
Selective Targeting of Class I Histone Deacetylases in a Model of Human Osteosarcoma

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Treatment
2.2. Western Blot Analysis
2.3. Cell Viability and Colony Formation Assays
2.4. Wound Healing Assay
2.5. Boyden Chamber-Based Cell Migration and Invasion Assays
2.6. Osteoblast Differentiation Assay
2.7. Analysis of Cell Cycle and Apoptosis by Flow Cytometry
2.8. RNA Sequencing, Pathway Analysis, and Data Availability
2.9. In Vivo Studies
2.10. Statistical Analysis
3. Results
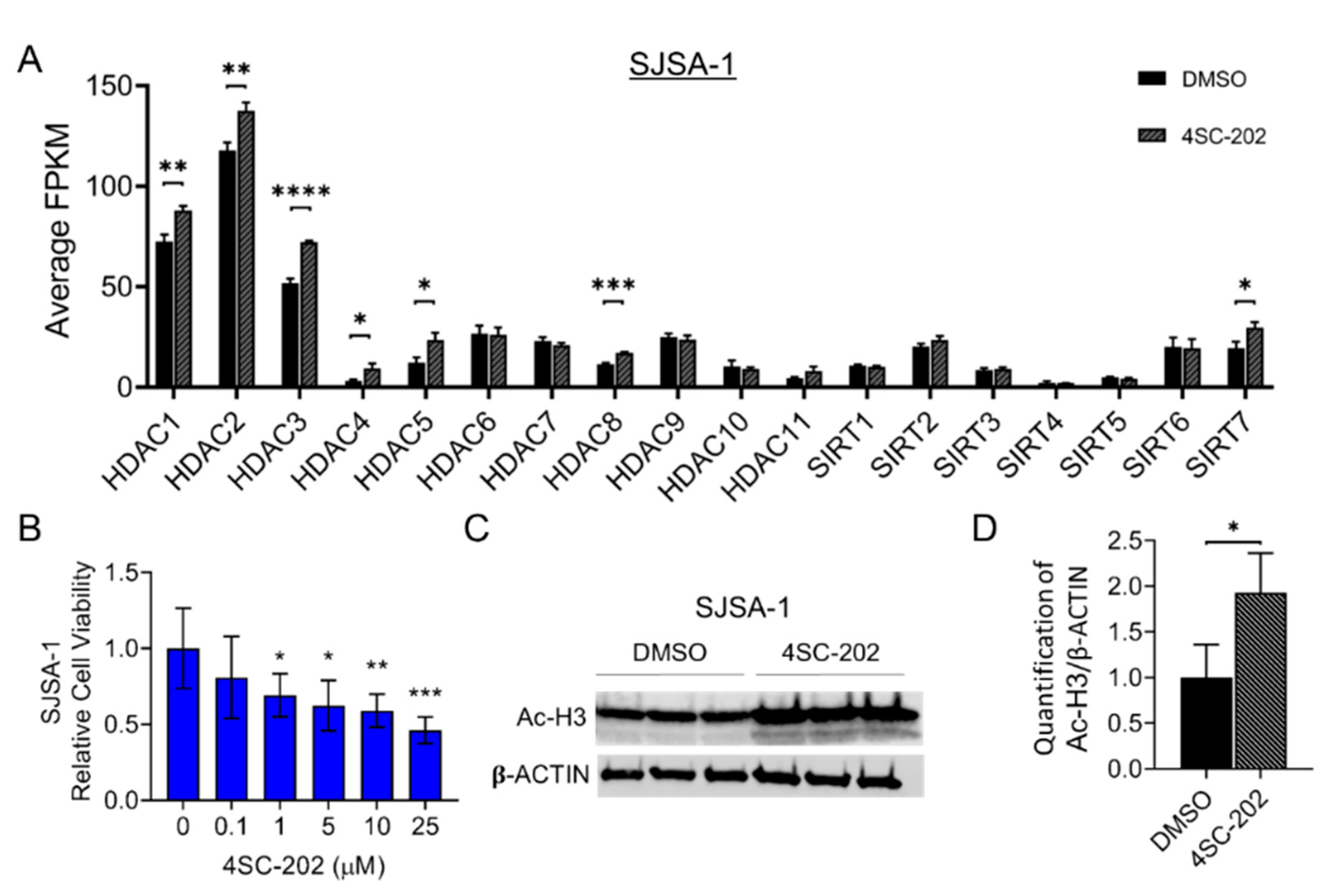
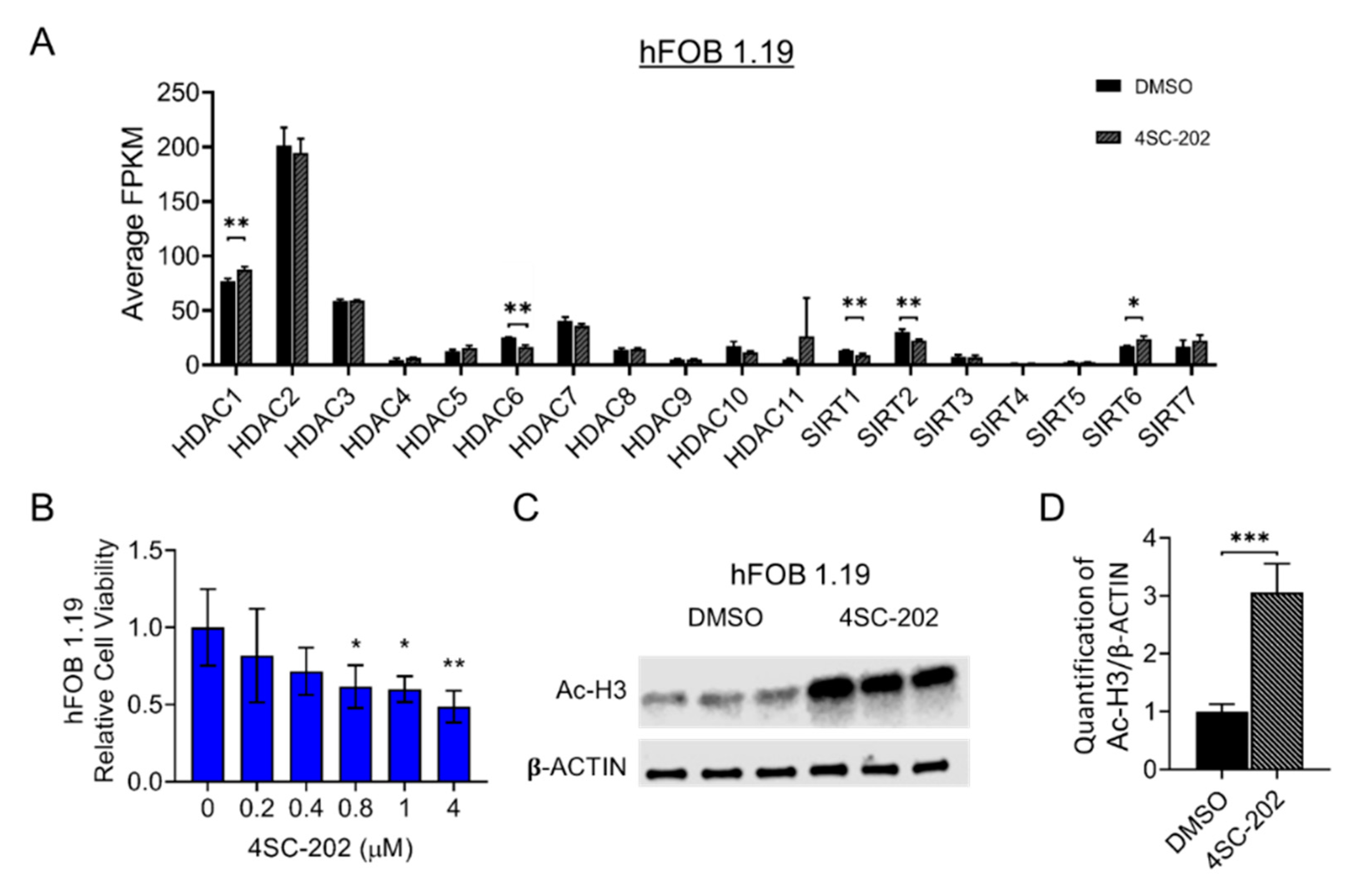
3.1. HDAC1-3 Have the Highest Expression among All Isoforms in Human OS Cells
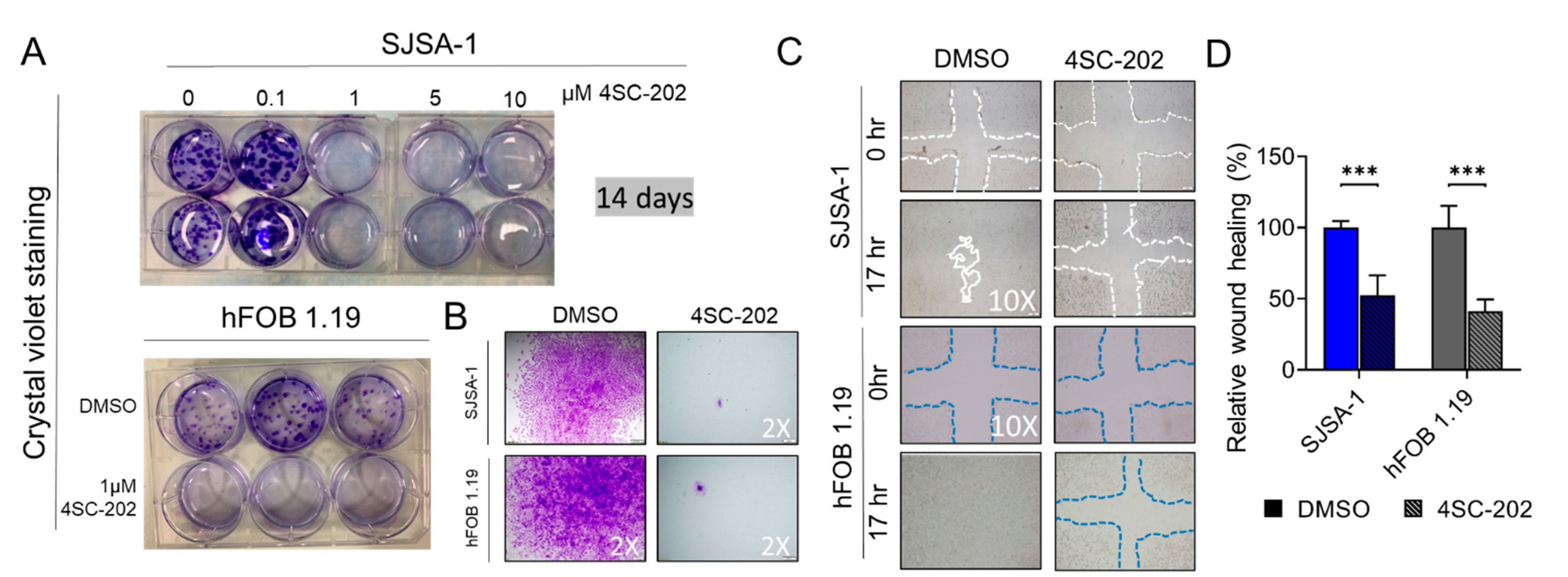
3.2. 4SC-202 Impairs Human Osteosarcoma Cell Growth and Clonogenicity In Vitro
3.3. Cell Migration and Invasion In Vitro Are Partially Suppressed by 4SC-202
3.4. Cell Cycle Is Arrested at the G2/M-Phase Checkpoint by 4SC-202
