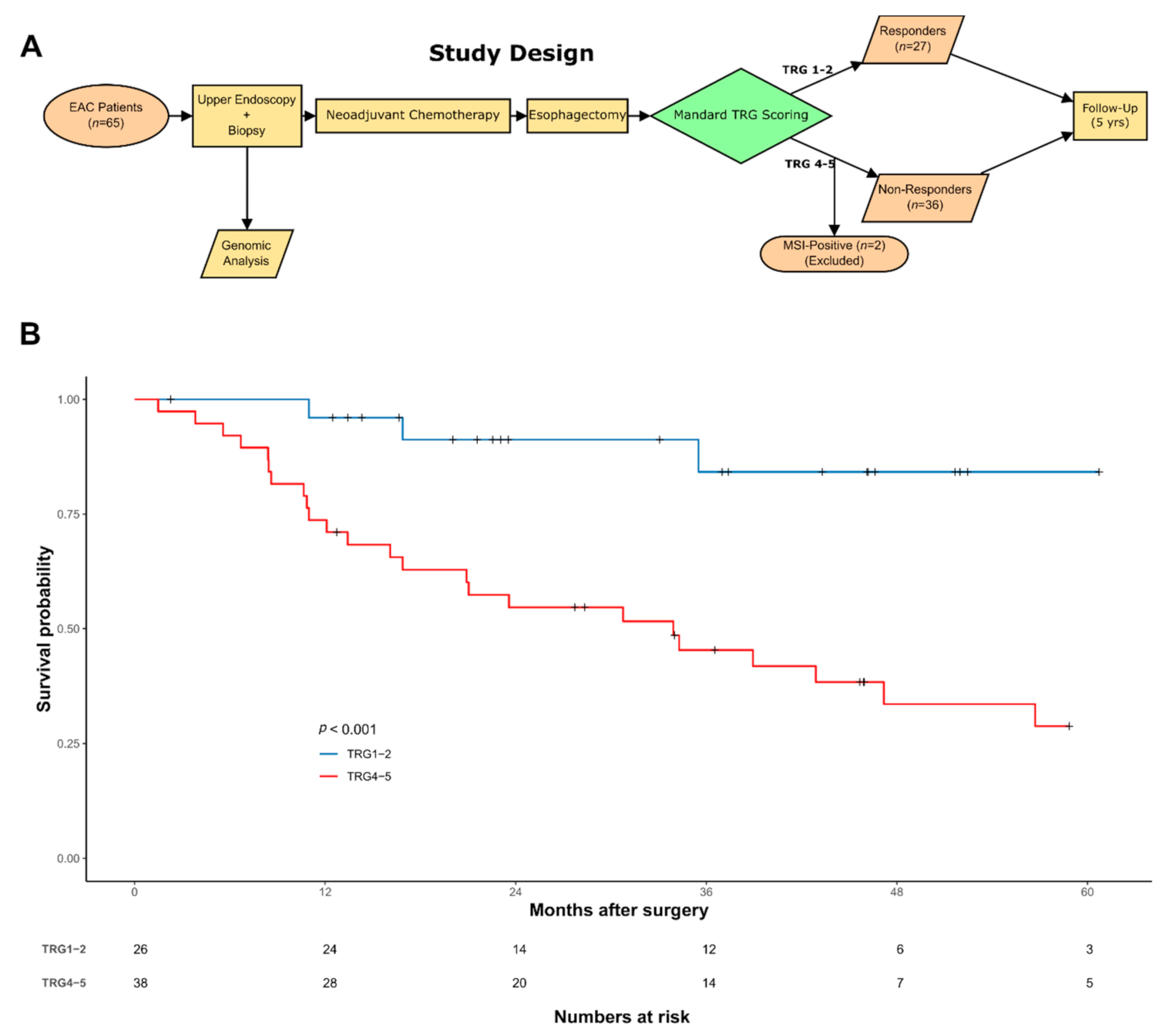
3.1. Patient Characteristics and Overall Study Design
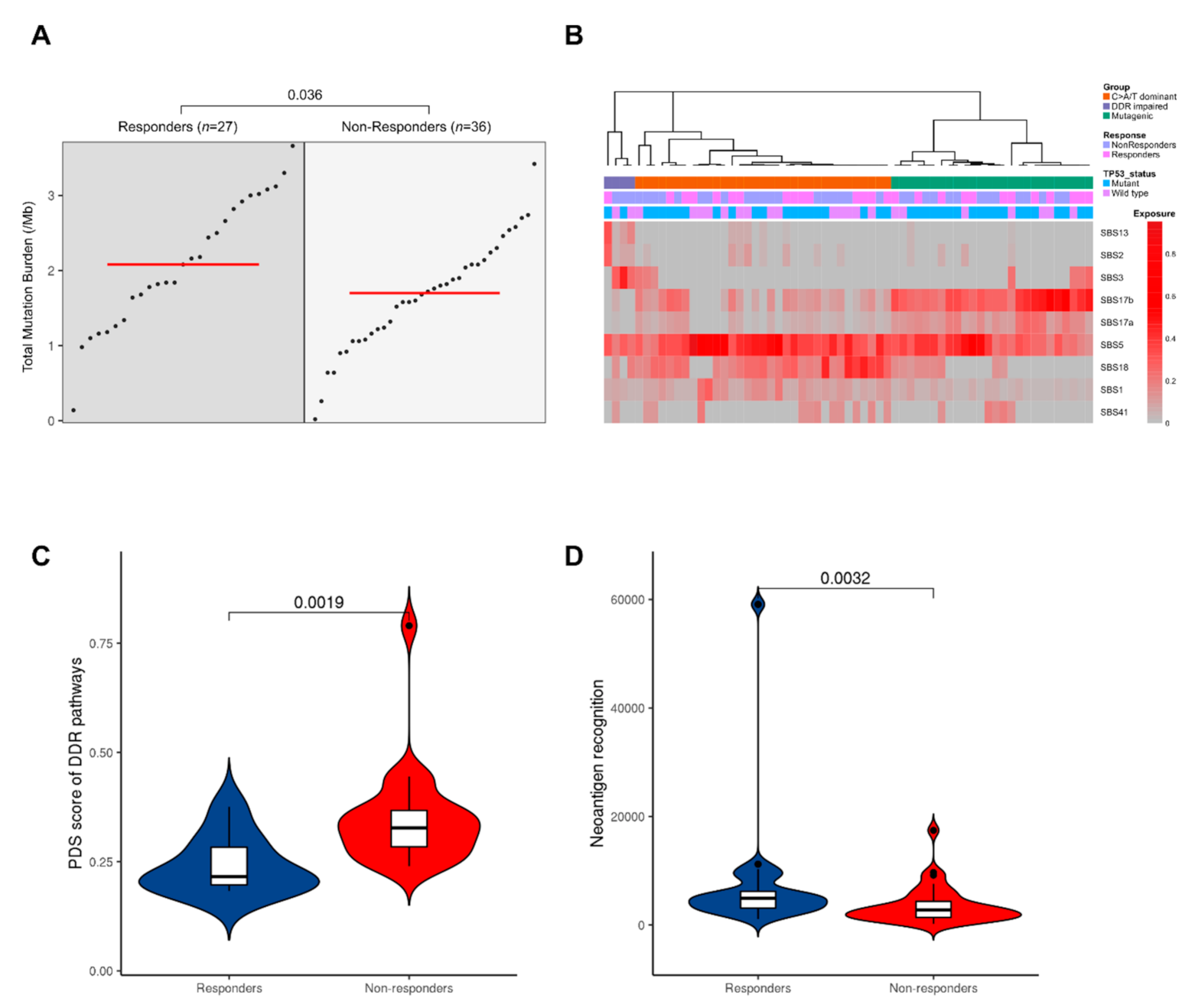
3.2. Mutational Profiles Associated with Response to NAC
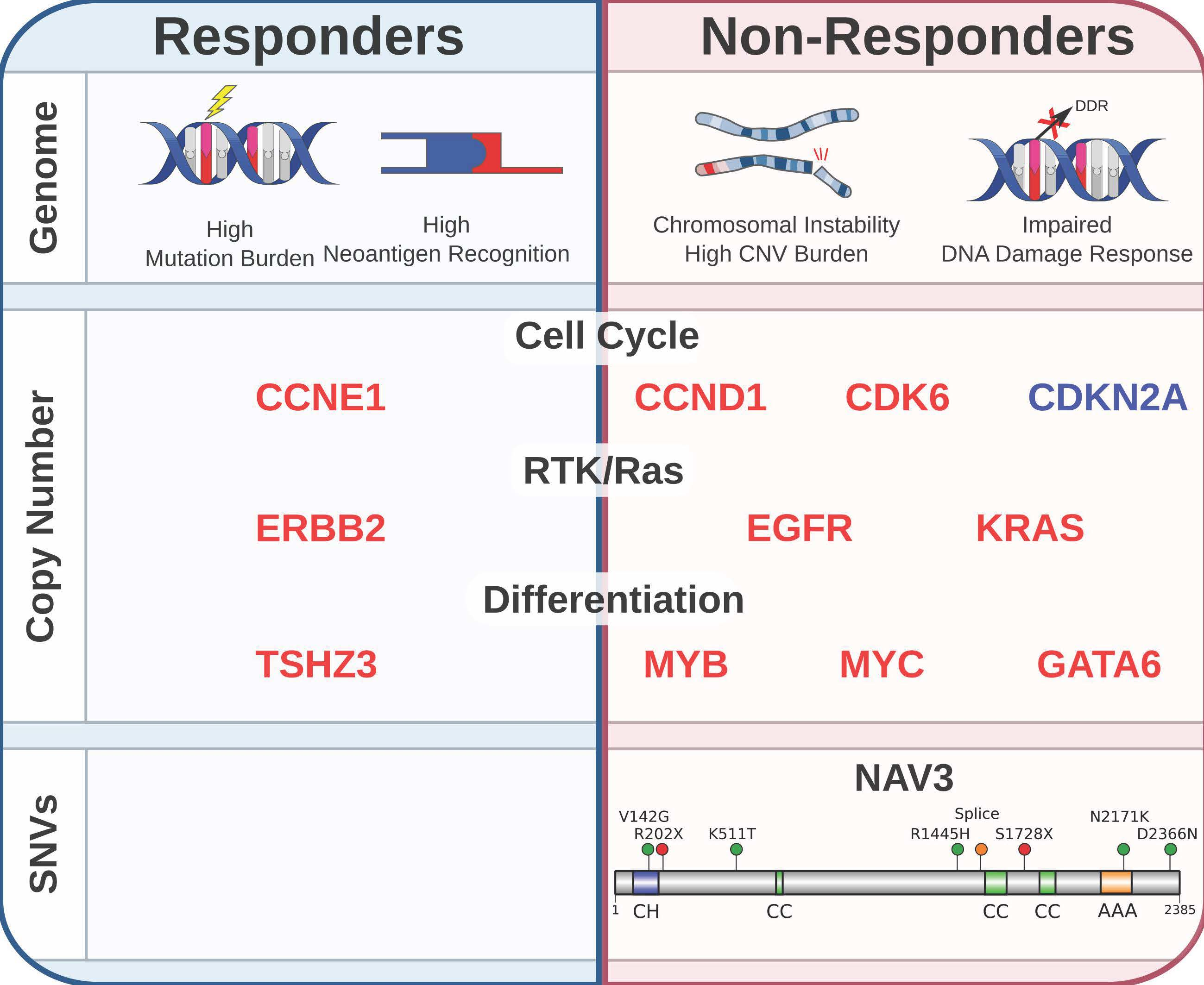
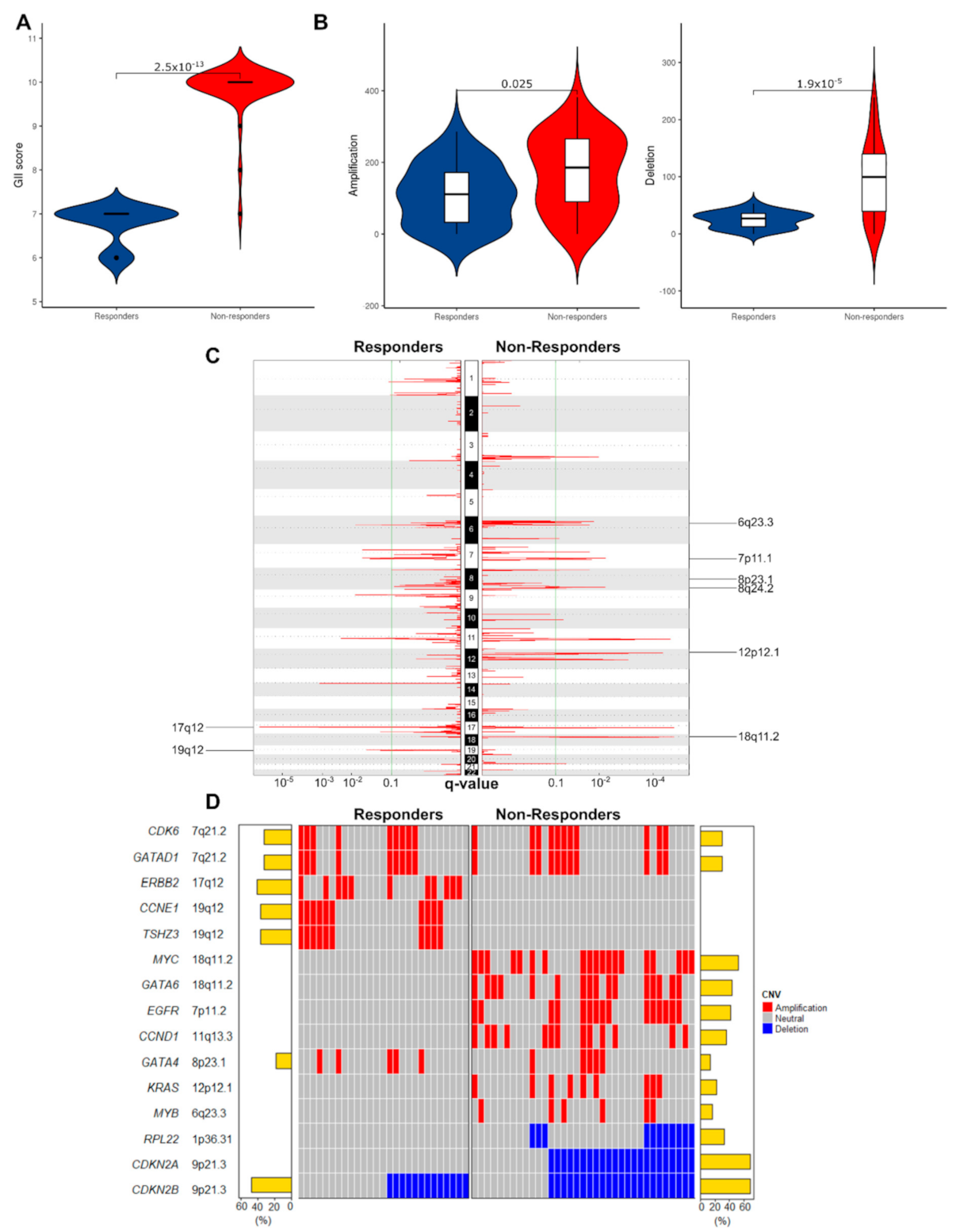
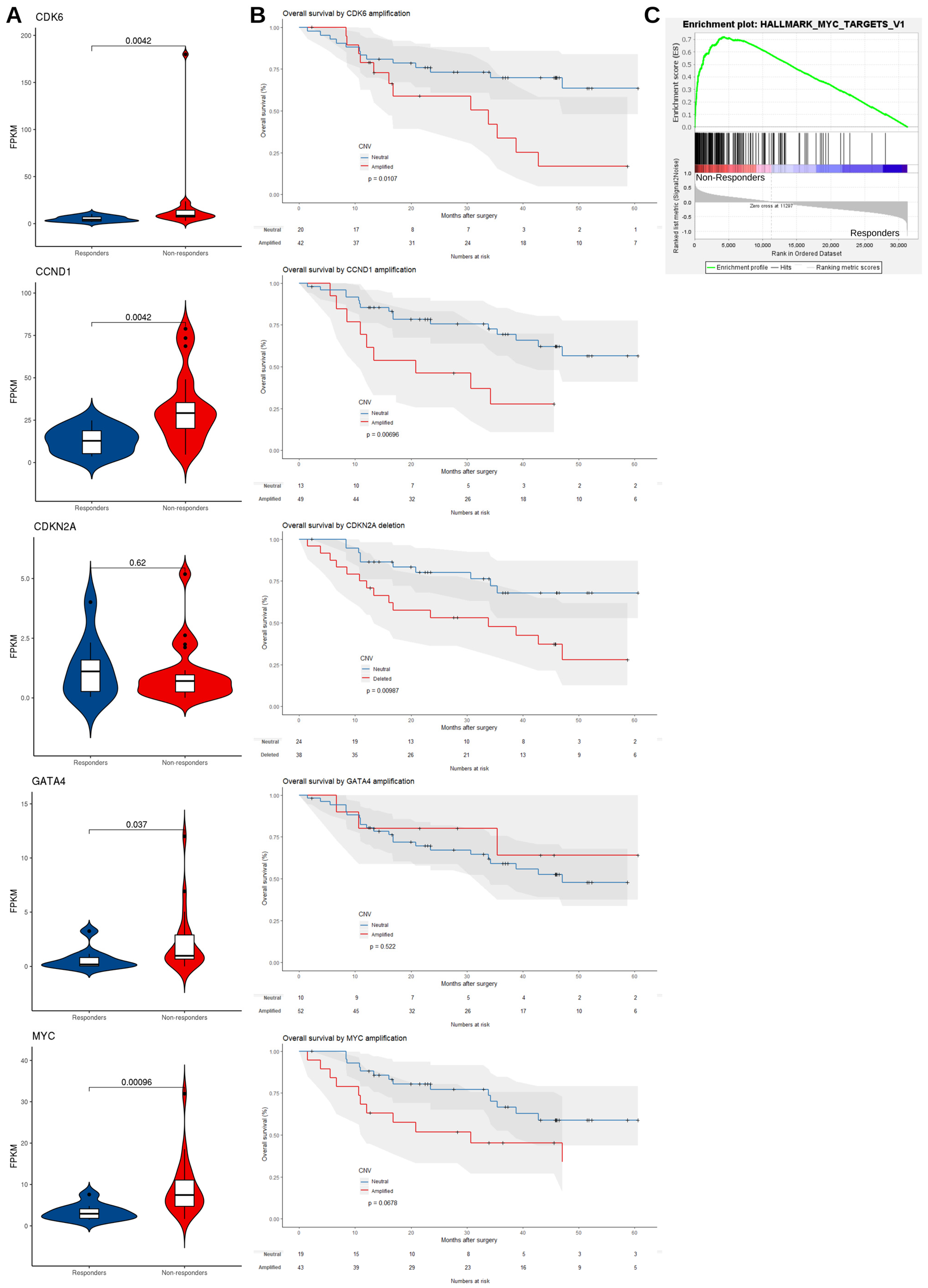
3.2.1. Non-Responders Have More Chromosomal Instability and Unique Copy Number Alterations
3.2.2. mRNA Expression Level Supports the Dysregulation of EAC Driver Genes in Non-Responders
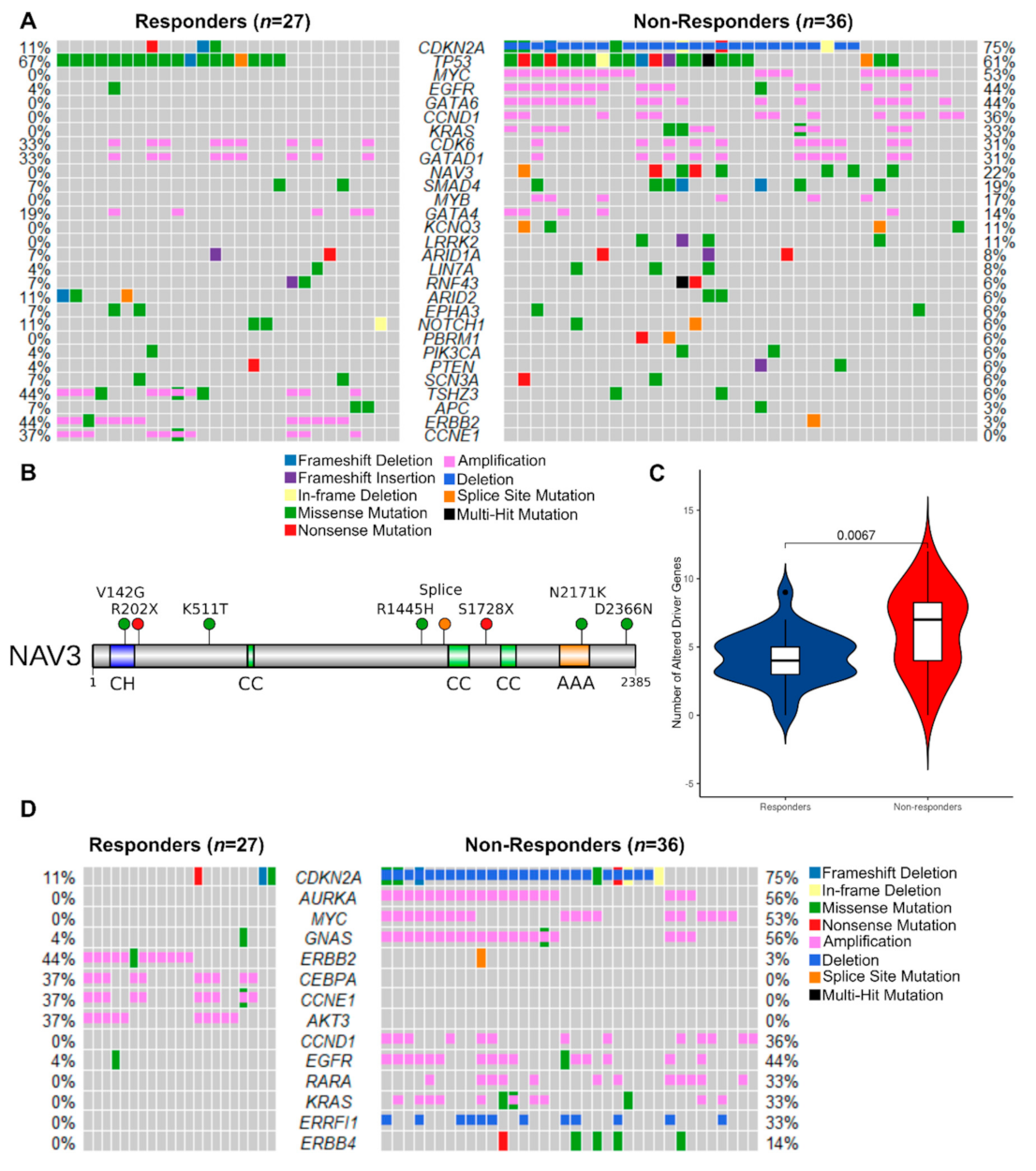
3.2.3. Mutated Driver Genes Differ Between Responders and Non-Responders
3.2.4. Potentially Targetable Alterations in Non-Responders
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ronellenfitsch, U.; Schwarzbach, M.; Hofheinz, R.; Kienle, P.; Kieser, M.; Slanger, T.E.; Jensen, K.; Burmeister, B.; Kelsen, D.; Niedzwiecki, D.; et al. Perioperative chemo(radio)therapy versus primary surgery for resectable adenocarcinoma of the stomach, gastroesophageal junction, and lower esophagus. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girling, D.J.; Bancewicz, J.; Clark, P.I.; Smith, D.B.; Donnelly, R.J.; Fayers, P.M.; Weeden, S.; Girling, D.J.; Hutchinson, T.; Harvey, A.; et al. Surgical resection with or without preoperative chemotherapy in oesophageal cancer: A randomised controlled trial. Lancet 2002, 359, 1727–1733. [Google Scholar] [CrossRef]
- Cunningham, D.; Allum, W.H.; Stenning, S.P.; Thompson, J.N.; van de Velde, C.J.H.; Nicolson, M.; Scarffe, J.H.; Lofts, F.J.; Falk, S.J.; Iveson, T.J.; et al. Perioperative Chemotherapy versus Surgery Alone for Resectable Gastroesophageal Cancer. N. Engl. J. Med. 2006, 355, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Allum, W.H.; Stenning, S.P.; Bancewicz, J.; Clark, P.I.; Langley, R.E. Long-term results of a randomized trial of surgery with or without preoperative chemotherapy in esophageal cancer. J. Clin. Oncol. 2009, 27, 5062–5067. [Google Scholar] [CrossRef] [PubMed]
- Al-Batran, S.E.; Homann, N.; Pauligk, C.; Goetze, T.O.; Meiler, J.; Kasper, S.; Kopp, H.G.; Mayer, F.; Haag, G.M.; Luley, K.; et al. Perioperative chemotherapy with fluorouracil plus leucovorin, oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus cisplatin and epirubicin for locally advanced, resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4): A randomised, phase 2/3 trial. Lancet 2019, 393, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Lagergren, J.; Fitzgerald, R.C.; Lordick, F.; Shah, M.A.; Lagergren, P.; Cunningham, D. Oesophageal cancer. Nat. Rev. Dis. Prim. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Noble, F.; Lloyd, M.A.; Turkington, R.; Griffiths, E.; O’Donovan, M.; O’Neill, J.R.; Mercer, S.; Parsons, S.L.; Fitzgerald, R.C.; Underwood, T.J.; et al. Multicentre cohort study to define and validate pathological assessment of response to neoadjuvant therapy in oesophagogastric adenocarcinoma. Br. J. Surg. 2017. [Google Scholar] [CrossRef] [Green Version]
