
Selective Inhibition of Aurora Kinase A by AK-01/LY3295668 Attenuates MCC Tumor Growth by Inducing MCC Cell Cycle Arrest and Apoptosis

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Critical Reagents and Compounds
2.2. Generation of MCC Cell Line Derived Xenograft Models in Mice
2.3. Cell Culture
2.4. Cell Proliferation and Viability Assay
2.5. Cell Cycle Analysis by Flow Cytometry
2.6. Immunoblotting
2.7. Statistical Analysis
3. Results
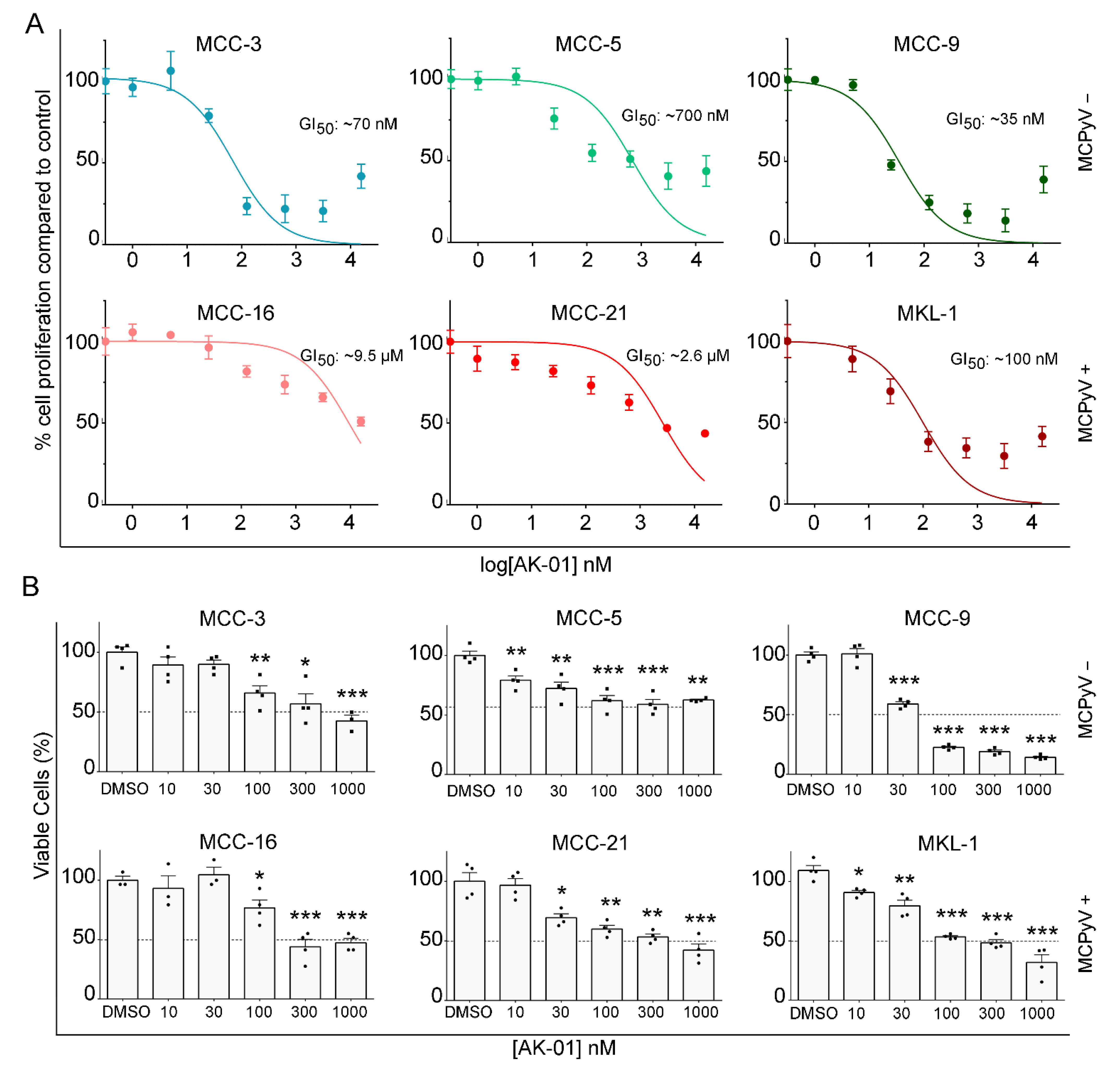
3.1. AK-01 Suppresses MCC Cell Proliferation and Viability In Vitro
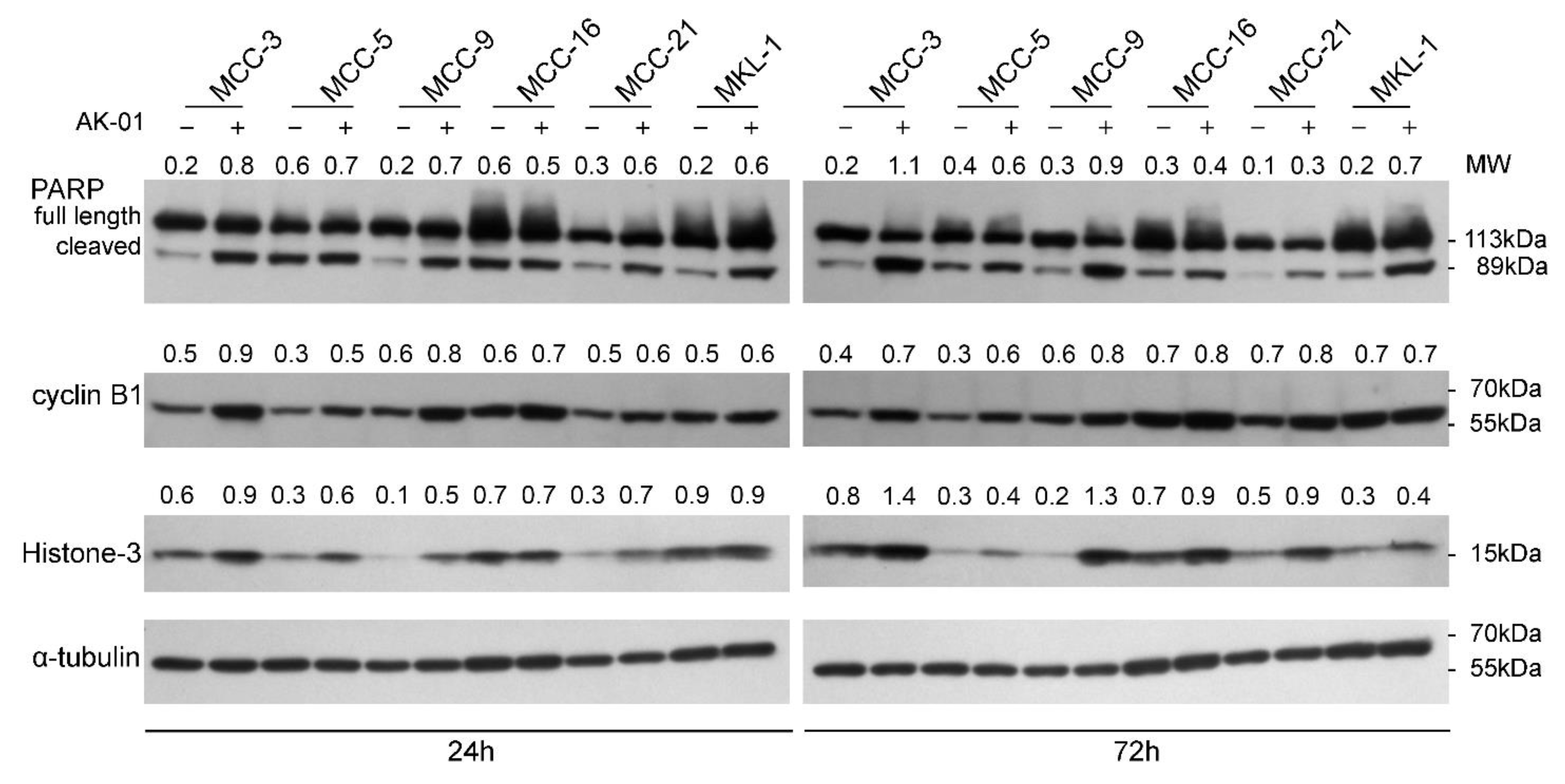
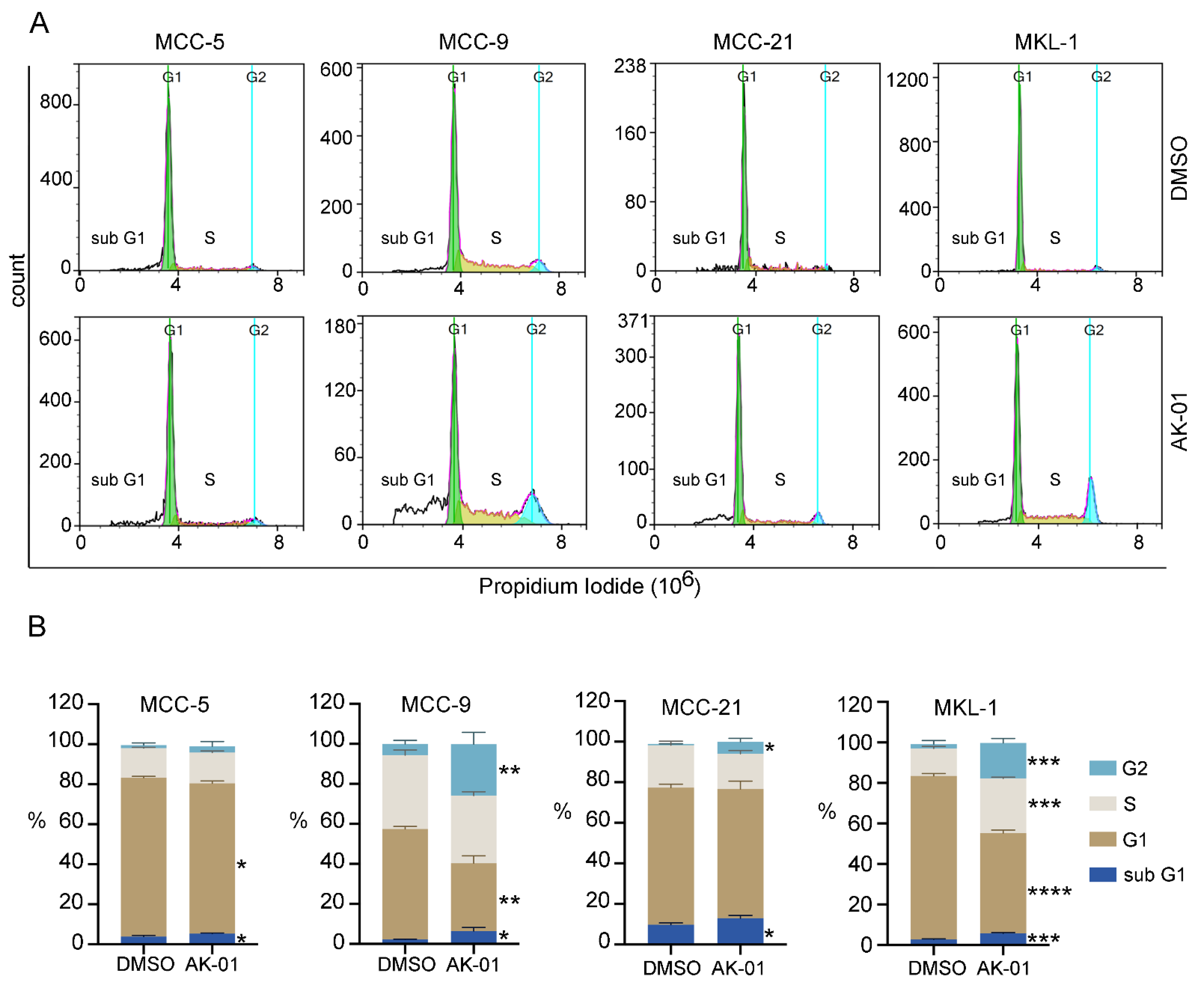
3.2. AK-01 Induces Cell-Cycle Arrest and Apoptosis in MCC Cells In Vitro
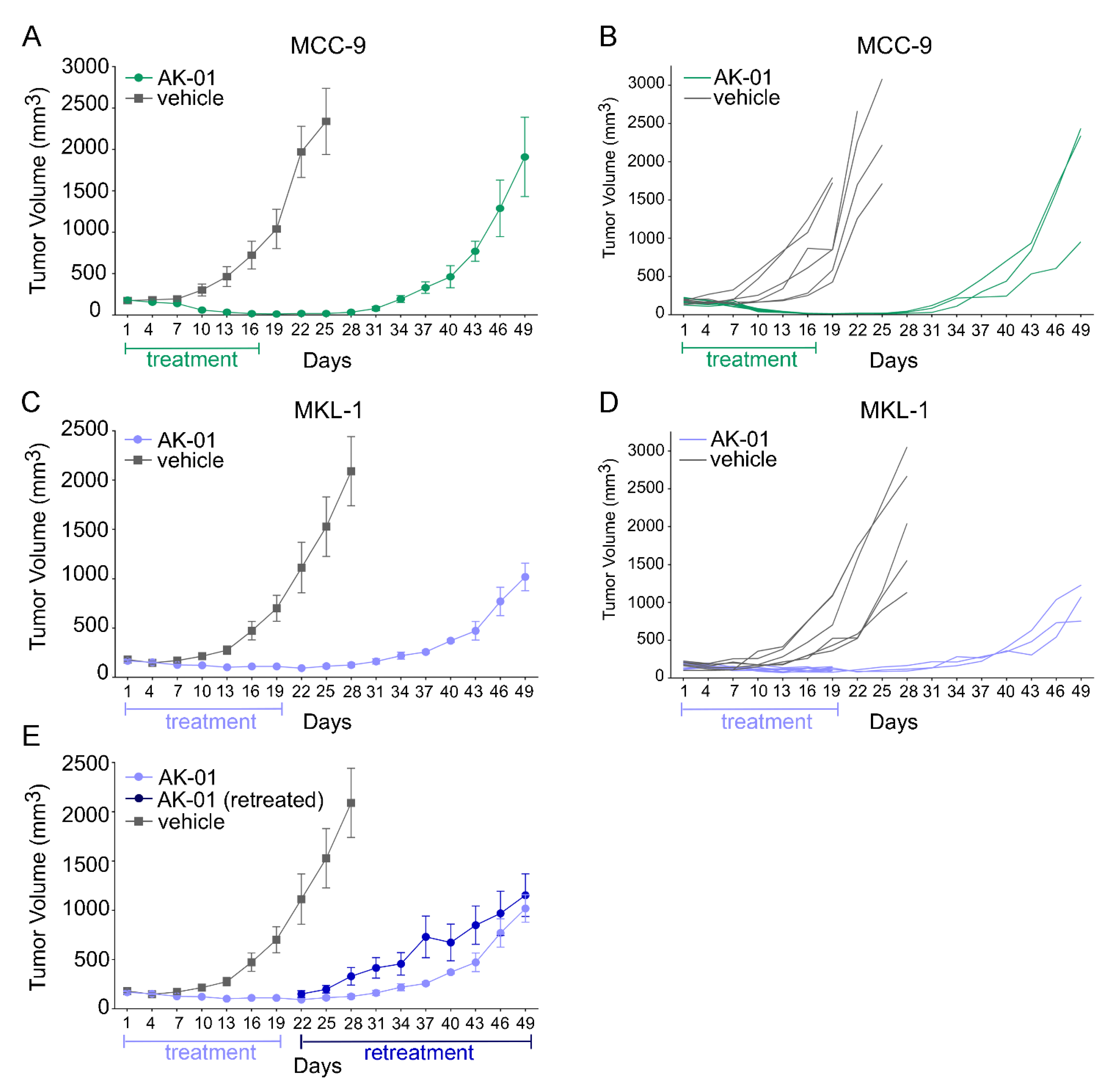
3.3. Aurora Kinase A Inhibition by AK-01 Attenuates MCC Xenograft Tumor Growth
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel cell carcinoma. Nat. Rev. Dis. Prim. 2017, 3, 1–17. [Google Scholar] [CrossRef]
- Harms, P.W.; Harms, K.L.; Moore, P.; DeCaprio, J.A.; Nghiem, P.; Wong, M.K.K.; Brownell, I.; International Workshop on Merkel Cell Carcinoma Research (IWMCC) Working Group. The biology and treatment of Merkel cell carcinoma: Current understanding and research priorities. Nat. Rev. Clin. Oncol. 2018, 15, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.W.; Rabkin, C.S. Merkel cell carcinoma and melanoma: Etiological similarities and differences. Cancer Epidemiol. Biomark. Prev. 1999, 8, 153–158. [Google Scholar]
- Paulson, K.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A.; et al. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463.e2. [Google Scholar] [CrossRef]
- Wong, S.Q.; Waldeck, K.; Vergara, I.; Schröder, J.; Madore, J.; Wilmott, J.S.; Colebatch, A.J.; De Paoli-Iseppi, R.; Li, J.; Lupat, R.; et al. UV-Associated Mutations Underlie the Etiology of MCV-Negative Merkel Cell Carcinomas. Cancer Res. 2015, 75, 5228–5234. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Cook, M.; Baker, K.; Redman, M.; Lachance, K.; Nguyen, M.H.; Parvathaneni, U.; Bhatia, S.; Nghiem, P.; Tseng, Y.D. Differential outcomes among immunosuppressed patients with Merkel cell carcinoma. Am. J. Clin. Oncol. 2019, 42, 82–88. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with pembrolizumab in advanced Merkel-cell carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Colunga, A.; Pulliam, T.; Nghiem, P. Merkel cell carcinoma in the age of immunotherapy: Facts and hopes. Clin. Cancer Res. 2017, 24, 2035–2043. [Google Scholar] [CrossRef] [Green Version]
