
PRKAR1A and Thyroid Tumors



1
Division of Intramural Population Health Research, Eunice Kennedy Shriver National Institutes of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892, USA
2
Section on Endocrinology and Genetics, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892, USA
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(15), 3834; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153834
Submission received: 16 June 2021
/
Revised: 24 July 2021
/
Accepted: 27 July 2021
/
Published: 30 July 2021
(This article belongs to the Special Issue Advances in the Diagnosis and Treatment of Thyroid Carcinoma)
Abstract
:Simple Summary
In 2021 it is estimated that there will be 44,280 new cases of thyroid cancer in the United States and the incidence rate is higher in women than in men by almost 3 times. Well-differentiated thyroid cancer is the most common subtype of thyroid cancer and includes follicular (FTC) and papillary (PTC) carcinomas. Over the last decade, researchers have been able to better understand the molecular mechanisms involved in thyroid carcinogenesis, identifying genes including but not limited to RAS, BRAF, PAX8/PPARγ chromosomal rearrangements and others, as well as several tumor genes involved in major signaling pathways regulating cell cycle, differentiation, growth, or proliferation. Patients with Carney complex (CNC) have increased incidence of thyroid tumors, including cancer, yet little is known about this association. CNC is a familial multiple neoplasia and lentiginosis syndrome cause by inactivating mutations in the PRKAR1A gene which encodes the regulatory subunit type 1α of protein kinase A. This work summarizes what we know today about PRKAR1A defects in humans and mice and their role in thyroid tumor development, as the first such review on this issue.
Abstract
Thyroid cancer is the most common type of endocrine malignancy and the incidence is rapidly increasing. Follicular (FTC) and papillary thyroid (PTC) carcinomas comprise the well-differentiated subtype and they are the two most common thyroid carcinomas. Multiple molecular genetic and epigenetic alterations have been identified in various types of thyroid tumors over the years. Point mutations in BRAF, RAS as well as RET/PTC and PAX8/PPARγ chromosomal rearrangements are common. Thyroid cancer, including both FTC and PTC, has been observed in patients with Carney Complex (CNC), a syndrome that is inherited in an autosomal dominant manner and predisposes to various tumors. CNC is caused by inactivating mutations in the tumor-suppressor gene encoding the cyclic AMP (cAMP)-dependent protein kinase A (PKA) type 1α regulatory subunit (PRKAR1A) mapped in chromosome 17 (17q22–24). Growth of the thyroid is driven by the TSH/cAMP/PKA signaling pathway and it has been shown in mouse models that PKA activation through genetic ablation of the regulatory subunit Prkar1a can cause FTC. In this review, we provide an overview of the molecular mechanisms contributing to thyroid tumorigenesis associated with inactivation of the RRKAR1A gene.
1. Introduction
1.1. Incidence of Thyroid Cancer
Thyroid cancer is the most common endocrine tumor in the general population and the incidence continues to rise in the United States [1]. The American Cancer Society estimates that there will be 44,280 new cases of thyroid cancer (12,150 in men and 32,130 in women) and about 2200 deaths (1050 in men and 1150 in women) in the United States in 2021 [2]. The increased incidence could be possibly attributed to the increased detection of these tumors with imaging technics (like ultrasound and computed tomography (CT)) that better characterize incidental findings of small thyroid nodules [3].
1.2. Subtypes of Thyroid Cancer
In the majority of patients (about 90%), well-differentiated epithelial thyroid cancer is present; this is further categorized into papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC), based on histological criteria [3,4]. The long-term survival of those patients is excellent, with 5-year relative survival rate (as of 2010–2016) being as high as 98% in all stages (>99% for local tumors and 55% for tumors with distant metastases) [2]. However, FTC tends to behave more aggressively with distant metastases and vascular invasion [5,6] being more common and thus its prognosis is poorer than PTC [7]. The rest of the thyroid carcinomas (~2–3%) include medullary thyroid carcinomas (MTCs) that originate from the calcitonin-producing parafollicular C cells, while anaplastic carcinomas (ATCs) and poorly differentiated carcinomas account for the remaining 7–8% [4]. In addition to the above tumors, benign thyroid tumors that usually present as thyroid nodules as well, include benign hyperplasia or benign follicular adenomas [3].
1.3. Evaluation of a Thyroid Nodule
Thyroid nodules are quite common and are found either clinically or as an incidental finding on imaging studies [8]. The majority of them are benign [9]; only a small percentage harbors thyroid cancer [8]. The initial steps in the evaluation of a thyroid nodule consist of medical history including symptoms (recent onset of hoarseness, neck discomfort or dysphagia), history of head/neck radiation and personal/family history of cancer, followed by physical examination and measurement of serum thyrotropin levels. Ultrasonography (US) is the next step in order to determine the size of the nodule, its characteristics and to assess for cervical lymphadenopathy [10]. If thyrotropin levels are normal or elevated and the nodule size is >1 cm, then fine needle aspiration (FNA) is indicated, according to the American Thyroid Association guidelines [11,12]. If thyrotropin levels are low, then Iodine-123 or technetium-99m thyroid scanning is recommended. In the case that the nodule is nonfunctioning and bigger than 1cm, FNA is the next step. If the cytological interpretation is benign, then repeated FNA is not required unless suspicious features appear in the follow up [12,13]. Currently, US-guided FNA is the gold standard in the diagnosis; however, in about 25% of the cases, the diagnosis remains indeterminate [9,14,15,16,17,18,19,20]. If cytologic results are interpreted as atypia of underdetermined significance or follicular lesion of underdetermined significance, then in the case of high suspicion, assessment of the aspirate for molecular abnormalities (e.g., mutations or rearrangements) is indicated [21].
1.4. Thyroid Cancer as Part of Genetic Syndromes
Thyroid malignancies are also associated with at least two syndromes with inherited tumor predisposition, Cowden syndrome (CS, OMIM# 158350) and Carney Complex (CNC, OMIM #160980). CS is a multiple hamartoma syndrome, including FTC, brain and breast cancer. It is caused by inactivating mutations in the PTEN gene, a dual-specificity phosphatase that negatively regulates PI3 Kinase/AKT pathway; mutations in this gene have been detected in 5% of FTCs [22]; however, a mouse harboring a deletion of Pten in the thyroid developed thyroid hyperplasia and not FTC [23].
In this review, we will focus on CNC, which is a multiple neoplasia syndrome that presents as the complex of myxomas, spotty skin pigmentation and endocrine tumors (Table 1) [24]. CNC is caused by inactivating mutations in the PRKAR1A gene (mapped in 17q22–24) [25]; somatic mutations in this gene have been reported in sporadic cases of thyroid cancer [26]. In tumors associated with CNC as well as in thyroid and adrenal tumors with downregulation of PRKAR1A, allelic losses of the 17q22–24 PRKAR1A chromosomal locus are frequently identified and are associated with changes in PKA activity [26,27,28,29].
2. PRKAR1A Structure and Function
Cyclic adenosine monophosphate (cAMP)-dependent protein kinase type 1-alpha regulatory subunit is encoded by the PRKAR1A gene. PRKAR1A consists of 11 exons; ten of them (2–11) are coding. Protein kinase A (PKA) (Figure 1), a serine/threonine kinase, is a second messenger-dependent enzyme and it is involved in G-protein coupled intracellular pathways. It is the main mediator of cAMP actions for various cellular processes in mammals, including cell differentiation, proliferation, and apoptosis [30,31,32].
The PKA holoenzyme is a hetero-tetramer composed of two regulatory (R) subunits and each is bound to one catalytic (C) subunit [33]. Four subtypes of R (RIα, RIβ, RIIα, RIIβ) and four subtypes of C (Cα, Cβ, Cγ and Prkx) subunits have been identified so far. A gene is coding each R (PRKR1A, PRKR1B, PRKR2A, PRKR2B) and each C (PRKACA, PRKACB, PRKACG, PRKX) subunit, respectively [33,34]. Two major isozymes have been identified, type I and type II PKA, based on their chromatographic elution patterns [32]; they are comprised of homodimers of either RIα and RIβ or RIIα and RIIβ, respectively [31,35]. In the basal state, the catalytic subunits bind mostly to type II subunits [31,35,36,37]. When cAMP binds to the R subunits, it alters their conformation; this causes the dissociation of each active C subunit from the dimer with the corresponding R subunit. Following that, the free C subunits phosphorylate threonine and serine residues of proteins that are critical to the activation of downstream processes [38,39,40].
RIα haploinsufficiency, as shown by mice and human studies, predisposes to the development of tumors [29,41]. The majority of PRKAR1A mutations result in premature stop codons with unstable mRNAs undergoing nonsense-mediated decay [25,42]. In the thyroid, PKA through the production of cAMP, signals downstream of thyrotropin (TSH) on cell proliferation and differentiation; increased levels of TSH in humans have been associated with thyroid tumors [43]. In addition, in a series of thyroid tumors, the Cα subunit was investigated but no mutations were detected [44].
3. Role of PRKAR1A in Thyroid Cancer
3.1. Mouse Studies
3.1.1. Mouse Models
In 2004, Griffin et al. generated a mouse model carrying an antisense transgene for Prkar1a exon 2 (X2AS) under the control of a tetracycline responsive promoter (the Tg(Prakr1a*x2as)1Stra, Tg(tTAhCMV)3Uh, or tTA/X2AS) [45]. Increased cAMP signaling was demonstrated due to significant Prkar1a downregulation and the mice exhibited a more severe phenotype with high incidence of thyroid lesions (thyroid follicular hyperplasia and adenomas). This was an extremely rare finding in wild type animals but quite common in those with the genetic defect (as it is common among patients with CNC). Furthermore, the lesions were associated with allelic loss of the Prkar1a locus on chromosome 11 as it happens in thyroid tumors with PRKAR1A mutations. Moreover, tumor tissues demonstrated an increase in the activity of type II PKA and higher RIIβ levels, an abnormal cAMP response.