- Sjoquist, K.M.; Burmeister, B.H.; Smithers, B.M.; Zalcberg, J.R.; Simes, R.J.; Barbour, A.; Gebski, V. Survival after neoadjuvant chemotherapy or chemoradiotherapy for resectable oesophageal carcinoma: An updated meta-analysis. Lancet Oncol. 2011. [Google Scholar] [CrossRef]
- Alderson, D.; Cunningham, D.; Nankivell, M.; Blazeby, J.M.; Griffin, S.M.; Crellin, A.; Grabsch, H.I.; Langer, R.; Pritchard, S.; Okines, A.; et al. Neoadjuvant cisplatin and fluorouracil versus epirubicin, cisplatin, and capecitabine followed by resection in patients with oesophageal adenocarcinoma (UK MRC OE05): An open-label, randomised phase 3 trial. Lancet Oncol. 2017, 18, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, D.; Stenning, S.P.; Smyth, E.C.; Okines, A.F.; Allum, W.H.; Rowley, S.; Stevenson, L.; Grabsch, H.I.; Alderson, D.; Crosby, T.; et al. Peri-operative chemotherapy with or without bevacizumab in operable oesophagogastric adenocarcinoma (UK Medical Research Council ST03): Primary analysis results of a multicentre, open-label, randomised phase 2–3 trial. Lancet Oncol. 2017, 18, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Al-Batran, S.E.; Hofheinz, R.D.; Pauligk, C.; Kopp, H.G.; Haag, G.M.; Luley, K.B.; Meiler, J.; Homann, N.; Lorenzen, S.; Schmalenberg, H.; et al. Histopathological regression after neoadjuvant docetaxel, oxaliplatin, fluorouracil, and leucovorin versus epirubicin, cisplatin, and fluorouracil or capecitabine in patients with resectable gastric or gastro-oesophageal junction adenocarcinoma (FLOT4-AIO): Results from the phase 2 part of a multicentre, open-label, randomised phase 2/3 trial. Lancet Oncol. 2016, 17, 1697–1708. [Google Scholar] [CrossRef]
- Mandard, A.-M.; Dalibard, F.; Mandard, J.-C.; Marnay, J.; Henry-Amar, M.; Petiot, J.-F.; Roussel, A.; Jacob, J.-H.; Segol, P.; Samama, G.; et al. Pathologic assessment of tumor regression after preoperative chemoradiotherapy of esophageal carcinoma. Clinicopathologic correlations. Cancer 1994. [Google Scholar] [CrossRef]
- Tan, C.; Qian, X.; Guan, Z.; Yang, B.; Ge, Y.; Wang, F.; Cai, J. Potential biomarkers for esophageal cancer. Springerplus 2016, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Tao, C.J.; Lin, G.; Xu, Y.P.; Mao, W.M. Predicting the response of neoadjuvant therapy for patients with esophageal carcinoma: An in-depth literature review. J. Cancer 2015, 6, 1179–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, C.; Law, S. Predictive factors in the evaluation of treatment response to neoadjuvant chemoradiotherapy in patients with advanced esophageal squamous cell cancer. J. Thorac. Dis. 2017, 