- Gallo, M.; Guarnotta, V.; De Cicco, F.; Rubino, M.; Faggiano, A.; Colao, A.; NIKE Group. Immune checkpoint blockade for Merkel cell carcinoma: Actual findings and unanswered. Questions 2019, 145, 429–443. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Bhatia, S.; Brohl, A.S.; Hamid, O.; Mehnert, J.M.; Terheyden, P.; Shih, K.C.; Brownell, I.; Lebbé, C.; Lewis, K.D.; et al. Avelumab in patients with previously treated metastatic Merkel cell carcinoma: Long-term data and biomarker analyses from the single-arm phase 2 JAVELIN Merkel 200 trial. J. Immunother. Cancer 2019, 8, e000674. [Google Scholar] [CrossRef]
- Houben, R.; Michel, B.; Vetter-Kauczok, C.S.; Pföhler, C.; Laetsch, B.; Wolter, M.D.; Leonard, J.H.; Trefzer, U.; Ugurel, S.; Schrama, D.; et al. Absence of Classical MAP Kinase Pathway Signalling in Merkel Cell Carcinoma. J. Investig. Dermatol. 2006, 126, 1135–1142. [Google Scholar] [CrossRef] [Green Version]
- Rabinowits, G.; Lezcano, C.; Catalano, P.J.; McHugh, P.; Becker, H.; Reilly, M.M.; Huang, J.; Tyagi, A.; Thakuria, M.; Bresler, S.C.; et al. Cabozantinib in Patients with Advanced Merkel Cell Carcinoma. Oncology 2018, 23, 814–821. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.C.; Schrama, D.; Houben, R. Merkel cell carcinoma. Cell. Mol. Life Sci. 2008, 66, 1. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A. Molecular pathogenesis of Merkel cell carcinoma. Annu. Rev. Pathol. Mech. Dis. 2021, 16, 69–91. [Google Scholar] [CrossRef] [PubMed]
- Nardi, V.; Song, Y.C.; Santamaria-Barria, J.; Cosper, A.K.; Lam, Q.; Faber, A.C.; Boland, G.M.; Yeap, B.Y.; Bergethon, K.; Scialabba, V.L.; et al. Activation of PI3K signaling in Merkel cell carcinoma. Clin. Cancer Res. 2012, 18, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Hafner, C.; Houben, R.; Baeurle, A.; Ritter, C.; Schrama, D.; Landthaler, M.; Becker, J.C. Activation of the PI3K/AKT pathway in Merkel cell carcinoma. PLoS ONE 2012, 7, e31255. [Google Scholar] [CrossRef]
- Lin, Z.; McDermott, A.; Shao, L.; Kannan, A.; Morgan, M.; Stack, B.C.; Moreno, M.; Davis, D.A.; Cornelius, L.A.; Gao, L. Chronic mTOR activation promotes cell survival in Merkel cell carcinoma. Cancer Lett. 2014, 344, 272–281. [Google Scholar] [CrossRef] [Green Version]