In a later study, Prkar1a haploinsufficiency in mice was investigated. It was shown that Prkar1a haploinsufficiency leads to tumor development arising in cAMP-responsive tissues, including among others, benign and malignant thyroid neoplasms [41]. Mice heterozygous for a conventional null allele of Prkar1a (Prkar1aΔ2/+ mice) were generated. These mice developed tumors in the same spectrum as CNC patients. Thyroid neoplasms were present in 10% of Prkar1aΔ2/+ mice [41]. In addition, allelic loss occurred in a portion of tumor cells, as indicated by genetic analysis, suggesting that complete loss of Prkar1a plays a vital role in tumor formation.
A different mouse model, carrying a thyroid-specific deletion of Prkar1a (Tpo-R1αKO) was studied [46]. In 43% of mice, FTC was observed by 1 year of age. However, distant hematogenous metastases were not present, which is a key feature of FTC in humans [46]; this could potentially suggest that metastases may be triggered by another genetic mutation in the case of Prkar1a mutation in the thyroid. An interesting observation by the authors was that thyroid ablation of PRKAR1A/Prkar1a is the only genetic change that has been described that results in FTC in both mice and humans.
3.1.2. Activation of mTOR Pathway
The role of PKA as a key regulator of FTC has also been suggested by a recent study demonstrating a concurrent activation of PKA and mTOR. In this study a double Prkar1a-Pten knockout mouse (DRP-TpoKO mice) with thyroid-specific deletion of both genes was generated and was compared to signaling alterations to human FTCs [1]; they found that mice developed aggressive FTC that exhibited 100% penetrance by 8 weeks of age. In addition, well-differentiated lung metastases appeared to be common in these mice (approximately one third of them), mimicking the human disease. The signaling pathways were analyzed and it was shown that PKA and the mammalian target of rapamycin (mTOR) pathways were consistently activated. mTOR has an essential role in promoting the metabolic changes that occur during tumorigenesis and is regulated by the AMP-dependent protein kinase (AMPK) [47]. AMPK is activated under nutrition restriction or increase in the AMP/ATP ration in order to increase energy production [48].
It has been suggested before that mTOR could be activated by Prkar1a deletion and that it could possibly interact with Prkar1a directly [49], but the data remain controversial [50]. Furthermore, activation of mTOR by TSH has been suggested to be partly due to PKA phosphorylation of the target of rapamycin complex 1 complex member PRAS40 [51]. Mouse models have been developed over the years that recapitulate how human FTCs progress from benign follicular adenoma (at one year of age) in the Pten-TpoKO [23] to locally invasive FTC as in the R1a-TpoKO [46] and subsequently to invasive and distantly metastatic FTC. The authors identified PKA and mTOR as essential signaling pathways and showed that activation of mTOR can occur independently of Akt [1]. Further, the concurrent activation of PKA and mTOR that was observed in human FTCs led to the conclusion that PKA activates mTOR/p70S6K that results in thyroid cancer, indicating that PKA is a vital component regulating FTC in both mice and humans [1].
The same group reported that, in FTCs, both in mice and humans, AMPK and mTOR pathways are activated concomitantly [52]. They showed that the tumor suppressor that causes Peutz–Jeghers syndrome, LKB1, mediates the signaling from PKA to AMPK in driving tumorigenesis [53,54]. The role of AMPK in the development of cancer has not been determined yet; according to the literature, it can act either as tumor promoter or tumor suppressor [55,56]. LKB1, like AMPK, can act as tumor promoter/suppressor as well, depending on the context [55,57,58,59,60,61]. Even though it typically suppresses the activity of mTOR [56,62], there is evidence that it can also act as a tumor promoter [63,64,65], which means that its functions depend on the type of tissue and other intracellular signals that may be present.
3.1.3. Targeting Downstream Effectors of cAMP
Because of the various effects of cAMP in physiological responses, therapies targeting cAMP signaling result in side effects; thus, understanding downstream targets of cAMP signaling has been attempted in a number of studies [66,67]. The roles of Rap1 and Epac1 in Prkar1a-associated thyroid carcinogenesis have been studied [68]. Rap1 is a small GTPase essential for effective signal transduction. There are two isoforms and each one is encoded by a separate gene, Rap1a and Rap1b, respectively. The activity of Rap1 has been shown to be regulated by both PKA and cAMP though signaling by TSH [69]. Increased Rap activity has been linked to various cancers, including thyroid cancer, while dysregulation of Rap1 has been postulated to contribute to the development of malignancy [70,71,72,73,74]. Epac (Exchange protein directly activated by cAMP) proteins are intracellular sensors for cAMP and mediate its effects to activate Rap1 [75,76]. The two isoforms include Epac1 which is ubiquitously expressed, with particularly high levels in the thyroid, among other tissues, and Epac2 which is not detected in the thyroid; however, it is expressed in a limited number of other tissues [76,77]. Epac regulates Rap activity in concert with and independently of PKA, and the effects—either stimulatory or inhibitory—seem to depend on the cellular context and the type of stimuli [69,75,78,79]. In addition, it has been shown that Epac1 plays a role in cell migration and invasion in other types of cancer [78,80]. Loss of Rap, specifically of the Rap1b isoform, in Prkar1a KO thyroids in the setting of overactivation of the PKA pathway, resulted in reduced risk of developing thyroid cancer by 65%; this occurred independently of Epac1 as its deletion did not have any effect in PKA-Rap1 associated thyroid tumorigenesis, underlying the essential role of PKA-Rap1 signaling in the development of FTC [68]. However, even though tumor suppression happened to a significant extent, the carcinogenic phenotype was not completely rescued, which led to the speculation that more complex signaling interactions may be involved [68].
These findings were further supported by other studies that showed that Rap proteins can be directly regulated by PKA using a specific phosphorylation site at serine 180 on Rap1a and serine 179 on Rap1b [81]. When PKA phosphorylates Rap, it regulates its subcellular localization, and its downstream effectors such as ERK and Rap-dependent regulation of cell migration [82,83]. These previous studies indicate that PKA can control Rap action and downstream cellular processes directly suggesting that PKA-Rap1 pathway is independent of Epac1 in thyroid cancer. On the other hand, previous studies have shown, that both PKA and Epac signal to Rap1 downstream of TSH [69,75], but it seems to be tissue-dependent [78,79].
In combination, these studies demonstrated that cAMP or PKA signaling or both play an important role in tumor development and that additional factors may contribute to Prkar1a haploinsufficiency in causing those tumors. Trp53+/− mice and other animal models for diseases like CNC, including Peutz–Jeghers and neurofibromatosis type 1, did not exhibit the same phenotype as in humans; it only occurred when one or more tumor suppressor genes were knocked out as well [84,85,86]. Prkar1a haploinsufficiency in addition to either Trp53 or Rb1 haploinsufficiency resulted in more tumors and decreased survival compared to Trp53+/− or Rb1+/− mice [87]. Specifically, Rb1+/− Prkar1a+/− mice developed more MTCs than Rb1+/− mice [87].
3.2. Studies in Humans
Further evidence to support the involvement of PKA in thyroid tumors was demonstrated by studying the PRKAR1A gene in thyroid tissue from patients with CNC [29]. The involvement of the thyroid in the syndrome was reported for the first time twelve years after CNC was first described [88], in 1997 [89]. In a cohort of 53 individuals with familial CNC, thyroid disease was identified in 11% of patients; of them, three were studied in detail, two with thyroid carcinomas (one PTC, one FTC) and one patient with a benign follicular adenoma. [89]. In addition, 60% of patients with the sporadic form of the complex exhibited thyroid gland lesions of follicular origin [89]. The authors concluded that thyroid carcinomas may develop in situ from precursor benign lesions in these patients. It is important to note that patients’ ethnicity does not seem to play a role in CNC phenotype that include thyroid carcinomas.
Since the PRKAR1A gene was identified as causal in CNC, many disease-causing mutations have been identified [24,90]. Sandrini et al. showed that in thyroid cancer the activity of PKA is greater than in adenomas, partly due to genetic defects in the PRKAR1A gene and/or locus [26]. The region 17q22–24 was frequently lost in cancer but not in benign tumors. In addition, it was shown that RIα, the most abundant regulatory subunit of cAMP-dependent PKA [91], in thyroid cells, possibly exhibits a tumor-suppressor function, as indicated by decreased expression of the RIα subunit in carcinomas and by the losses of PRKAR1A 17q22–24 locus in about 50% of all informative cancers. It has been known that the activation of cAMP/PKA pathway is involved in normal thyroid cell growth [92]; the same appears to be true for thyroid adenomas, while in the case of PTCs inhibition is induced [93]. The results suggested that PRKAR1A is indeed involved in sporadic thyroid tumors, along with other genes [94,95,96,97], some of which could be associated with the PKA pathway [91]. Any disruption of that, because of deficiency of the RIα subunit, could lead to cAMP-dependent PKA mediated cell proliferation and/or stimulation of other pathways linked to proliferation of thyroid cells [91,98].
In a recent series of 353 CNC patients from 185 families, patients from various ethnicities and with a wide range of clinical manifestations were studied [99]. More than 60% of them harbored mutations in the PRKAR1A gene. In 25% of patients, thyroid tumors were present while thyroid cancer (either FTC or PTC or both) was present in 2.5% of cases. In addition, thyroid tumors (p = 0.016) were more frequent in PRKAR1A carriers and presented at a younger age (p = 0.03). Moreover, they were more commonly associated with the ‘hot spot’ c.491–492delTG mutation in comparison with all other PRKAR1A defects. It was also observed that patients with no mutations of the PRKAR1A gene or its genomic locus 17q22–24, were less likely to develop thyroid tumors. In a review of 26 patients, in 61% of them benign lesions (including follicular adenoma, follicular hyperplasia or nodular hyperplasia) were detected, while 38% of them had thyroid carcinomas (seven with FTC and three with PTC). The majority of patients presented with an asymptomatic thyroid nodule and included middle-aged women [100].