9, S773–S780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakiba, C.; Lagarde, P.; Pissaloux, D.; Neuville, A.; Brulard, C.; Pérot, G.; Coindre, J.M.; Terrier, P.; Ranchere-Vince, D.; Ferrari, A.; et al. Response to chemotherapy is not related to chromosome instability in synovial sarcoma. Ann. Oncol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.; Simone, C.B.; Busch, T.M.; Cengel, K.A. Adjuvant, neoadjuvant, and definitive radiation therapy for malignant pleural mesothelioma. J. Thorac. Dis. 2018, 10, S2565. [Google Scholar] [CrossRef]
- Greenbaum, A.; Martin, D.R.; Bocklage, T.; Lee, J.H.; Ness, S.A.; Rajput, A. Tumor Heterogeneity as a Predictor of Response to Neoadjuvant Chemotherapy in Locally Advanced Rectal Cancer. Clin. Colorectal Cancer 2019, 18, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Höglander, E.K.; Nord, S.; Wedge, D.C.; Lingjærde, O.C.; Silwal-Pandit, L.; Gythfeldt, H.V.L.; Vollan, H.K.M.; Fleischer, T.; Krohn, M.; Schlitchting, E.; et al. Time series analysis of neoadjuvant chemotherapy and bevacizumab-treated breast carcinomas reveals a systemic shift in genomic aberrations. Genome Med. 2018. [Google Scholar] [CrossRef]
- Lesurf, R.; Griffith, O.L.; Griffith, M.; Hundal, J.; Trani, L.; Watson, M.A.; Aft, R.; Ellis, M.J.; Ota, D.; Suman, V.J.; et al. Genomic characterization of HER2-positive breast cancer and response to neoadjuvant trastuzumab and chemotherapy-results from the ACOSOG Z1041 (Alliance) trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2017. [Google Scholar] [CrossRef]
- Li, Z.; Gao, X.; Peng, X.; Chen, M.J.M.; Li, Z.; Wei, B.; Wen, X.; Wei, B.; Dong, Y.; Bu, Z.; et al. Multi-omics characterization of molecular features of gastric cancer correlated with response to neoadjuvant chemotherapy. Sci. Adv. 2020. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Muskhelishvili, L.; Tong, W.; Borlak, J.; Chen, M. Cancer genomics predicts disease relapse and therapeutic response to neoadjuvant chemotherapy of hormone sensitive breast cancers. Sci. Rep. 2020. [Google Scholar] [CrossRef]
- Nones, K.; Waddell, N.; Wayte, N.; Patch, A.M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat. Commun. 2014. [Google Scholar] [CrossRef] [Green Version]
- Secrier, M.; Li, X.; De Silva, N.; Eldridge, M.D.; Contino, G.; Bornschein, J.; Macrae, S.; Grehan, N.; O’Donovan, M.; Miremadi, A.; et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat. Genet. 2016, 48, 1131–1141. [Google Scholar] [CrossRef] [Green Version]
- Frankell, A.M.; Jammula, S.G.; Li, X.; Contino, G.; Killcoyne, S.; Abbas, S.; Perner, J.; Bower, L.; Devonshire, G.; Ococks, E.; et al. The landscape of selection in 551 esophageal adenocarcinomas defines genomic biomarkers for the clinic. Nat. Genet. 2019, 51, 506–516. [Google Scholar] [CrossRef]