- Fang, B.; Kannan, A.; Zhao, S.; Nguyen, Q.H.; Ejadi, S.; Yamamoto, M.; Barreto, J.C.; Zhao, H.; Gao, L. Inhibition of PI3K by copanlisib exerts potent antitumor effects on Merkel cell carcinoma cell lines and mouse xenografts. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.; Lin, Z.; Shao, Q.; Zhao, S.; Fang, B.; Moreno, M.A.; Vural, E.; Stack, B.C.; Suen, J.Y.; Kannan, K.; et al. Dual mTOR inhibitor MLN0128 suppresses Merkel cell carcinoma (MCC) xenograft tumor growth. Oncotarget 2015, 7, 6576–6592. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Kannan, A.; Lin, Z.; Stack, B.C.; Suen, J.Y.; Gao, L. BET Protein inhibitor JQ1 attenuates Myc-Amplified MCC tumor growth in vivo. Cancer Res. 2014, 74, 7090–7102. [Google Scholar] [CrossRef] [Green Version]
- Park, D.E.; Cheng, J.; Berrios, C.; Montero, J.; Cortés-Cros, M.; Ferretti, S.; Arora, R.; Tillgren, M.L.; Gokhale, P.C.; DeCaprio, J.A. Dual inhibition of MDM2 and MDM4 in virus-positive Merkel cell carcinoma enhances the p53 response. Proc. Natl. Acad. Sci. USA 2018, 116, 1027–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Bretz, A.C.; Gravemeyer, J.; Spassova, I.; Muminova, S.; Gambichler, T.; Sriram, A.; Ferrone, S.; Becker, J.C. The HDAC inhibitor domatinostat promotes cell-cycle arrest, induces apoptosis, and increases immunogenicity of Merkel cell carcinoma cells. J. Investig. Dermatol. 2021, 141, 903–912.e4. [Google Scholar] [CrossRef]
- Giet, R.; Glover, D.M. Drosophila Aurora B Kinase is required for histone H3 phosphorylation and condensin recruitment during chromosome condensation and to organize the central spindle during cytokinesis. J. Cell Biol. 2001, 152, 669–682. [Google Scholar] [CrossRef] [Green Version]
- Goos, J.; Coupe, V.; Diosdado, B.; Delis-van Diemen, P.M.; Karga, C.; Beliën, J.A.M.; Carvalho, B.; van den Tol, M.P.; Verheul, H.; Geldof, A.A.; et al. Aurora kinase A (AURKA) expression in colorectal cancer liver metastasis is associated with poor prognosis. Br. J. Cancer 2013, 109, 2445–2452. [Google Scholar] [CrossRef] [Green Version]
- Al-Khafaji, A.; Marcus, M.W.; Davies, M.P.A.; Risk, J.M.; Shaw, R.J.; Field, J.; Liloglou, T. AURKA mRNA expression is an independent predictor of poor prognosis in patients with non-small cell lung cancer. Oncol. Lett. 2017, 13, 4463–4468. [Google Scholar] [CrossRef] [Green Version]