4. Other Molecular Events in Thyroid Cancer
A significant number of mutations in thyroid cancer involves encoding genes of the MAPK and PI3K/AKT pathways. Mutated genes that affect these pathways encode the signal transduction molecules RAS, BRAF and NTRK1 and RET receptor tyrosine kinases. These mutations are present in approximately 70% of patients with PTCs and they exhibit particular clinical manifestations as well as specific histopathological characteristics in the tumor level [101,102,103,104]. Among FTCs, RAS mutations and PAX8/PPARγ rearrangements are common [105]. Because PAX8 is important for the development of the thyroid, it has been speculated that the fusion of PPARγ and PAX8 can lead to cancer by activation of aberrant gene transcription [3]. PAX8/PPARγ is found in FTCs with a frequency of 30–35% and in a very small percentage of the follicular variant PTCs and follicular adenomas [106,107,108,109]. Thyroid tumors harboring RAS mutations, most commonly NRAS and HRAS mutations, include FTCs in 40–50%, PTCs in 10–20% and 20–40% of anaplastic and poorly differentiated carcinomas [110,111,112,113,114,115,116]. Because PTCs that harbor RAS mutations form neoplastic follicles and no papillary structures, they are known as follicular variant of PTC [101,117]. Benign follicular adenomas have also been found to harbor RAS mutations in 20–40%, indicating that they may be precursors of RAS-positive FTCs and follicular variant of PTCs [118,119,120,121]. Furthermore, BRAFV600E mutation represents 98–99% of all BRAF mutations in thyroid cancer [122,123,124]. It accounts for 40–45% of classic PTCs, 30–40% of ATCs, and 20–40% of poorly differentiated thyroid carcinomas [125,126,127,128].
Mutations in BRAF and RAS are thought to represent an early event in the progression of thyroid cancer, given that they are present in poorly and well-differentiated thyroid cancer, as well as in ATCs. On the other hand, additional genetic alterations are usually present in poorly differentiated carcinomas and ATCs; these constitute late events that may be necessary for tumor dedifferentiation. These genetic alterations include mutations in the TP53 and CTNNB1 genes and encoding genes of the PI3K/AKT signaling pathway [105]. In 50–80% of ATCs, point mutations that lead to loss-of-function of TP53 have been identified; these are very rare in well-differentiated thyroid cancer [129,130]. CTNNB1 mutations occur in approximately 60% of ATCs [131,132].
5. Medullary Thyroid Cancer as Part of MEN2 Syndromes
About one-third of MTCs are hereditary, presenting as multicentric and bilateral, in contrast with sporadic cases that are a single unilateral tumor [133,134]. They present as part of MEN2A (70–80%), MEN2B (5%), or familial MTC (FMTC) (10–20%). The first inherited subtype of MTC, MEN2A, consists of primary hyperparathyroidism, pheochromocytoma and MTC in which it can occur early in life (approximately 5 years of age) in contrast with sporadic cases that presents between 15 and 20 years [134,135]. MEN2B is characterized by pheochromocytoma, MTC and non-endocrine diseases such as mucosal neuromas, intestinal tumors (most commonly ganglioneuromas) and Marfanoid habitus [135]. In the case of FMTC, only the thyroid gland is affected, but in a significant number of relatives in the same family, usually between the ages of 20 and 40 [135,136,137]. Activating germline RET mutations have been identified as the main cause of up to 98% of hereditary MTCs and up to half of sporadic cases [138]. Depending on the mutated residue within the RET protein, the phenotype may differ [139,140,141,142]. Families with two or more members with MTC are referred for genetic counseling and screening, if positive they undergo further testing for hyperparathyroidism and pheochromocytoma [2,143,144]. In the case of sporadic MTCs, somatic RET mutations, particularly M918T, has been shown to be associated with more aggressive disease and worse prognosis [144,145].
6. Anaplastic Thyroid Carcinoma
ATC is a rare (1–2%) but very aggressive type of thyroid cancer [146] with average age at diagnosis over 70 years [147]. It is considered to evolve from dedifferentiation of a pre-existing DTC caused by accumulation of several genetic alterations that lead to disruption of two signaling pathways that are involved in cell proliferation, PI3K-AKT and MAPK [148,149,150]. The most common mutations include TP53, which is considered a genetic hallmark of ATC, as well as RAS, BRAF, PIK3CA [151,152], mutations that have also been identified in DTC [153]. Median survival is usually less than 6 months after diagnosis and the mortality rate is >90% [154,155]. Due to its extremely aggressive nature, it is critical to be diagnosed promptly. Clinical symptoms are usually used for the diagnosis, in contrast with DTC in which diagnosis is made by FNA of a suspicious nodule [147]. The symptoms can last from 4 weeks to 11 months and usually consist of a rapidly enlarging neck mass along with vocal cord paralysis and dyspnea [147].
7. Systemic Treatments for Thyroid Cancer
Two multikinase inhibitors (MKI), lenvatinib and sorafenib, are currently approved by the US Food and Drug Administration (FDA) for the treatment of advanced DTC. Sorafenib was approved based on the favorable results of a placebo-controlled phase 3 clinical trial (DECISION) [156]. The positive results of the lenvatinib phase 3 SELECT trial [157] as well as a phase 2 study led to the approval of that drug [158]. Cabozatinib and vandetanib are approved by the FDA for the treatment of MTC. Vandetanib is approved for symptomatic, unresectable, locally advanced, or metastatic MTC in patients based on a phase 3 trial (ZETA) [159]. Cabozantinib was studied in a phase 3 clinical trial (EXAM) [160] and showed good results while another clinical trial in MTC patients is still active (EXAMINER, NCT01896479). RET-inhibitors have been studied as well for thyroid cancers that harbor RET mutations (NCT03157128, NCT04211337, NCT03906331, NCT04280081, NCT03037385).
7.1. Immunotherapy
In the recent years, immunotherapy has emerged as a new transformative approach into the body’s natural antitumor defenses. To date, there is no approved immunotherapy for advanced thyroid cancer. A few clinical trials using novel immunotherapy agents like programmed cell death protein 1 (PD-1) checkpoint inhibitors are ongoing. Pembrolizumab in an Ib phase trial (KEYNOTE) showed a tumor size reduction of 35–50% in PTC and FTC. The use of another anti-PD1 agent (spartalizumab) was evaluated in progressive ATC that responded to therapy [161]. In an ongoing phase 2 clinical trial (NCT03246958), the efficacy of the combination of nivolumab (anti-PD1-1) and ipilimumab (anti-CTLA-4- cytotoxic T-lymphocyte-associated protein 4) was evaluated in patients with aggressive thyroid cancer. In addition, multiple clinical trials with VEGF and/or VEGF inhibitor and immune checkpoint inhibitors have been designed. Pemproblizumab plus lenvatinib was investigated in a phase 2 trial for unresectable ATC (NCT04171622) as well as in a randomized study in a small group of advanced ATC and PDTC [162]. The same combination is under study in DTC and PDTC naïve or progressing after lenvatinib patients (NCT02973997). Triple combined therapy (cabozantinib plus nivolumab and ipilimumab) is under evaluation for DTC and PDTC (NCT03914300).
7.2. Treatment for PRKAR1A-Associated Thyroid Tumors
To date, there is no medical treatment targeting cAMP/PKA signaling in CNC. Surgical treatment is the treatment of choice in patients with PRKAR1A-associated thyroid tumor [163].
8. Clinical Surveillance in Patients with PRKAR1A-Associated Thyroid Tumors
Human studies in CNC underly the importance of investigating thyroid nodules in these patients. Multiple thyroid nodules are present in up to 75% of patients with CNC on thyroid ultrasound; the majority of them are non-functioning follicular adenomas [164]. However, thyroid carcinomas are common as well. Early detection is vital and CNC patients should be followed with long-term clinical and/or ultrasound surveillance with biopsy of suspicious nodules, for early detection of carcinomas [164].
Because CNC is inherited in an autosomal dominant manner, each child of an affected individual has a 50% chance of inheriting the pathogenic variant. Most of the affected patients (approximately 70%) have an affected parent. In the case that the pathogenic variant is known in a family, prenatal testing may be recommended [164].
9. Conclusions
In summary, recent advances in molecular mechanisms of thyroid cancer have improved cancer prognosis and detection. PRKAR1A, a regulator of PKA activity, is possibly involved in the molecular events that contribute to thyroid cancer. Identifying the genetic basis of PRKAR1A-associated thyroid tumors is important as it will provide better clinical management to these patients.
Funding
This work was supported by the project Z01-HD008920 (Principal Investigator: C.A.S.) of the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD), National Institutes of Health (NIH), Bethesda, MD, USA.
Conflicts of Interest
C.A.S. holds patent on the PRKAR1A, PDE11A, and GPR101 genes and/or their function and his laboratory has received research funding from Pfizer, Inc. F.R.F. holds patent on the GPR101 gene and/or its function. C.A.S. is receiving compensation by ELPEN, Inc. Neither Pfizer, Inc. nor ELPEN, Inc. had any role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. All authors declare no conflict of interest.