- Findlay, J.M.; Castro-Giner, F.; Makino, S.; Rayner, E.; Kartsonaki, C.; Cross, W.; Kovac, M.; Ulahannan, D.; Palles, C.; Gillies, R.S.; et al. Differential clonal evolution in oesophageal cancers in response to neo-adjuvant chemotherapy. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Murugaesu, N.; Wilson, G.A.; Birkbak, N.J.; Watkins, T.B.K.; McGranahan, N.; Kumar, S.; Abbassi-Ghadi, N.; Salm, M.; Mitter, R.; Horswell, S.; et al. Tracking the genomic evolution of esophageal adenocarcinoma through neoadjuvant chemotherapy. Cancer Discov. 2015, 5, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noorani, A.; Bornschein, J.; Lynch, A.G.; Secrier, M.; Achilleos, A.; Eldridge, M.; Bower, L.; Weaver, J.M.J.; Crawte, J.; Ong, C.A.; et al. A comparative analysis of whole genome sequencing of esophageal adenocarcinoma pre-and post-chemotherapy. Genome Res. 2017, 27, 902–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, C.T.; Wong, W.S.W.; Swamy, S.; Becq, J.; Murray, L.J.; Cheetham, R.K. Strelka: Accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 2012, 28, 1811–1817. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Mourikis, T.P.; Benedetti, L.; Foxall, E.; Temelkovski, D.; Nulsen, J.; Perner, J.; Cereda, M.; Lagergren, J.; Howell, M.; Yau, C.; et al. Patient-specific cancer genes contribute to recurrently perturbed pathways and establish therapeutic vulnerabilities in esophageal adenocarcinoma. Nat. Commun. 2019. [Google Scholar] [CrossRef] [Green Version]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Van Loo, P.; Nordgard, S.H.; Lingjærde, O.C.; Russnes, H.G.; Rye, I.H.; Sun, W.; Weigman, V.J.; Marynen, P.; Zetterberg, A.; Naume, B.; et al. Allele-specific copy number analysis of tumors. Proc. Natl. Acad. Sci. USA 2010. [Google Scholar] [CrossRef] [Green Version]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020. [Google Scholar] [CrossRef] [Green Version]
- Luksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017. [Google Scholar] [CrossRef]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drier, Y.; Sheffer, M.; Domany, E. Pathway-based personalized analysis of cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 6388–6393. [Google Scholar] [CrossRef] [Green Version]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M.G. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A Precision Oncology Knowledge Base. JCO Precis. Oncol. 2017, 2017, 1–16. [Google Scholar] [CrossRef]
- Ainscough, B.J.; Griffith, M.; Coffman, A.C.; Wagner, A.H.; Kunisaki, J.; Choudhary, M.N.K.; McMichael, J.F.; Fulton, R.S.; Wilson, R.K.; Griffith, O.L.; et al. DoCM: A database of curated mutations in cancer. Nat. Methods 2016, 13, 806–807. [Google Scholar] [CrossRef] [Green Version]
- Mancini, R.; Pattaro, G.; Diodoro, M.G.; Sperduti, I.; Garufi, C.; Stigliano, V.; Perri, P.; Grazi, G.L.; Cosimelli, M. Tumor Regression Grade After Neoadjuvant Chemoradiation and Surgery for Low Rectal Cancer Evaluated by Multiple Correspondence Analysis: Ten Years as Minimum Follow-up. Clin. Colorectal Cancer 2018. [Google Scholar] [CrossRef] [Green Version]