- Landen, C.N.; Lin, Y.G.; Immaneni, A.; Deavers, M.T.; Merritt, W.M.; Spannuth, W.A.; Bodurka, D.; Gershenson, D.M.; Brinkley, W.R.; Sood, A.K. Overexpression of the centrosomal protein Aurora-A Kinase is associated with poor prognosis in epithelial ovarian cancer patients. Clin. Cancer Res. 2007, 13, 4098–4104. [Google Scholar] [CrossRef] [Green Version]
- Iacono, M.L.; Monica, V.; Saviozzi, S.; Ceppi, P.; Bracco, E.; Papotti, M.; Scagliotti, G.V. Aurora Kinase A expression is associated with lung cancer histological-subtypes and with tumor de-differentiation. J. Transl. Med. 2011, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Chuang, T.-P.; Wang, J.-Y.; Jao, S.-W.; Wu, C.-C.; Chen, J.-H.; Hsiao, K.-H.; Lin, C.-Y.; Chen, S.-H.; Su, S.-Y.; Chen, Y.-J.; et al. Over-expression of AURKA, SKA3 and DSN1 contributes to colorectal adenoma to carcinoma progression. Oncotarget 2016, 7, 45803–45818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeng, Y.-M.; Peng, S.-Y.; Lin, C.-Y.; Hsu, H.-C. Overexpression and amplification of Aurora-A in hepatocellular carcinoma. Clin. Cancer Res. 2004, 10, 2065–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, K.N.; Bandyopadhyay, S. Targeting the evolution of drug resistance in lung cancer. Mol. Cell. Oncol. 2019, 6, e1603092. [Google Scholar] [CrossRef]
- Murga-Zamalloa, C.; Inamdar, K.V.; Wilcox, R.A. The role of Aurora A and polo-like kinases in high-risk lymphomas. Blood Adv. 2019, 3, 1778–1787. [Google Scholar] [CrossRef] [Green Version]
- Görgün, G.; Calabrese, E.; Hideshima, T.; Ecsedy, J.; Perrone, G.; Mani, M.; Ikeda, H.; Bianchi, G.; Hu, Y.; Cirstea, D.; et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood 2010, 115, 5202–5213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, R.W.; Odedra, R.; Heaton, S.P.; Wedge, S.R.; Keen, N.J.; Crafter, C.; Foster, J.R.; Brady, M.C.; Bigley, A.; Brown, E.; et al. AZD1152, a selective inhibitor of Aurora B Kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin. Cancer Res. 2007, 13, 3682–3688. [Google Scholar] [CrossRef] [Green Version]