References
- Pringle, D.R.; Vasko, V.V.; Yu, L.; Manchanda, P.K.; Lee, A.A.; Zhang, X.; Kirschner, J.M.; Parlow, A.F.; Saji, M.; Jarjoura, D.; et al. Follicular thyroid cancers demonstrate dual activation of PKA and mTOR as modeled by thyroid-specific deletion of Prkar1a and Pten in mice. J. Clin. Endocrinol. Metab. 2014, 99, E804–E812. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.; Mulligan, L.M.; Smith, D.P.; Healey, C.S.; Frilling, A.; Raue, F.; Neumann, H.P.; Ponder, M.A.; Ponder, B.A. Low frequency of germline mutations in the RET proto-oncogene in patients with apparently sporadic medullary thyroid carcinoma. Clin. Endocrinol. 1995, 43, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Raman, P.; Koenig, R.J. Pax-8-PPAR-gamma fusion protein in thyroid carcinoma. Nat. Rev. Endocrinol. 2014, 10, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Younis, E. Oncogenesis of Thyroid Cancer. Asian Pac. J. Cancer Prev. 2017, 18, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Besic, N.; Auersperg, M.; Golouh, R. Prognostic factors in follicular carcinoma of the thyroid—A multivariate survival analysis. Eur. J. Surg. Oncol. 1999, 25, 599–605. [Google Scholar] [CrossRef]
- Nguyen, X.V.; Roy Choudhury, K.; Tessler, F.N.; Hoang, J.K. Effect of Tumor Size on Risk of Metastatic Disease and Survival for Thyroid Cancer: Implications for Biopsy Guidelines. Thyroid 2018, 28, 295–300. [Google Scholar] [CrossRef]
- Sugino, K.; Ito, K.; Nagahama, M.; Kitagawa, W.; Shibuya, H.; Ohkuwa, K.; Yano, Y.; Uruno, T.; Akaishi, J.; Kameyama, K.; et al. Prognosis and prognostic factors for distant metastases and tumor mortality in follicular thyroid carcinoma. Thyroid 2011, 21, 751–757. [Google Scholar] [CrossRef]
- Burman, K.D.; Wartofsky, L. Clinical Practice. Thyroid Nodules. N. Engl. J. Med. 2015, 373, 2347–2356. [Google Scholar] [CrossRef]
- Yassa, L.; Cibas, E.S.; Benson, C.B.; Frates, M.C.; Doubilet, P.M.; Gawande, A.A.; Moore, F.D., Jr.; Kim, B.W.; Nose, V.; Marqusee, E.; et al. Long-term assessment of a multidisciplinary approach to thyroid nodule diagnostic evaluation. Cancer 2007, 111, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Cooper, D.S. American Thyroid Association Guidelines Taskforce on Thyroid Nodules and Differentiated Thyroid Cancer. Revised American Thyroid Association management guidelines for patients with thyroid nodules and differentiated thyroid cancer. Thyroid 2009, 19, 1167–1214. [Google Scholar] [CrossRef] [Green Version]
- Frates, M.C.; Benson, C.B.; Charboneau, J.W.; Cibas, E.S.; Clark, O.H.; Coleman, B.G.; Cronan, J.J.; Doubilet, P.M.; Evans, D.B.; Goellner, J.R.; et al. Management of thyroid nodules detected at US: Society of Radiologists in Ultrasound consensus conference statement. Radiology 2005, 237, 794–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M.; et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oertel, Y.C.; Miyahara-Felipe, L.; Mendoza, M.G.; Yu, K. Value of repeated fine needle aspirations of the thyroid: An analysis of over ten thousand FNAs. Thyroid 2007, 17, 1061–1066. [Google Scholar] [CrossRef]
- Brander, A.; Viikinkoski, P.; Nickels, J.; Kivisaari, L. Thyroid gland: US screening in a random adult population. Radiology 1991, 181, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Papini, E.; Guglielmi, R.; Bianchini, A.; Crescenzi, A.; Taccogna, S.; Nardi, F.; Panunzi, C.; Rinaldi, R.; Toscano, V.; Pacella, C.M. Risk of malignancy in nonpalpable thyroid nodules: Predictive value of ultrasound and color-Doppler features. J. Clin. Endocrinol. Metab. 2002, 87, 1941–1946. [Google Scholar] [CrossRef]
- Carroll, B.A. Asymptomatic thyroid nodules: Incidental sonographic detection. AJR Am. J. Roentgenol. 1982, 138, 499–501. [Google Scholar] [CrossRef]
- Sclabas, G.M.; Staerkel, G.A.; Shapiro, S.E.; Fornage, B.D.; Sherman, S.I.; Vassillopoulou-Sellin, R.; Lee, J.E.; Evans, D.B. Fine-needle aspiration of the thyroid and correlation with histopathology in a contemporary series of 240 patients. Am. J. Surg. 2003, 186, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Greaves, T.S.; Olvera, M.; Florentine, B.D.; Raza, A.S.; Cobb, C.J.; Tsao-Wei, D.D.; Groshen, S.; Singer, P.; Lopresti, J.; Martin, S.E. Follicular lesions of thyroid: A 5-year fine-needle aspiration experience. Cancer 2000, 90, 335–341. [Google Scholar] [CrossRef]
- Tomimori, E.; Pedrinola, F.; Cavaliere, H.; Knobel, M.; Medeiros-Neto, G. Prevalence of incidental thyroid disease in a relatively low iodine intake area. Thyroid 1995, 5, 273–276. [Google Scholar] [CrossRef]
- Wiest, P.W.; Hartshorne, M.F.; Inskip, P.D.; Crooks, L.A.; Vela, B.S.; Telepak, R.J.; Williamson, M.R.; Blumhardt, R.; Bauman, J.M.; Tekkel, M. Thyroid palpation versus high-resolution thyroid ultrasonography in the detection of nodules. J. Ultrasound Med. 1998, 17, 487–496. [Google Scholar] [CrossRef]
- Burman, K.D.; Wartofsky, L. Thyroid Nodules. N. Engl. J. Med. 2016, 374, 1294–1295. [Google Scholar] [CrossRef] [PubMed]
- Nagy, R.; Ganapathi, S.; Comeras, I.; Peterson, C.; Orloff, M.; Porter, K.; Eng, C.; Ringel, M.D.; Kloos, R.T. Frequency of germline PTEN mutations in differentiated thyroid cancer. Thyroid 2011, 21, 505–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeager, N.; Klein-Szanto, A.; Kimura, S.; Di Cristofano, A. Pten loss in the mouse thyroid causes goiter and follicular adenomas: Insights into thyroid function and Cowden disease pathogenesis. Cancer Res. 2007, 67, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratakis, C.A.; Kirschner, L.S.; Carney, J.A. Clinical and molecular features of the Carney complex: Diagnostic criteria and recommendations for patient evaluation. J. Clin. Endocrinol. Metab. 2001, 86, 4041–4046. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, L.S.; Sandrini, F.; Monbo, J.; Lin, J.P.; Carney, J.A.; Stratakis, C.A. Genetic heterogeneity and spectrum of mutations of the PRKAR1A gene in patients with the carney complex. Hum. Mol. Genet. 2000, 9, 3037–3046. [Google Scholar] [CrossRef]
- Sandrini, F.; Matyakhina, L.; Sarlis, N.J.; Kirschner, L.S.; Farmakidis, C.; Gimm, O.; Stratakis, C.A. Regulatory subunit type I-alpha of protein kinase A (PRKAR1A): A tumor-suppressor gene for sporadic thyroid cancer. Genes Chromosomes Cancer 2002, 35, 182–192. [Google Scholar] [CrossRef]
- Bertherat, J.; Groussin, L.; Sandrini, F.; Matyakhina, L.; Bei, T.; Stergiopoulos, S.; Papageorgiou, T.; Bourdeau, I.; Kirschner, L.S.; Vincent-Dejean, C.; et al. Molecular and functional analysis of PRKAR1A and its locus (17q22-24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res. 2003, 63, 5308–5319. [Google Scholar]
- Bossis, I.; Voutetakis, A.; Bei, T.; Sandrini, F.; Griffin, K.J.; Stratakis, C.A. Protein kinase A and its role in human neoplasia: The Carney complex paradigm. Endocr. Relat. Cancer 2004, 11, 265–280. [Google Scholar] [CrossRef]
- Kirschner, L.S.; Carney, J.A.; Pack, S.D.; Taymans, S.E.; Giatzakis, C.; Cho, Y.S.; Cho-Chung, Y.S.; Stratakis, C.A. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat. Genet. 2000, 26, 89–92. [Google Scholar] [CrossRef]
- McKnight, G.S.; Clegg, C.H.; Uhler, M.D.; Chrivia, J.C.; Cadd, G.G.; Correll, L.A.; Otten, A.D. Analysis of the cAMP-dependent protein kinase system using molecular genetic approaches. Recent Prog. Horm. Res. 1988, 44, 307–335. [Google Scholar] [CrossRef]
- Cho-Chung, Y.S.; Pepe, S.; Clair, T.; Budillon, A.; Nesterova, M. cAMP-dependent protein kinase: Role in normal and malignant growth. Crit. Rev. Oncol. Hematol. 1995, 21, 33–61. [Google Scholar] [CrossRef]
- Skalhegg, B.S.; Tasken, K. Specificity in the cAMP/PKA signaling pathway. Differential expression, regulation, and subcellular localization of subunits of PKA. Front. Biosci. 2000, 5, D678–D693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossis, I.; Stratakis, C.A. Minireview: PRKAR1A: Normal and abnormal functions. Endocrinology 2004, 145, 5452–5458. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.Q.; Stratakis, C.A. How does cAMP/protein kinase A signaling lead to tumors in the adrenal cortex and other tissues? Mol. Cell Endocrinol. 2011, 336, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Amieux, P.S.; McKnight, G.S. The essential role of RI alpha in the maintenance of regulated PKA activity. Ann. N. Y. Acad. Sci. 2002, 968, 75–95. [Google Scholar] [CrossRef]