- Rice, T.W.; Patil, D.T.; Blackstone, E.H. 8th edition AJCC/UICC staging of cancers of the esophagus and esophagogastric junction: Application to clinical practice. Ann. Cardiothorac. Surg. 2017, 6, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Francies, H.E.; Secrier, M.; Perner, J.; Miremadi, A.; Galeano-Dalmau, N.; Barendt, W.J.; Letchford, L.; Leyden, G.M.; Goffin, E.K.; et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Van Velzen, M.J.M.; Derks, S.; van Grieken, N.C.T.; Haj Mohammad, N.; van Laarhoven, H.W.M. MSI as a predictive factor for treatment outcome of gastroesophageal adenocarcinoma. Cancer Treat. Rev. 2020, 86, 102024. [Google Scholar] [CrossRef]
- Schenck, R.O.; Lakatos, E.; Gatenbee, C.; Graham, T.A.; Anderson, A.R.A. NeoPredPipe: High-throughput neoantigen prediction and recognition potential pipeline. BMC Bioinformatics 2019. [Google Scholar] [CrossRef] [Green Version]
- Noble, F.; Mellows, T.; McCormick Matthews, L.H.; Bateman, A.C.; Harris, S.; Underwood, T.J.; Byrne, J.P.; Bailey, I.S.; Sharland, D.M.; Kelly, J.J.; et al. Tumour infiltrating lymphocytes correlate with improved survival in patients with oesophageal adenocarcinoma. Cancer Immunol. Immunother. 2016, 65, 651–662. [Google Scholar] [CrossRef]
- Do Canto, L.M.; Larsen, S.J.; Kupper, B.E.C.; De Souza Begnami, M.D.F.; Scapulatempo-Neto, C.; Petersen, A.H.; Aagaard, M.M.; Baumbach, J.; Aguiar, S.; Rogatto, S.R. Increased levels of genomic instability and mutations in homologous recombination genes in locally advanced rectal carcinomas. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.L.; Eklund, A.C.; Kohane, I.S.; Harris, L.N.; Szallasi, Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 2006. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.; Wang, Y.; Cao, L.; Ouellette, M.M.; Freeman, J.W. Expression of oncogenic K-ras and loss of Smad4 cooperate to induce the expression of EGFR and to promote invasion of immortalized human pancreas ductal cells. Int. J. Cancer 2010. [Google Scholar] [CrossRef] [Green Version]
- Turkington, R.C.; Knight, L.A.; Blayney, J.K.; Secrier, M.; Douglas, R.; Parkes, E.E.; Sutton, E.K.; Stevenson, L.; McManus, D.; Halliday, S.; et al. Immune activation by DNA damage predicts response to chemotherapy and survival in oesophageal adenocarcinoma. Gut 2019. [Google Scholar] [CrossRef]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Liu, Z.; Li, M.; Chen, C.; Wang, X. Immunogenomics Analysis Reveals that TP53 Mutations Inhibit Tumor Immunity in Gastric Cancer. Transl. Oncol. 2018. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015. [Google Scholar] [CrossRef] [Green Version]