- Nair, J.S.; Ho, A.L.; Tse, A.N.; Coward, J.; Cheema, H.; Ambrosini, G.; Keen, N.; Schwartz, G.K. Aurora B Kinase regulates the postmitotic endoreduplication checkpoint via phosphorylation of the retinoblastoma protein at serine 780. Mol. Biol. Cell 2009, 20, 2218–2228. [Google Scholar] [CrossRef] [Green Version]
- Nair, J.S.; Ho, A.L.; Schwartz, G.K. The induction of polyploidy or apoptosis by the Aurora A kinase inhibitor MK8745 is p53-dependent. Cell Cycle 2012, 11, 807–817. [Google Scholar] [CrossRef] [Green Version]
- Nair, J.S.; Schwartz, G.K. MLN-8237: A dual inhibitor of aurora A and B in soft tissue sarcomas. Oncotarget 2016, 7, 12893–12903. [Google Scholar] [CrossRef] [Green Version]
- De Groot, C.O.; Hsia, J.E.; Anzola, J.V.; Motamedi, A.; Yoon, M.; Wong, Y.L.; Jenkins, D.; Lee, H.J.; Martinez, M.B.; Davis, R.L.; et al. A Cell Biologist’s Field Guide to Aurora Kinase Inhibitors. Front. Oncol. 2015, 5, 285. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Parameswaran, J.; Sandoval-Schaefer, T.; Eoh, K.J.; Yang, D.H.; Zhu, F.; Mehra, R.; Sharma, R.; Gaffney, S.G.; Perry, E.B.; et al. Combined Aurora Kinase A (AURKA) and WEE1 inhibition demonstrates synergistic antitumor effect in squamous cell carcinoma of the head and neck. Clin. Cancer Res. 2019, 25, 3430–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ommer, J.; Selfe, J.L.; Wachtel, M.; O´Brien, E.M.; Laubscher, D.; Roemmele, M.; Kasper, S.; Delattre, O.; Surdez, D.; Petts, G.; et al. Aurora A Kinase inhibition destabilizes PAX3-FOXO1 and MYCN and synergizes with navitoclax to induce rhabdomyosarcoma cell death. Cancer Res. 2019, 80, 832–842. [Google Scholar] [CrossRef] [Green Version]
- Čančer, M.; Drews, L.F.; Bengtsson, J.; Bolin, S.; Rosén, G.; Westermark, B.; Nelander, S.; Forsberg-Nilsson, K.; Uhrbom, L.; Weishaupt, H.; et al. BET and Aurora Kinase A inhibitors synergize against MYCN-positive human glioblastoma cells. Cell Death Dis. 2019, 10, 881. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yan, L.; Torres, R.; Gong, X.; Bian, H.; Marugán, C.; Boehnke, K.; Baquero, C.; Hui, Y.-H.; Chapman, S.C.; et al. Aurora A–Selective inhibitor LY3295668 leads to dominant mitotic arrest, apoptosis in cancer cells, and shows potent preclinical antitumor efficacy. Mol. Cancer Ther. 2019, 18, 2207–2219. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Du, J.; Parsons, S.H.; Merzoug, F.F.; Webster, Y.; Iversen, P.W.; Chio, L.-C.; Van Horn, R.D.; Lin, X.; Blosser, W.; et al. Aurora A Kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov. 2018, 9, 248–263. [Google Scholar] [CrossRef] [Green Version]