- Cho-Chung, Y.S.; Nesterova, M.; Pepe, S.; Lee, G.R.; Noguchi, K.; Srivastava, R.K.; Srivastava, A.R.; Alper, O.; Park, Y.G.; Lee, Y.N. Antisense DNA-targeting protein kinase A-RIA subunit: A novel approach to cancer treatment. Front. Biosci. 1999, 4, D898–D907. [Google Scholar] [CrossRef] [Green Version]
- Scott, J.D. Cyclic nucleotide-dependent protein kinases. Pharmacol. Ther. 1991, 50, 123–145. [Google Scholar] [CrossRef]
- Griffioen, G.; Thevelein, J.M. Molecular mechanisms controlling the localisation of protein kinase A. Curr. Genet. 2002, 41, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Colledge, M.; Scott, J.D. AKAPs: From structure to function. Trends Cell Biol. 1999, 9, 216–221. [Google Scholar] [CrossRef]
- Wang, L.; Sunahara, R.K.; Krumins, A.; Perkins, G.; Crochiere, M.L.; Mackey, M.; Bell, S.; Ellisman, M.H.; Taylor, S.S. Cloning and mitochondrial localization of full-length D-AKAP2, a protein kinase A anchoring protein. Proc. Natl. Acad. Sci. USA 2001, 98, 3220–3225. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, L.S.; Kusewitt, D.F.; Matyakhina, L.; Towns, W.H., 2nd; Carney, J.A.; Westphal, H.; Stratakis, C.A. A mouse model for the Carney complex tumor syndrome develops neoplasia in cyclic AMP-responsive tissues. Cancer Res. 2005, 65, 4506–4514. [Google Scholar] [CrossRef] [Green Version]
- Robinson-White, A.; Hundley, T.R.; Shiferaw, M.; Bertherat, J.; Sandrini, F.; Stratakis, C.A. Protein kinase-A activity in PRKAR1A-mutant cells, and regulation of mitogen-activated protein kinases ERK1/2. Hum. Mol. Genet. 2003, 12, 1475–1484. [Google Scholar] [CrossRef] [Green Version]
- Haymart, M.R.; Repplinger, D.J.; Leverson, G.E.; Elson, D.F.; Sippel, R.S.; Jaume, J.C.; Chen, H. Higher serum thyroid stimulating hormone level in thyroid nodule patients is associated with greater risks of differentiated thyroid cancer and advanced tumor stage. J. Clin. Endocrinol. Metab. 2008, 93, 809–814. [Google Scholar] [CrossRef] [Green Version]
- Esapa, C.T.; Harris, P.E. Mutation analysis of protein kinase A catalytic subunit in thyroid adenomas and pituitary tumours. Eur. J. Endocrinol. 1999, 141, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, K.J.; Kirschner, L.S.; Matyakhina, L.; Stergiopoulos, S.G.; Robinson-White, A.; Lenherr, S.M.; Weinberg, F.D.; Claflin, E.S.; Batista, D.; Bourdeau, I.; et al. A transgenic mouse bearing an antisense construct of regulatory subunit type 1A of protein kinase A develops endocrine and other tumours: Comparison with Carney complex and other PRKAR1A induced lesions. J. Med. Genet. 2004, 41, 923–931. [Google Scholar] [CrossRef] [Green Version]
- Pringle, D.R.; Yin, Z.; Lee, A.A.; Manchanda, P.K.; Yu, L.; Parlow, A.F.; Jarjoura, D.; La Perle, K.M.; Kirschner, L.S. Thyroid-specific ablation of the Carney complex gene, PRKAR1A, results in hyperthyroidism and follicular thyroid cancer. Endocr. Relat. Cancer 2012, 19, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavrakis, M.; Lippincott-Schwartz, J.; Stratakis, C.A.; Bossis, I. mTOR kinase and the regulatory subunit of protein kinase A (PRKAR1A) spatially and functionally interact during autophagosome maturation. Autophagy 2007, 3, 151–153. [Google Scholar] [CrossRef] [Green Version]
- Day, M.E.; Gaietta, G.M.; Sastri, M.; Koller, A.; Mackey, M.R.; Scott, J.D.; Perkins, G.A.; Ellisman, M.H.; Taylor, S.S. Isoform-specific targeting of PKA to multivesicular bodies. J. Cell Biol. 2011, 193, 347–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blancquaert, S.; Wang, L.; Paternot, S.; Coulonval, K.; Dumont, J.E.; Harris, T.E.; Roger, P.P. cAMP-dependent activation of mammalian target of rapamycin (mTOR) in thyroid cells. Implication in mitogenesis and activation of CDK4. Mol. Endocrinol. 2010, 24, 1453–1468. [Google Scholar] [CrossRef]
- Kari, S.; Vasko, V.V.; Priya, S.; Kirschner, L.S. PKA Activates AMPK Through LKB1 Signaling in Follicular Thyroid Cancer. Front. Endocrinol. 2019, 10, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenne, D.E.; Reimann, H.; Nezu, J.; Friedel, W.; Loff, S.; Jeschke, R.; Muller, O.; Back, W.; Zimmer, M. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 1998, 18, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, A.; Markie, D.; Tomlinson, I.; Avizienyte, E.; Roth, S.; Loukola, A.; Bignell, G.; Warren, W.; Aminoff, M.; Hoglund, P.; et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998, 391, 184–187. [Google Scholar] [CrossRef]
- Monteverde, T.; Muthalagu, N.; Port, J.; Murphy, D.J. Evidence of cancer-promoting roles for AMPK and related kinases. FEBS J. 2015, 282, 4658–4671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunton, J.; Steele, S.; Ziehr, B.; Moorman, N.; Kawula, T. Feeding uninvited guests: mTOR and AMPK set the table for intracellular pathogens. PLoS Pathog. 2013, 9, e1003552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, T.G.; Wang, L.; Li, W.; Li, J.Z.; Li, J. miR-143 inhibits oncogenic traits by degrading NUAK2 in glioblastoma. Int. J. Mol. Med. 2016, 37, 1627–1635. [Google Scholar] [CrossRef]
- Tang, L.; Tong, S.J.; Zhan, Z.; Wang, Q.; Tian, Y.; Chen, F. Expression of NUAK2 in gastric cancer tissue and its effects on the proliferation of gastric cancer cells. Exp. Ther. Med. 2017, 13, 676–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda, F.; Mannion, D.; Liu, S.; Zheng, Y.; Mangala, L.S.; Redondo, C.; Herrero-Gonzalez, S.; Xu, R.; Taylor, C.; Chedom, D.F.; et al. Salt-Inducible Kinase 2 Couples Ovarian Cancer Cell Metabolism with Survival at the Adipocyte-Rich Metastatic Niche. Cancer Cell 2016, 30, 273–289. [Google Scholar] [CrossRef]
- Hubaux, R.; Thu, K.L.; Vucic, E.A.; Pikor, L.A.; Kung, S.H.; Martinez, V.D.; Mosslemi, M.; Becker-Santos, D.D.; Gazdar, A.F.; Lam, S.; et al. Microtubule affinity-regulating kinase 2 is associated with DNA damage response and cisplatin resistance in non-small cell lung cancer. Int. J. Cancer 2015, 137, 2072–2082. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Goransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Makela, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Zang, M.; Guo, W. AMPK as a metabolic tumor suppressor: Control of metabolism and cell growth. Future Oncol. 2010, 6, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, B.; Chhipa, R.R. Evolving Lessons on the Complex Role of AMPK in Normal Physiology and Cancer. Trends Pharmacol. Sci. 2016, 37, 192–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.M.; Hay, N. The dark face of AMPK as an essential tumor promoter. Cell Logist. 2012, 2, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Saunders, M.P.; Salisbury, A.J.; O’Byrne, K.J.; Long, L.; Whitehouse, R.M.; Talbot, D.C.; Mawer, E.B.; Harris, A.L. A novel cyclic adenosine monophosphate analog induces hypercalcemia via production of 1,25-dihydroxyvitamin D in patients with solid tumors. J. Clin. Endocrinol. Metab. 1997, 82, 4044–4048. [Google Scholar] [CrossRef] [PubMed]
- Propper, D.J.; Saunders, M.P.; Salisbury, A.J.; Long, L.; O’Byrne, K.J.; Braybrooke, J.P.; Dowsett, M.; Taylor, M.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I study of the novel cyclic AMP (cAMP) analogue 8-chloro-cAMP in patients with cancer: Toxicity, hormonal, and immunological effects. Clin. Cancer Res. 1999, 5, 1682–1689. [Google Scholar]
- Huk, D.J.; Ashtekar, A.; Magner, A.; La Perle, K.; Kirschner, L.S. Deletion of Rap1b, but not Rap1a or Epac1, Reduces Protein Kinase A-Mediated Thyroid Cancer. Thyroid 2018, 28, 1153–1161. [Google Scholar] [CrossRef]