- Van Allen, E.M.; Miao, D.; Schilling, B.; Shukla, S.A.; Blank, C.; Zimmer, L.; Sucker, A.; Hillen, U.; Foppen, M.H.G.; Goldinger, S.M.; et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016. [Google Scholar] [CrossRef] [Green Version]
- Kawakubo, H.; Ozawa, S.; Ando, N.; Kitagawa, Y.; Mukai, M.; Ueda, M.; Kitajima, M. Alterations of p53, cyclin D1 and pRB expression in the carcinogenesis of esophageal squamous cell carcinoma. Oncol. Rep. 2005. [Google Scholar] [CrossRef]
- Arber, N.; Gammon, M.D.; Hibshoosh, H.; Britton, J.A.; Zhang, Y.; Schonberg, J.B.; Roterdam, H.; Fabian, I.; Holt, P.R.; Weinstein, I.B. Overexpression of cyclin D1 occurs in both squamous carcinomas and adenocarcinomas of the esophagus and in adenocarcinomas of the stomach. Hum. Pathol. 1999. [Google Scholar] [CrossRef]
- Morgan, R.J.; Newcomb, P.V.; Hardwick, R.H.; Alderson, D. Amplification of cyclin D1 and MDM-2 in oesophageal carcinoma. Eur. J. Surg. Oncol. 1999. [Google Scholar] [CrossRef]
- Miller, C.T.; Moy, J.R.; Lin, L.; Schipper, M.; Normolle, D.; Brenner, D.E.; Iannettoni, M.D.; Orringer, M.B.; Beer, D.G. Gene Amplification in Esophageal Adenocarcinomas and Barrett’s with High-Grade Dysplasia. Clin. Cancer Res. 2003, 9, 4819–4825. [Google Scholar]
- Bani-Hani, K.; Martin, I.G.; Hardie, L.J.; Mapstone, N.; Briggs, J.A.; Forman, D.; Wild, C.P. Prospective study of cyclin D1 overexpression in Barrett’s esophagus: Association with increased risk of adenocarcinoma. J. Natl. Cancer Inst. 2000. [Google Scholar] [CrossRef] [Green Version]
- Milas, L.; Akimoto, T.; Hunter, N.R.; Mason, K.A.; Buchmiller, L.; Yamakawa, M.; Muramatsu, H.; Ang, K.K. Relationship between cyclin D1 expression and poor radioresponse of murine carcinomas. Int. J. Radiat. Oncol. Biol. Phys. 2002. [Google Scholar] [CrossRef]
- Kosovec, J.E.; Zaidi, A.H.; Omstead, A.N.; Matsui, D.; Biedka, M.J.; Cox, E.J.; Campbell, P.T.; Biederman, R.W.W.; Kelly, R.J.; Jobe, B.A. CDK4/6 dual inhibitor abemaciclib demonstrates compelling preclinical activity against esophageal adenocarcinoma: A novel therapeutic option for a deadly disease. Oncotarget 2017, 8, 100421–100432. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and safety of Abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non–small cell lung cancer, and other solid tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maitland, M.L.; Piha-Paul, S.; Falchook, G.; Kurzrock, R.; Nguyen, L.; Janisch, L.; Karovic, S.; McKee, M.; Hoening, E.; Wong, S.; et al. Clinical pharmacodynamic/exposure characterisation of the multikinase inhibitor ilorasertib (ABT-348) in a phase 1 dose-escalation trial. Br. J. Cancer 2018, 118, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M. Ilorasertib in Treating Patients with CDKN2A-deficient Advanced or Metastatic Solid Cancers that Cannot Be Removed by Surgery—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02540876 (accessed on 19 January 2021).