- Chu, Q.S.-C.; Bouganim, N.; Fortier, C.; Zaknoen, S.; Stille, J.R.; Kremer, J.D.; Yuen, E.; Hui, Y.-H.; de la Peña, A.; Lithio, A.; et al. Aurora kinase A inhibitor, LY3295668 erbumine: A phase 1 monotherapy safety study in patients with locally advanced or metastatic solid tumors. Investig. New Drugs 2021, 1–10. [Google Scholar] [CrossRef]
- Goh, G.; Walradt, T.; Markarov, V.; Blom, A.; Riaz, N.; Doumani, R.; Stafstrom, K.; Moshiri, A.; Yelistratova, L.; Levinsohn, J.; et al. Mutational landscape of MCPyV-positive and MCPyV-negative Merkel cell carcinomas with implications for immunotherapy. Oncotarget 2015, 7, 3403–3415. [Google Scholar] [CrossRef] [Green Version]
- Hesbacher, S.; Pfitzer, L.; Wiedorfer, K.; Angermeyer, S.; Borst, A.; Haferkamp, S.; Scholz, C.-J.; Wobser, M.; Schrama, D.; Houben, R. RB1 is the crucial target of the Merkel cell polyomavirus Large T antigen in Merkel cell carcinoma cells. Oncotarget 2016, 7, 32956–32968. [Google Scholar] [CrossRef]
- Cohen, P.R.; Tomson, B.N.; Elkin, S.K.; Marchlik, E.; Carter, J.L.; Kurzrock, R. Genomic portfolio of Merkel cell carcinoma as determined by comprehensive genomic profiling: Implications for targeted therapeutics. Oncotarget 2016, 7, 23454–23467. [Google Scholar] [CrossRef] [Green Version]
- Shultz, L.D.; Schweitzer, P.A.; Christianson, S.W.; Gott, B.; Schweitzer, I.B.; Tennent, B.; McKenna, S.; Mobraaten, L.; Rajan, T.V.; Greiner, D.L. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J. Immunol. 1995, 154, 180–191. [Google Scholar] [PubMed]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Creation of “Humanized” Mice to Study Human Immunity. Curr. Protoc. Immunol. 2008, 81, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yao, L.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.; Shi, W.; Ma, A.; White, R.W.D.V.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, J.; Yang, E.J.; Zhang, B.; Wu, C.; Pardeshi, L.; Shi, C.; Mou, P.K.; Liu, Y.; Tan, K.; Shim, J.S. Synthetic lethality of RB1 and aurora A is driven by stathmin-mediated disruption of microtubule dynamics. Nat. Commun. 2020, 11, 5105. [Google Scholar] [CrossRef]