- Tsygankova, O.M.; Saavedra, A.; Rebhun, J.F.; Quilliam, L.A.; Meinkoth, J.L. Coordinated regulation of Rap1 and thyroid differentiation by cyclic AMP and protein kinase A. Mol. Cell. Biol. 2001, 21, 1921–1929. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Neto, F.; Leon, A.; Urbani-Brocard, J.; Lou, L.; Nyska, A.; Altschuler, D.L. cAMP-dependent oncogenic action of Rap1b in the thyroid gland. J. Biol. Chem. 2004, 279, 46868–46875. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, D.H.; Saporito-Irwin, S.; DeClue, J.E.; Wienecke, R.; Guha, A. Alterations in the rap1 signaling pathway are common in human gliomas. Oncogene 1997, 15, 1611–1616. [Google Scholar] [CrossRef] [Green Version]
- Chevillard, S.; Ugolin, N.; Vielh, P.; Ory, K.; Levalois, C.; Elliott, D.; Clayman, G.L.; El-Naggar, A.K. Gene expression profiling of differentiated thyroid neoplasms: Diagnostic and clinical implications. Clin. Cancer Res. 2004, 10, 6586–6597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattori, M.; Minato, N. Rap1 GTPase: Functions, regulation, and malignancy. J. Biochem. 2003, 134, 479–484. [Google Scholar] [CrossRef]
- Chen, C.H.; Chuang, H.C.; Huang, C.C.; Fang, F.M.; Huang, H.Y.; Tsai, H.T.; Su, L.J.; Shiu, L.Y.; Leu, S.; Chien, C.Y. Overexpression of Rap-1A indicates a poor prognosis for oral cavity squamous cell carcinoma and promotes tumor cell invasion via Aurora-A modulation. Am. J. Pathol. 2013, 182, 516–528. [Google Scholar] [CrossRef]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dremier, S.; Milenkovic, M.; Blancquaert, S.; Dumont, J.E.; Doskeland, S.O.; Maenhaut, C.; Roger, P.P. Cyclic adenosine 3’,5’-monophosphate (cAMP)-dependent protein kinases, but not exchange proteins directly activated by cAMP (Epac), mediate thyrotropin/cAMP-dependent regulation of thyroid cells. Endocrinology 2007, 148, 4612–4622. [Google Scholar] [CrossRef]
- Almahariq, M.; Tsalkova, T.; Mei, F.C.; Chen, H.; Zhou, J.; Sastry, S.K.; Schwede, F.; Cheng, X. A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol. 2013, 83, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Ji, Z.; Tsalkova, T.; Mei, F. Epac and PKA: A tale of two intracellular cAMP receptors. Acta Biochim. Biophys. Sin. 2008, 40, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Luo, C.; Cheng, X.; Lu, M. Lithium and an EPAC-specific inhibitor ESI-09 synergistically suppress pancreatic cancer cell proliferation and survival. Acta Biochim. Biophys. Sin. 2017, 49, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Li, Y.; Dillon, T.J.; Stork, P.J. Phosphorylation of Rap1 by cAMP-dependent Protein Kinase (PKA) Creates a Binding Site for KSR to Sustain ERK Activation by cAMP. J. Biol. Chem. 2017, 292, 1449–1461. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Dillon, T.J.; Pokala, V.; Mishra, S.; Labudda, K.; Hunter, B.; Stork, P.J. Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol. Cell. Biol. 2006, 26, 2130–2145. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Dillon, T.J.; Liu, C.; Kariya, Y.; Wang, Z.; Stork, P.J. Protein kinase A-dependent phosphorylation of Rap1 regulates its membrane localization and cell migration. J. Biol. Chem. 2013, 288, 27712–27723. [Google Scholar] [CrossRef] [Green Version]
- Vogel, K.S.; Klesse, L.J.; Velasco-Miguel, S.; Meyers, K.; Rushing, E.J.; Parada, L.F. Mouse tumor model for neurofibromatosis type 1. Science 1999, 286, 2176–2179. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Miyoshi, H.; Kojima, Y.; Oshima, M.; Taketo, M.M. Accelerated onsets of gastric hamartomas and hepatic adenomas/carcinomas in Lkb1+/-p53-/- compound mutant mice. Oncogene 2006, 25, 1816–1820. [Google Scholar] [CrossRef] [Green Version]
- Cichowski, K.; Shih, T.S.; Schmitt, E.; Santiago, S.; Reilly, K.; McLaughlin, M.E.; Bronson, R.T.; Jacks, T. Mouse models of tumor development in neurofibromatosis type 1. Science 1999, 286, 2172–2176. [Google Scholar] [CrossRef]
- Almeida, M.Q.; Muchow, M.; Boikos, S.; Bauer, A.J.; Griffin, K.J.; Tsang, K.M.; Cheadle, C.; Watkins, T.; Wen, F.; Starost, M.F.; et al. Mouse Prkar1a haploinsufficiency leads to an increase in tumors in the Trp53+/− or Rb1+/− backgrounds and chemically induced skin papillomas by dysregulation of the cell cycle and Wnt signaling. Hum. Mol. Genet. 2010, 19, 1387–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carney, J.A.; Gordon, H.; Carpenter, P.C.; Shenoy, B.V.; Go, V.L. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine 1985, 64, 270–283. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Courcoutsakis, N.A.; Abati, A.; Filie, A.; Doppman, J.L.; Carney, J.A.; Shawker, T. Thyroid gland abnormalities in patients with the syndrome of spotty skin pigmentation, myxomas, endocrine overactivity, and schwannomas (Carney complex). J. Clin. Endocrinol. Metab. 1997, 82, 2037–2043. [Google Scholar] [CrossRef]
- Casey, M.; Vaughan, C.J.; He, J.; Hatcher, C.J.; Winter, J.M.; Weremowicz, S.; Montgomery, K.; Kucherlapati, R.; Morton, C.C.; Basson, C.T. Mutations in the protein kinase A R1alpha regulatory subunit cause familial cardiac myxomas and Carney complex. J. Clin. Investig. 2000, 106, R31–R38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, J.E.; Jauniaux, J.C.; Roger, P.P. The cyclic AMP-mediated stimulation of cell proliferation. Trends Biochem. Sci. 1989, 14, 67–71. [Google Scholar] [CrossRef]
- Roger, P.; Taton, M.; Van Sande, J.; Dumont, J.E. Mitogenic effects of thyrotropin and adenosine 3’,5’-monophosphate in differentiated normal human thyroid cells in vitro. J. Clin. Endocrinol. Metab. 1988, 66, 1158–1165. [Google Scholar] [CrossRef]
- Hishinuma, A.; Yamanaka, T.; Kasai, K.; So, S.; Tseng, C.C.; Bamba, N.; Ohtake, H.; Shimoda, S. Different growth control of the two human thyroid cell lines of adenomatous goiter and papillary carcinoma. Thyroid 1995, 5, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Sarlis, N.J. Expression patterns of cellular growth-controlling genes in non-medullary thyroid cancer: Basic aspects. Rev. Endocr. Metab. Disord. 2000, 1, 183–196. [Google Scholar] [CrossRef]
- Eng, C. Familial papillary thyroid cancer--many syndromes, too many genes? J. Clin. Endocrinol. Metab. 2000, 85, 1755–1757. [Google Scholar] [CrossRef] [PubMed]
- van der Laan, B.F.; Freeman, J.L.; Asa, S.L. Expression of growth factors and growth factor receptors in normal and tumorous human thyroid tissues. Thyroid 1995, 5, 67–73. [Google Scholar] [CrossRef]
- Farid, N.R.; Zou, M.; Shi, Y. Genetics of follicular thyroid cancer. Endocrinol. Metab. Clin. N. Am. 1995, 24, 865–883. [Google Scholar] [CrossRef]
- Maenhaut, C.; Roger, P.P.; Reuse, S.; Dumont, J.E. Activation of the cyclic AMP cascade as an oncogenic mechanism: The thyroid example. Biochimie 1991, 73, 29–36. [Google Scholar] [CrossRef]
- Bertherat, J.; Horvath, A.; Groussin, L.; Grabar, S.; Boikos, S.; Cazabat, L.; Libe, R.; Rene-Corail, F.; Stergiopoulos, S.; Bourdeau, I.; et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5’-monophosphate-dependent protein kinase (PRKAR1A): Phenotype analysis in 353 patients and 80 different genotypes. J. Clin. Endocrinol. Metab. 2009, 94, 2085–2091. [Google Scholar] [CrossRef] [Green Version]
- Carney, J.A.; Lyssikatos, C.; Seethala, R.R.; Lakatos, P.; Perez-Atayde, A.; Lahner, H.; Stratakis, C.A. The Spectrum of Thyroid Gland Pathology in Carney Complex: The Importance of Follicular Carcinoma. Am. J. Surg. Pathol. 2018, 42, 587–594. [Google Scholar] [CrossRef]
- Adeniran, A.J.; Zhu, Z.; Gandhi, M.; Steward, D.L.; Fidler, J.P.; Giordano, T.J.; Biddinger, P.W.; Nikiforov, Y.E. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am. J. Surg. Pathol. 2006, 30, 216–222. [Google Scholar] [CrossRef]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar] [PubMed]
- Soares, P.; Trovisco, V.; Rocha, A.S.; Lima, J.; Castro, P.; Preto, A.; Maximo, V.; Botelho, T.; Seruca, R.; Sobrinho-Simoes, M. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003, 22, 4578–4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frattini, M.; Ferrario, C.; Bressan, P.; Balestra, D.; De Cecco, L.; Mondellini, P.; Bongarzone, I.; Collini, P.; Gariboldi, M.; Pilotti, S.; et al. Alternative mutations of BRAF, RET and NTRK1 are associated with similar but distinct gene expression patterns in papillary thyroid cancer. Oncogene 2004, 23, 7436–7440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Marques, A.R.; Espadinha, C.; Catarino, A.L.; Moniz, S.; Pereira, T.; Sobrinho, L.G.; Leite, V. Expression of PAX8-PPAR gamma 1 rearrangements in both follicular thyroid carcinomas and adenomas. J. Clin. Endocrinol. Metab. 2002, 87, 3947–3952. [Google Scholar] [CrossRef] [Green Version]