- Hong, D.S. Phase II Study of Ilorasertib (ABT348) in Patients with CDKN2A Deficient Solid Tumors—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02478320 (accessed on 19 January 2021).
- Catenacci, D.V.T.; Moya, S.; Lomnicki, S.; Chase, L.M.; Peterson, B.F.; Reizine, N.; Alpert, L.; Setia, N.; Xiao, S.-Y.; Hart, J.; et al. Personalized antibodies for gastroesophageal adenocarcinoma (PANGEA): A phase 2 study evaluating an individualized treatment strategy for metastatic disease. Cancer Discov. 2020. [Google Scholar] [CrossRef]
- Bang, Y.J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Hofheinz, R.; Hegewisch-Becker, S.; Kunzmann, V.; Thuss-Patience, P.; Fuchs, M.; Homann, N.; Graeven, U.; Schulte, N.; Merx, K.; Pohl, M.; et al. Trastuzumab in combination with 5-fluorouracil, leucovorin, oxaliplatin and docetaxel as perioperative treatment for patients with human epidermal growth factor receptor 2-positive locally advanced esophagogastric adenocarcinoma: A phase II trial of the Arbeitsgemeinschaft Internistische Onkologie Gastric Cancer Study Group. Int. J. Cancer 2021, ijc.33696. [Google Scholar] [CrossRef]
- Rivera, F.; Izquierdo-Manuel, M.; García-Alfonso, P.; Martínez de Castro, E.; Gallego, J.; Limón, M.L.; Alsina, M.; López, L.; Galán, M.; Falcó, E.; et al. Perioperative trastuzumab, capecitabine and oxaliplatin in patients with HER2-positive resectable gastric or gastro-oesophageal junction adenocarcinoma: NEOHX phase II trial. Eur. J. Cancer 2021, 145, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Luber, B.; Deplazes, J.; Keller, G.; Walch, A.; Rauser, S.; Eichmann, M.; Langer, R.; Höfler, H.; Hegewisch-Becker, S.; Folprecht, G.; et al. Biomarker analysis of cetuximab plus oxaliplatin/leucovorin/5-fluorouracil in first-line metastatic gastric and oesophago-gastric junction cancer: Results from a phase II trial of the Arbeitsgemeinschaft Internistische Onkologie (AIO). BMC Cancer 2011, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.A.; Blaszkowsky, L.S.; Enzinger, P.C.; Ryan, D.P.; Abrams, T.A.; Zhu, A.X.; Temel, J.S.; Schrag, D.; Bhargava, P.; Meyerhardt, J.A.; et al. A multicenter phase II trial of single-agent cetuximab in advanced esophageal and gastric adenocarcinoma. Ann. Oncol. 2011, 22, 1367–1373. [Google Scholar] [CrossRef] [PubMed]
- Ruhstaller, T.; Thuss-Patience, P.; Hayoz, S.; Schacher, S.; Knorrenschild, J.R.; Schnider, A.; Plasswilm, L.; Budach, W.; Eisterer, W.; Hawle, H.; et al. Neoadjuvant chemotherapy followed by chemoradiation and surgery with and without cetuximab in patients with resectable esophageal cancer: A randomized, open-label, phase III trial (SAKK 75/08). Ann. Oncol. 2018, 29, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Suntharalingam, M.; Winter, K.; Ilson, D.; Dicker, A.P.; Kachnic, L.; Konski, A.; Chakravarthy, A.B.; Anker, C.J.; Thakrar, H.; Horiba, N.; et al. Effect of the addition of cetuximab to paclitaxel, cisplatin, and radiation therapy for patients with esophageal cancer the NRG oncology rtog 0436 phase 3 randomized clinical trial. JAMA Oncol. 2017, 3, 1520–1528. [Google Scholar] [CrossRef]
- Crosby, T.; Hurt, C.N.; Falk, S.; Gollins, S.; Mukherjee, S.; Staffurth, J.; Ray, R.; Bashir, N.; Bridgewater, J.A.; Geh, J.I.; et al. Chemoradiotherapy with or without cetuximab in patients with oesophageal cancer (SCOPE1): A multicentre, phase 2/3 randomised trial. Lancet Oncol. 2013, 14, 627–637. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Lièvre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Côté, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [Green Version]
- Richards, D.; Kocs, D.M.; Spira, A.I.; David McCollum, A.; Diab, S.; Hecker, L.I.; Cohn, A.; Zhan, F.; Asmar, L. Results of docetaxel plus oxaliplatin (DOCOX) ± cetuximab in patients with metastatic gastric and/or gastroesophageal junction adenocarcinoma: Results of a randomised Phase 2 study. Eur. J. Cancer 2013, 49, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Dvashi, H.; Ben-Chetrit, N.; Russell, R.; Carvalho, S.; Lauriola, M.; Nisani, S.; Mancini, M.; Nataraj, N.; Kedmi, M.; Roth, L.; et al. Navigator-3, a modulator of cell migration, may act as a suppressor of breast cancer progression. EMBO Mol. Med. 2015, 7, 299–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Zhou, D.; Wu, W.; Tan, W.L.; Wang, J.; Zhou, C.; Lou, Y. MHC class II restricted neoantigen peptides predicted by clonal mutation analysis in lung adenocarcinoma patients: Implications on prognostic immunological biomarker and vaccine design. BMC Genomics 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Uboveja, A.; Satija, Y.K.; Siraj, F.; Sharma, I.; Saluja, D. p73-NAV3 axis plays a critical role in suppression of colon cancer metastasis. Oncogenesis 2020, 9, 12. [Google Scholar] [CrossRef]