- Wike, C.L.; Graves, H.K.; Hawkins, R.; Gibson, M.D.; Ferdinand, M.B.; Zhang, T.; Chen, Z.; Hudson, D.F.; Ottesen, J.; Poirier, M.G.; et al. Aurora-A mediated histone H3 phosphorylation of threonine 118 controls condensin I and cohesin occupancy in mitosis. eLife 2016, 5, e11402. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Lyu, J.; Yang, E.J.; Liu, Y.; Zhang, B.; Shim, J.S. Targeting AURKA-CDC25C axis to induce synthetic lethality in ARID1A-deficient colorectal cancer cells. Nat. Commun. 2018, 9, 3212. [Google Scholar] [CrossRef] [PubMed]
- Samlowski, W.E.; Moon, J.; Tuthill, R.J.; Heinrich, M.; Balzer-Haas, N.S.; Merl, S.A.; DeConti, R.C.; Thompson, J.A.; Witter, M.T.; Flaherty, L.E.; et al. A Phase II trial of imatinib mesylate in Merkel cell carcinoma (neuroendocrine carcinoma of the skin). Am. J. Clin. Oncol. 2010, 33, 495–499. [Google Scholar] [CrossRef] [Green Version]
- Shiver, M.B.; Mahmoud, F.; Gao, L. Response to idelalisib in a patient with Stage IV Merkel-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1580–1582. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Goel, S.; DeCristo, M.J.; Watt, A.C.; BrinJones, H.; Sceneay, J.; Li, B.B.; Khan, N.; Ubellacker, J.M.; Xie, S.; Metzger-Filho, O.; et al. CDK4/6 inhibition triggers anti-tumour immunity. Nat. Cell Biol. 2017, 548, 471–475. [Google Scholar] [CrossRef]
- Schaer, D.A.; Beckmann, R.P.; Dempsey, J.A.; Huber, L.; Forest, A.; Amaladas, N.; Li, Y.; Wang, Y.C.; Rasmussen, E.R.; Chin, D.; et al. The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep. 2018, 22, 2978–2994. [Google Scholar] [CrossRef] [Green Version]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.-C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; et al. Genomic aberrations that activate D-type cyclins are associated with enhanced sensitivity to the CDK4 and CDK6 inhibitor Abemaciclib. Cancer Cell 2017, 32, 761–776.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jhaveri, K.; Burris, H.A., 3rd; Yap, T.A.; Hamilton, E.; Rugo, H.S.; Goldman, J.W.; Dann, S.; Liu, F.; Wong, G.Y.; Krupka, H.; et al. The evolution of cyclin dependent kinase inhibitors in the treatment of cancer. Expert Rev. Anticancer Ther. 2021. [Google Scholar] [CrossRef]
- Suski, J.M.; Braun, M.; Strmiska, V.; Sicinski, P. Targeting cell-cycle machinery in cancer. Cancer Cell 2021, 39, 759–778. [Google Scholar] [CrossRef]