- Nikiforova, M.N.; Biddinger, P.W.; Caudill, C.M.; Kroll, T.G.; Nikiforov, Y.E. PAX8-PPARgamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am. J. Surg. Pathol. 2002, 26, 1016–1023. [Google Scholar] [CrossRef]
- Dwight, T.; Thoppe, S.R.; Foukakis, T.; Lui, W.O.; Wallin, G.; Hoog, A.; Frisk, T.; Larsson, C.; Zedenius, J. Involvement of the PAX8/peroxisome proliferator-activated receptor gamma rearrangement in follicular thyroid tumors. J. Clin. Endocrinol. Metab. 2003, 88, 4440–4445. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W., 2nd; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef] [Green Version]
- Ezzat, S.; Zheng, L.; Kolenda, J.; Safarian, A.; Freeman, J.L.; Asa, S.L. Prevalence of activating ras mutations in morphologically characterized thyroid nodules. Thyroid 1996, 6, 409–416. [Google Scholar] [CrossRef]
- Karga, H.; Lee, J.K.; Vickery, A.L., Jr.; Thor, A.; Gaz, R.D.; Jameson, J.L. Ras oncogene mutations in benign and malignant thyroid neoplasms. J. Clin. Endocrinol. Metab. 1991, 73, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Namba, H.; Rubin, S.A.; Fagin, J.A. Point mutations of ras oncogenes are an early event in thyroid tumorigenesis. Mol. Endocrinol. 1990, 4, 1474–1479. [Google Scholar] [CrossRef] [Green Version]
- Manenti, G.; Pilotti, S.; Re, F.C.; Della Porta, G.; Pierotti, M.A. Selective activation of ras oncogenes in follicular and undifferentiated thyroid carcinomas. Eur. J. Cancer 1994, 30A, 987–993. [Google Scholar] [CrossRef]
- Motoi, N.; Sakamoto, A.; Yamochi, T.; Horiuchi, H.; Motoi, T.; Machinami, R. Role of ras mutation in the progression of thyroid carcinoma of follicular epithelial origin. Pathol. Res. Pract. 2000, 196, 1–7. [Google Scholar] [CrossRef]
- Esapa, C.T.; Johnson, S.J.; Kendall-Taylor, P.; Lennard, T.W.; Harris, P.E. Prevalence of Ras mutations in thyroid neoplasia. Clin. Endocrinol. 1999, 50, 529–535. [Google Scholar] [CrossRef]
- Suarez, H.G.; du Villard, J.A.; Severino, M.; Caillou, B.; Schlumberger, M.; Tubiana, M.; Parmentier, C.; Monier, R. Presence of mutations in all three ras genes in human thyroid tumors. Oncogene 1990, 5, 565–570. [Google Scholar]
- Zhu, Z.; Gandhi, M.; Nikiforova, M.N.; Fischer, A.H.; Nikiforov, Y.E. Molecular profile and clinical-pathologic features of the follicular variant of papillary thyroid carcinoma. An unusually high prevalence of ras mutations. Am. J. Clin. Pathol. 2003, 120, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rostan, G.; Zhao, H.; Camp, R.L.; Pollan, M.; Herrero, A.; Pardo, J.; Wu, R.; Carcangiu, M.L.; Costa, J.; Tallini, G. ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J. Clin. Oncol. 2003, 21, 3226–3235. [Google Scholar] [CrossRef]
- Saavedra, H.I.; Knauf, J.A.; Shirokawa, J.M.; Wang, J.; Ouyang, B.; Elisei, R.; Stambrook, P.J.; Fagin, J.A. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene 2000, 19, 3948–3954. [Google Scholar] [CrossRef] [Green Version]
- Basolo, F.; Pisaturo, F.; Pollina, L.E.; Fontanini, G.; Elisei, R.; Molinaro, E.; Iacconi, P.; Miccoli, P.; Pacini, F. N-ras mutation in poorly differentiated thyroid carcinomas: Correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid 2000, 10, 19–23. [Google Scholar] [CrossRef]
- Fagin, J.A. Minireview: Branded from the start-distinct oncogenic initiating events may determine tumor fate in the thyroid. Mol. Endocrinol. 2002, 16, 903–911. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Liu, D.; Xing, M. Functional characterization of the T1799-1801del and A1799-1816ins BRAF mutations in papillary thyroid cancer. Cell Cycle 2007, 6, 377–379. [Google Scholar] [CrossRef] [PubMed]
- Carta, C.; Moretti, S.; Passeri, L.; Barbi, F.; Avenia, N.; Cavaliere, A.; Monacelli, M.; Macchiarulo, A.; Santeusanio, F.; Tartaglia, M.; et al. Genotyping of an Italian papillary thyroid carcinoma cohort revealed high prevalence of BRAF mutations, absence of RAS mutations and allowed the detection of a new mutation of BRAF oncoprotein (BRAF(V599lns)). Clin. Endocrinol. 2006, 64, 105–109. [Google Scholar] [CrossRef]
- Trovisco, V.; Vieira de Castro, I.; Soares, P.; Maximo, V.; Silva, P.; Magalhaes, J.; Abrosimov, A.; Guiu, X.M.; Sobrinho-Simoes, M. BRAF mutations are associated with some histological types of papillary thyroid carcinoma. J. Pathol. 2004, 202, 247–251. [Google Scholar] [CrossRef]
- Begum, S.; Rosenbaum, E.; Henrique, R.; Cohen, Y.; Sidransky, D.; Westra, W.H. BRAF mutations in anaplastic thyroid carcinoma: Implications for tumor origin, diagnosis and treatment. Mod. Pathol. 2004, 17, 1359–1363. [Google Scholar] [CrossRef] [Green Version]
- Nikiforova, M.N.; Kimura, E.T.; Gandhi, M.; Biddinger, P.W.; Knauf, J.A.; Basolo, F.; Zhu, Z.; Giannini, R.; Salvatore, G.; Fusco, A.; et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J. Clin. Endocrinol. Metab. 2003, 88, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- Namba, H.; Nakashima, M.; Hayashi, T.; Hayashida, N.; Maeda, S.; Rogounovitch, T.I.; Ohtsuru, A.; Saenko, V.A.; Kanematsu, T.; Yamashita, S. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J. Clin. Endocrinol. Metab. 2003, 88, 4393–4397. [Google Scholar] [CrossRef] [Green Version]
- Xing, M. BRAF mutation in thyroid cancer. Endocr. Relat. Cancer 2005, 12, 245–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Seyama, T.; Mizuno, T.; Tsuyama, N.; Hayashi, T.; Hayashi, Y.; Dohi, K.; Nakamura, N.; Akiyama, M. Unique association of p53 mutations with undifferentiated but not with differentiated carcinomas of the thyroid gland. Cancer Res. 1992, 52, 1369–1371. [Google Scholar]
- Dobashi, Y.; Sugimura, H.; Sakamoto, A.; Mernyei, M.; Mori, M.; Oyama, T.; Machinami, R. Stepwise participation of p53 gene mutation during dedifferentiation of human thyroid carcinomas. Diagn. Mol. Pathol. 1994, 3, 9–14. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Tallini, G.; Herrero, A.; D’Aquila, T.G.; Carcangiu, M.L.; Rimm, D.L. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999, 59, 1811–1815. [Google Scholar]
- Kurihara, T.; Ikeda, S.; Ishizaki, Y.; Fujimori, M.; Tokumoto, N.; Hirata, Y.; Ozaki, S.; Okajima, M.; Sugino, K.; Asahara, T. Immunohistochemical and sequencing analyses of the Wnt signaling components in Japanese anaplastic thyroid cancers. Thyroid 2004, 14, 1020–1029. [Google Scholar] [CrossRef]
- Block, M.A.; Jackson, C.E.; Greenawald, K.A.; Yott, J.B.; Tashjian, A.H., Jr. Clinical characteristics distinguishing hereditary from sporadic medullary thyroid carcinoma. Treatment implications. Arch. Surg. 1980, 115, 142–148. [Google Scholar] [CrossRef]
- Kouvaraki, M.A.; Shapiro, S.E.; Perrier, N.D.; Cote, G.J.; Gagel, R.F.; Hoff, A.O.; Sherman, S.I.; Lee, J.E.; Evans, D.B. RET proto-oncogene: A review and update of genotype-phenotype correlations in hereditary medullary thyroid cancer and associated endocrine tumors. Thyroid 2005, 15, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Brandi, M.L.; Gagel, R.F.; Angeli, A.; Bilezikian, J.P.; Beck-Peccoz, P.; Bordi, C.; Conte-Devolx, B.; Falchetti, A.; Gheri, R.G.; Libroia, A.; et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J. Clin. Endocrinol. Metab. 2001, 86, 5658–5671. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Pardi, E.; Cetani, F.; Elisei, R. Genetic and clinical features of multiple endocrine neoplasia types 1 and 2. J. Oncol. 2012, 2012, 705036. [Google Scholar] [CrossRef] [PubMed]
- Wohllk, N.; Schweizer, H.; Erlic, Z.; Schmid, K.W.; Walz, M.K.; Raue, F.; Neumann, H.P. Multiple endocrine neoplasia type 2. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.; Clayton, D.; Schuffenecker, I.; Lenoir, G.; Cote, G.; Gagel, R.F.; van Amstel, H.K.; Lips, C.J.; Nishisho, I.; Takai, S.I.; et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2: International RET mutation consortium analysis. JAMA 1996, 276, 1575–1579. [Google Scholar] [CrossRef]