- Carlsson, E.; Ranki, A.; Sipilä, L.; Karenko, L.; Abdel-Rahman, W.M.; Ovaska, K.; Siggberg, L.; Aapola, U.; Ssämäki, R.; Häyry, V.; et al. Potential role of a navigator gene NAV3 in colorectal cancer. Br. J. Cancer 2012, 106, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Pink, R.C.; Samuel, P.; Massa, D.; Caley, D.P.; Brooks, S.A.; Carter, D.R.F. The passenger strand, miR-21-3p, plays a role in mediating cisplatin resistance in ovarian cancer cells. Gynecol. Oncol. 2015, 137, 143–151. [Google Scholar] [CrossRef]
- Carlsson, E.; Krohn, K.; Ovaska, K.; Lindberg, P.; Häyry, V.; Maliniemi, P.; Lintulahti, A.; Korja, M.; Kivisaari, R.; Hussein, S.; et al. Neuron navigator 3 alterations in nervous system tumors associate with tumor malignancy grade and prognosis. Genes Chromosom. Cancer 2013, 52. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Category | Non-Responders (n = 38) | Responders (n = 27) | Overall (n = 65) | p-Value (X2 test) |
|---|---|---|---|---|---|
| Age | 66.25 (15.3) | 64.30 (12.2) | 65.00 (14) | 0.739 | |
| Gender | Female | 5 (13.2) | 2 (7.4) | 7 (10.8) | 0.741 |
| Male | 33 (86.8) | 25 (92.6) | 58 (89.2) | ||
| cT Stage | T1 | 0 (0.0) | 1 (3.7) | 1 (1.5) | 0.485 |
| T2 | 5 (13.2) | 5 (18.5) | 10 (15.4) | ||
| T3 | 31 (81.6) | 19 (70.4) | 50 (76.9) | ||
| T4 | 2 (5.3) | 1 (3.7) | 3 (4.6) | ||
| Missing | 0 (0.0) | 1 (3.7) | 1 (1.5) | ||
| cN Stage | N0 | 8 (21.1) | 7 (25.9) | 15 (23.1) | 0.494 |
| N1 | 24 (63.2) | 12 (44.4) | 36 (55.4) | ||
| N2 | 5 (13.2) | 6 (22.2) | 11 (16.9) | ||
| N3 | 1 (2.6) | 1 (3.7) | 2 (3.1) | ||
| Missing | 0 (0.0) | 1 (3.7) | 1 (1.5) | ||
| Tumor Location | GOJ | 19 (50.0) | 16 (59.3) | 35 (53.8) | 0.627 |
| Esophagus | 19 (50.0) | 11 (40.7) | 30 (46.2) | ||
| ypT Stage | T0 | 1 (2.6) | 17 (63.0) | 18 (27.7) | <0.001 * |
| T1 | 3 (7.9) | 5 (18.5) | 8 (12.3) | ||
| T2 | 4 (10.5) | 1 (3.7) | 5 (7.7) | ||
| T3 | 24 (63.2) | 4 (14.8) | 28 (43.1) | ||
| T4 | 6 (15.8) | 0 (0.0) | 6 (9.2) | ||
| ypN Stage | N0 | 9 (23.7) | 17 (63.0) | 26 (40.0) | <0.001 * |
| N1 | 6 (15.8) | 4 (14.8) | 10 (15.4) | ||
| N2 | 14 (36.8) | 2 (7.4) | 16 (24.6) | ||
| N3 | 8 (21.1) | 0 (0.0) | 8 (12.3) | ||
| Missing | 1 (2.6) | 4 (14.8) | 5 (7.7) | ||
| ypM Stage | M0 | 35 (92.1) | 27 (100.0) | 62 (95.4) | 0.371 |
| M1 | 3 (7.9) | 0 (0.0) | 3 (4.6) | ||
| Treatment Regimen | CarboTaxol | 2 (5.3) | 0 (0.0) | 2 (3.1) | <0.001 * |
| CF | 1 (2.6) | 1 (3.7) | 2 (3.1) | ||
| CX | 0 (0.0) | 4 (14.8) | 4 (6.2) | ||
| CROSS | 0 (0.0) | 2 (7.4) | 2 (3.1) | ||
| ECarboX | 0 (0.0) | 1 (3.7) | 1 (1.5) | ||
| ECF | 0 (0.0) | 1 (3.7) | 1 (1.5) | ||
| ECOx | 1 (2.6) | 0 (0.0) | 1 (1.5) | ||
| ECX | 29 (76.3) | 11 (41.0) | 39 (60.0) | ||
| ECX + Bevacizumab | 2 (5.3) | 0 (0.0) | 2 (3.1) | ||
| EOX | 1 (2.6) | 3 (11.1) | 4 (6.2) | ||
| LEO | 1 (2.6) | 0 (0.0) | 1 (1.5) | ||
| CAPOX | 0 (0.0) | 1 (3.7) | 1 (1.5) | ||
| Missing | 3 (7.9) | 1 (3.7) | 4 (6.2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izadi, F.; Sharpe, B.P.; Breininger, S.P.; Secrier, M.; Gibson, J.; Walker, R.C.; Rahman, S.; Devonshire, G.; Lloyd, M.A.; Walters, Z.S.; et al. Genomic Analysis of Response to Neoadjuvant Chemotherapy in Esophageal Adenocarcinoma. Cancers 2021, 13, 3394. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143394
Izadi F, Sharpe BP, Breininger SP, Secrier M, Gibson J, Walker RC, Rahman S, Devonshire G, Lloyd MA, Walters ZS, et al. Genomic Analysis of Response to Neoadjuvant Chemotherapy in Esophageal Adenocarcinoma. Cancers. 2021; 13(14):3394. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143394
Chicago/Turabian StyleIzadi, Fereshteh, Benjamin P. Sharpe, Stella P. Breininger, Maria Secrier, Jane Gibson, Robert C. Walker, Saqib Rahman, Ginny Devonshire, Megan A. Lloyd, Zoë S. Walters, and et al. 2021. "Genomic Analysis of Response to Neoadjuvant Chemotherapy in Esophageal Adenocarcinoma" Cancers 13, no. 14: 3394. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13143394