- Iwasaki, T.; Matsushita, M.; Nonaka, D.; Kuwamoto, S.; Kato, M.; Murakami, I.; Nagata, K.; Nakajima, H.; Sano, S.; Hayashi, K. Comparison of Akt/mTOR/4E-BP1 pathway signal activation and mutations of PIK3CA in Merkel cell polyomavirus–positive and Merkel cell polyomavirus–negative carcinomas. Hum. Pathol. 2015, 46, 210–216. [Google Scholar] [CrossRef]
- Quartuccio, S.M.; Schindler, K. Functions of Aurora kinase C in meiosis and cancer. Front. Cell Dev. Biol. 2015, 3, 50. [Google Scholar] [CrossRef] [Green Version]
- D’Assoro, A.B.; Ehaddad, T.; Egalanis, E. Aurora-A Kinase as a Promising Therapeutic Target in Cancer. Front. Oncol. 2016, 5, 295. [Google Scholar] [CrossRef] [Green Version]
- Tagal, V.; Wei, S.; Zhang, W.; Brekken, R.A.; Posner, B.A.; Peyton, M.; Girard, L.; Hwang, T.; Wheeler, D.A.; Minna, J.D.; et al. SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX-680 in non-small cell lung cancers. Nat. Commun. 2017, 8, 14098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, F.A.; Li, S.S.-C. Drugging RB1 Deficiency: Synthetic Lethality with Aurora Kinases. Cancer Discov. 2019, 9, 169–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Martinez-Ledesma, E.; Zhang, C.; Gao, F.; Zheng, S.; Ding, J.; Wu, S.; Nguyen, N.; Clifford, S.C.; Wen, P.Y.; et al. Tie2-FGFR1 interaction induces adaptive PI3K inhibitor resistance by upregulating Aurora A/PLK1/CDK1 signaling in glioblastoma. Cancer Res. 2019, 79, 5088–5101. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, B.K.; Kannan, A.; Nguyen, Q.; Gogoi, J.; Zhao, H.; Gao, L. Selective Inhibition of Aurora Kinase A by AK-01/LY3295668 Attenuates MCC Tumor Growth by Inducing MCC Cell Cycle Arrest and Apoptosis. Cancers 2021, 13, 3708. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153708
Das BK, Kannan A, Nguyen Q, Gogoi J, Zhao H, Gao L. Selective Inhibition of Aurora Kinase A by AK-01/LY3295668 Attenuates MCC Tumor Growth by Inducing MCC Cell Cycle Arrest and Apoptosis. Cancers. 2021; 13(15):3708. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153708
Chicago/Turabian StyleDas, Bhaba K., Aarthi Kannan, Quy Nguyen, Jyoti Gogoi, Haibo Zhao, and Ling Gao. 2021. "Selective Inhibition of Aurora Kinase A by AK-01/LY3295668 Attenuates MCC Tumor Growth by Inducing MCC Cell Cycle Arrest and Apoptosis" Cancers 13, no. 15: 3708. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153708