- Lodish, M.B.; Stratakis, C.A. RET oncogene in MEN2, MEN2B, MTC and other forms of thyroid cancer. Expert Rev. Anticancer Ther. 2008, 8, 625–632. [Google Scholar] [CrossRef]
- Wells, S.A., Jr. Advances in the management of MEN2: From improved surgical and medical treatment to novel kinase inhibitors. Endocr. Relat. Cancer 2018, 25, T1–T13. [Google Scholar] [CrossRef] [Green Version]
- Romei, C.; Ciampi, R.; Elisei, R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat. Rev. Endocrinol. 2016, 12, 192–202. [Google Scholar] [CrossRef]
- Mulligan, L.M. RET revisited: Expanding the oncogenic portfolio. Nat. Rev. Cancer 2014, 14, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Elisei, R.; Tacito, A.; Ramone, T.; Ciampi, R.; Bottici, V.; Cappagli, V.; Viola, D.; Matrone, A.; Lorusso, L.; Valerio, L.; et al. Twenty-Five Years Experience on RET Genetic Screening on Hereditary MTC: An Update on The Prevalence of Germline RET Mutations. Genes 2019, 10, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romei, C.; Ugolini, C.; Cosci, B.; Torregrossa, L.; Vivaldi, A.; Ciampi, R.; Tacito, A.; Basolo, F.; Materazzi, G.; Miccoli, P.; et al. Low prevalence of the somatic M918T RET mutation in micro-medullary thyroid cancer. Thyroid 2012, 22, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Casella, F.; Tacito, A.; Bottici, V.; Valerio, L.; Viola, D.; Cappagli, V.; Matrone, A.; Ciampi, R.; Piaggi, P.; et al. New insights in the molecular signature of advanced medullary thyroid cancer: Evidence of a bad outcome of cases with double RET mutations. J. Med. Genet. 2016, 53, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Molinaro, E.; Romei, C.; Biagini, A.; Sabini, E.; Agate, L.; Mazzeo, S.; Materazzi, G.; Sellari-Franceschini, S.; Ribechini, A.; Torregrossa, L.; et al. Anaplastic thyroid carcinoma: From clinicopathology to genetics and advanced therapies. Nat. Rev. Endocrinol. 2017, 13, 644–660. [Google Scholar] [CrossRef]
- Deeken-Draisey, A.; Yang, G.Y.; Gao, J.; Alexiev, B.A. Anaplastic thyroid carcinoma: An epidemiologic, histologic, immunohistochemical, and molecular single-institution study. Hum. Pathol. 2018, 82, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Wreesmann, V.B.; Ghossein, R.A.; Patel, S.G.; Harris, C.P.; Schnaser, E.A.; Shaha, A.R.; Tuttle, R.M.; Shah, J.P.; Rao, P.H.; Singh, B. Genome-wide appraisal of thyroid cancer progression. Am. J. Pathol. 2002, 161, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Charles, R.P.; Silva, J.; Iezza, G.; Phillips, W.A.; McMahon, M. Activating BRAF and PIK3CA mutations cooperate to promote anaplastic thyroid carcinogenesis. Mol. Cancer Res. 2014, 12, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Rostan, G.; Costa, A.M.; Pereira-Castro, I.; Salvatore, G.; Hernandez, R.; Hermsem, M.J.; Herrero, A.; Fusco, A.; Cameselle-Teijeiro, J.; Santoro, M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005, 65, 10199–10207. [Google Scholar] [CrossRef] [Green Version]
- Gauchotte, G.; Philippe, C.; Lacomme, S.; Leotard, B.; Wissler, M.P.; Allou, L.; Toussaint, B.; Klein, M.; Vignaud, J.M.; Bressenot, A. BRAF, p53 and SOX2 in anaplastic thyroid carcinoma: Evidence for multistep carcinogenesis. Pathology 2011, 43, 447–452. [Google Scholar] [CrossRef]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [Green Version]
- Tiedje, V.; Ting, S.; Herold, T.; Synoracki, S.; Latteyer, S.; Moeller, L.C.; Zwanziger, D.; Stuschke, M.; Fuehrer, D.; Schmid, K.W. NGS based identification of mutational hotspots for targeted therapy in anaplastic thyroid carcinoma. Oncotarget 2017, 8, 42613–42620. [Google Scholar] [CrossRef] [Green Version]
- Baloch, Z.W.; LiVolsi, V.A. Special types of thyroid carcinoma. Histopathology 2018, 72, 40–52. [Google Scholar] [CrossRef]
- Aschebrook-Kilfoy, B.; Ward, M.H.; Sabra, M.M.; Devesa, S.S. Thyroid cancer incidence patterns in the United States by histologic type, 1992–2006. Thyroid 2011, 21, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabanillas, M.E.; Schlumberger, M.; Jarzab, B.; Martins, R.G.; Pacini, F.; Robinson, B.; McCaffrey, J.C.; Shah, M.H.; Bodenner, D.L.; Topliss, D.; et al. A phase 2 trial of lenvatinib (E7080) in advanced, progressive, radioiodine-refractory, differentiated thyroid cancer: A clinical outcomes and biomarker assessment. Cancer 2015, 121, 2749–2756. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A., Jr.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elisei, R.; Schlumberger, M.J.; Muller, S.P.; Schoffski, P.; Brose, M.S.; Shah, M.H.; Licitra, L.; Jarzab, B.; Medvedev, V.; Kreissl, M.C.; et al. Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol. 2013, 31, 3639–3646. [Google Scholar] [CrossRef] [Green Version]
- Capdevila, J.; Wirth, L.J.; Ernst, T.; Ponce Aix, S.; Lin, C.C.; Ramlau, R.; Butler, M.O.; Delord, J.P.; Gelderblom, H.; Ascierto, P.A.; et al. PD-1 Blockade in Anaplastic Thyroid Carcinoma. J. Clin. Oncol. 2020, 38, 2620–2627. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Seufert, J.; Aumann, K.; Ruf, J.; Klein, C.; Kiefer, S.; Rassner, M.; Boerries, M.; Zielke, A.; la Rosee, P.; et al. Combination of Lenvatinib and Pembrolizumab Is an Effective Treatment Option for Anaplastic and Poorly Differentiated Thyroid Carcinoma. Thyroid 2021, 31, 1076–1085. [Google Scholar] [CrossRef] [PubMed]
- Bouys, L.; Bertherat, J. Management of Endocrine Disease: Carney complex: Clinical and genetic update 20 years after the identification of the CNC1 (PRKAR1A) gene. Eur. J. Endocrinol. 2021, 184, R99–R109. [Google Scholar] [CrossRef] [PubMed]
- Stratakis, C.A.; Raygada, M. Carney Complex. In GeneReviews(R); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
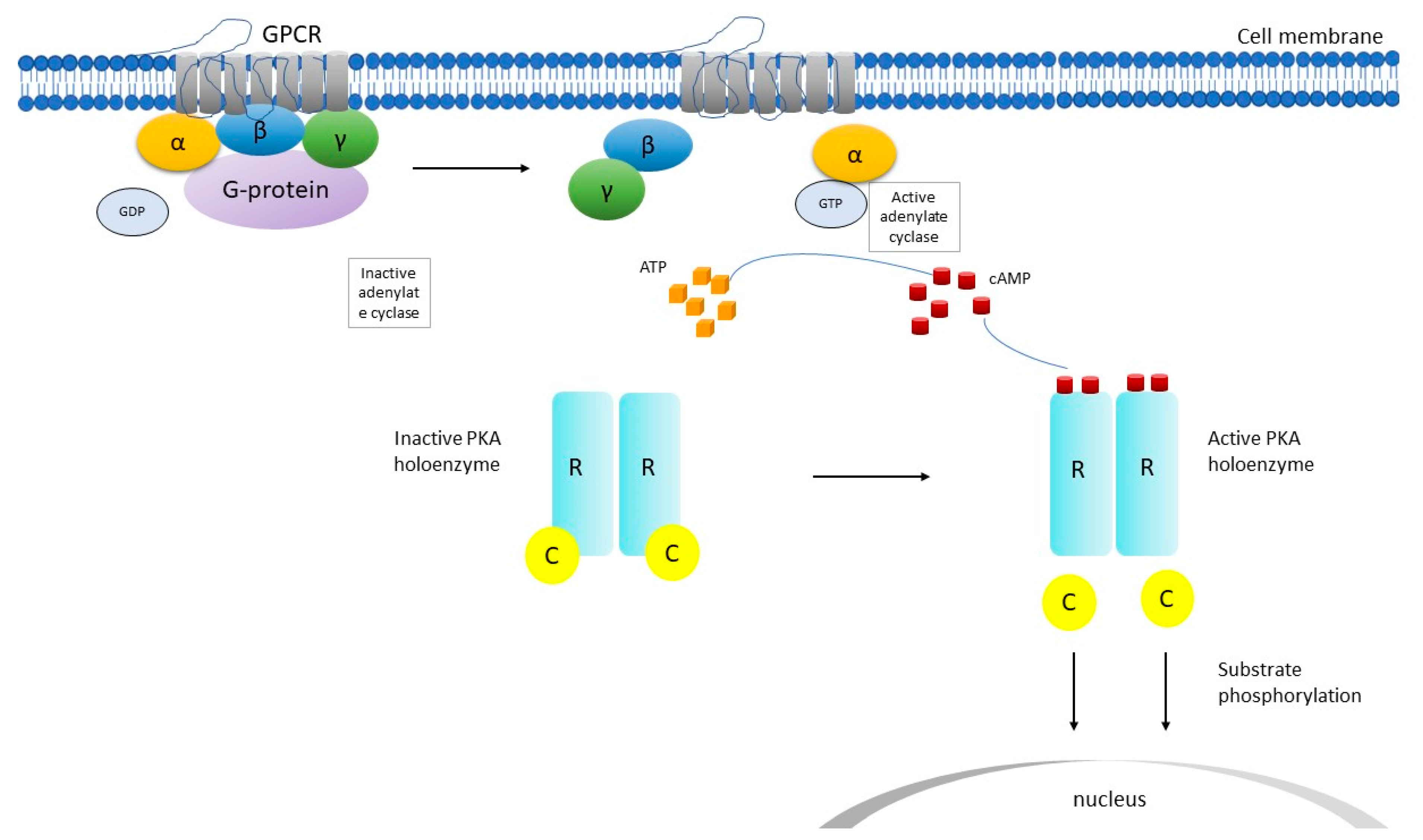
Figure 1.
Schematic representation of cyclic adenosine monophosphate (cAMP) signaling pathway. C catalytic subunit of PKA, GDP guanosine diphosphate, GPCR G-protein coupled receptor, GTP guanosine triphosphate, PKA protein kinase, R regulatory subunit of PKA, α, β, γ subunits. After the GPCR is activated, adenylate cyclase is activated and produces cAMP, which binds to the R subunit and activates PKA. Then, conformational changes ensue and the C subunits are released and phosphorylate cytoplasmic targets.
Figure 1.
Schematic representation of cyclic adenosine monophosphate (cAMP) signaling pathway. C catalytic subunit of PKA, GDP guanosine diphosphate, GPCR G-protein coupled receptor, GTP guanosine triphosphate, PKA protein kinase, R regulatory subunit of PKA, α, β, γ subunits. After the GPCR is activated, adenylate cyclase is activated and produces cAMP, which binds to the R subunit and activates PKA. Then, conformational changes ensue and the C subunits are released and phosphorylate cytoplasmic targets.


{kind=link}
Table 1.
Diagnostic criteria for Carney Complex [24].
Table 1.
Diagnostic criteria for Carney Complex [24].
| Main Diagnostic Criteria |
|---|
|
|
|
|
|
|
|
|
|
|
|
|
| Supplementary Criteria |
|
|
a with histologic confirmation; LCCSCT large cell calcifying Sertoli cell tumor, PPNAD primary pigmented nodular adrenocortical disease; To make a diagnosis of CNC, a patient must either: (1) exhibit two of the manifestations of the disease listed, or (2) exhibit one of these manifestations and meet one of the supplemental criteria.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pitsava, G.; Stratakis, C.A.; Faucz, F.R. PRKAR1A and Thyroid Tumors. Cancers 2021, 13, 3834. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153834
AMA Style
Pitsava G, Stratakis CA, Faucz FR. PRKAR1A and Thyroid Tumors. Cancers. 2021; 13(15):3834. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153834
Chicago/Turabian StylePitsava, Georgia, Constantine A. Stratakis, and Fabio R. Faucz. 2021. "PRKAR1A and Thyroid Tumors" Cancers 13, no. 15: 3834. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13153834
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.