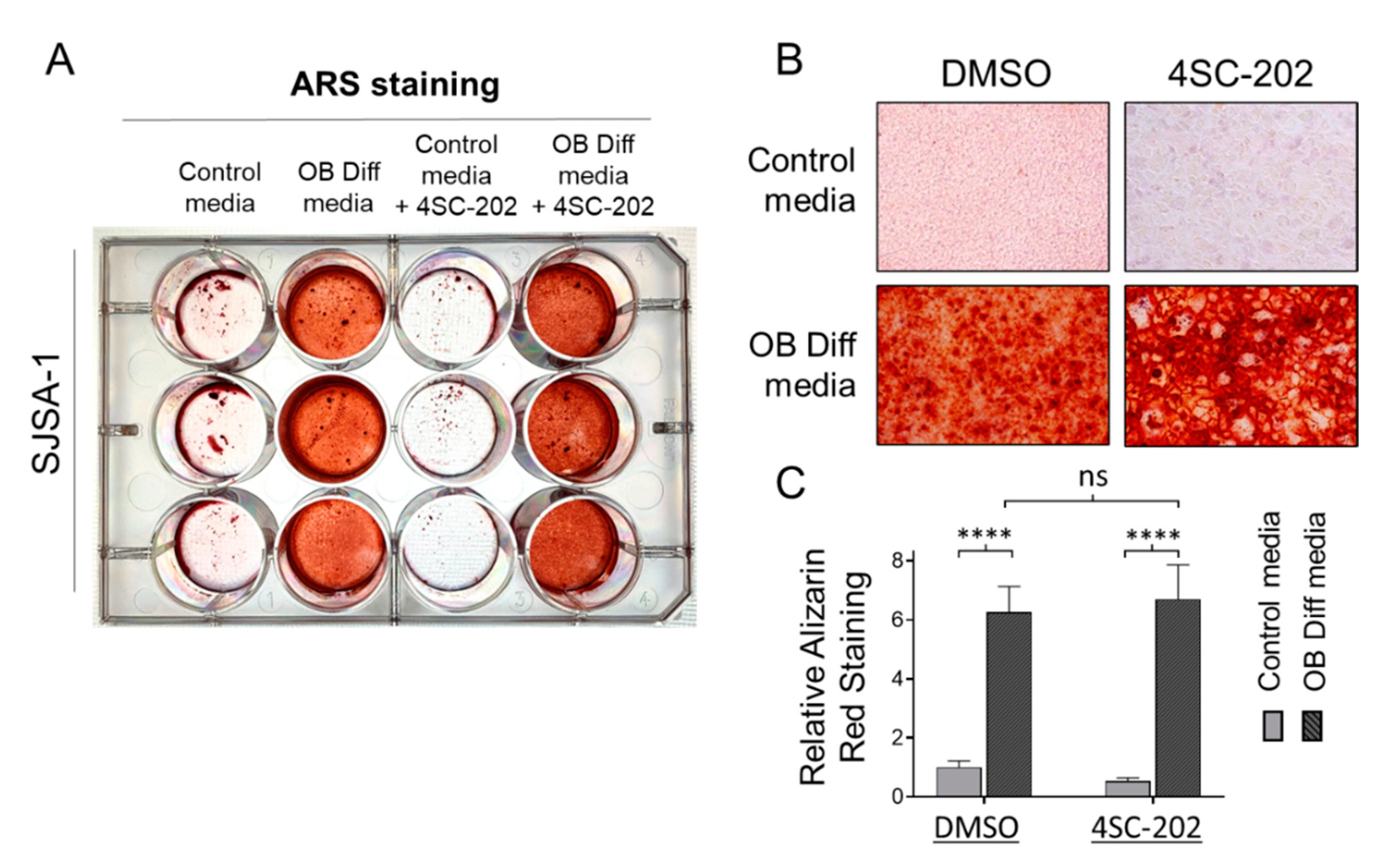
3.5. 4SC-202 Is Incompetent at Initiating and Enhancing Osteogenic Differentiation of Osteosarcoma Cells into Mineralizing Osteoblast-Like Cells
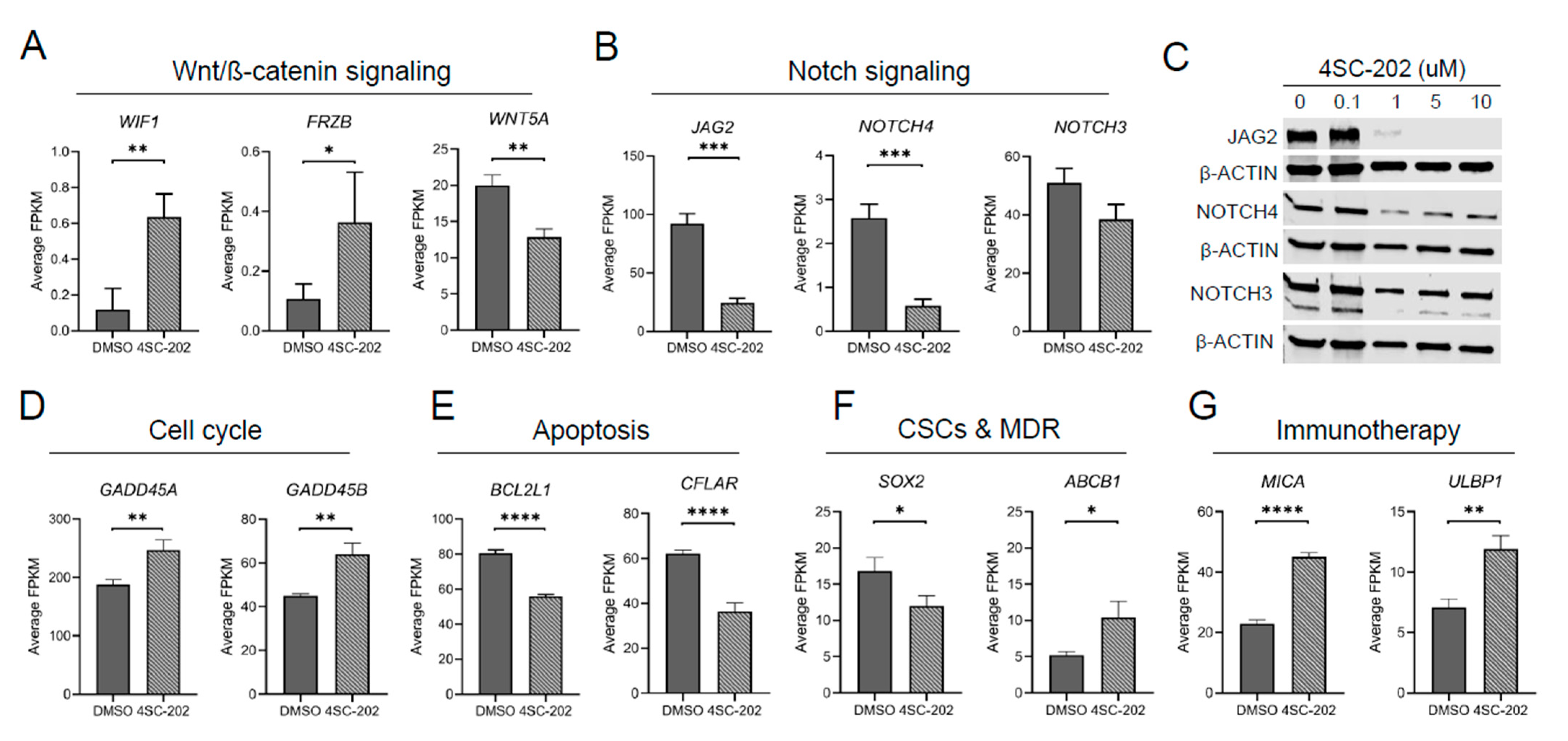
3.6. In Vitro 4SC-202 Treatment Revises Global Transcriptomic Profiling and Induces Distinct Gene Expression Signatures in Human Osteosarcoma Cells
3.7. In Vivo 4SC-202 Treatment Reduces the Tumor Growth of Osteosarcoma in Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roberts, R.D.; Lizardo, M.M.; Reed, D.R.; Hingorani, P.; Glover, J.; Allen-Rhoades, W.; Fan, T.; Khanna, C.; Sweet-Cordero, E.A.; Cash, T.; et al. Provocative questions in osteosarcoma basic and translational biology: A report from the Children’s Oncology Group. Cancer 2019, 125, 3514–3525. [Google Scholar] [CrossRef]
- Yelin, E.; Weinstein, S.; King, T. The burden of musculoskeletal diseases in the United States. Semin. Arthritis Rheum. 2016, 46, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Winkler, K.; Beron, G.; Delling, G.; Heise, U.; Kabisch, H.; Purfürst, C.; Berger, J.; Ritter, J.; Jürgens, H.; Gerein, V. Neoadjuvant chemotherapy of osteosarcoma: Results of a randomized cooperative trial (COSS-82) with salvage chemotherapy based on histological tumor response. J. Clin. Oncol. 1988, 6, 329–337. [Google Scholar] [CrossRef]
- Khanna, C.; Fan, T.M.; Gorlick, R.; Helman, L.J.; Kleinerman, E.S.; Adamson, P.C.; Houghton, P.J.; Tap, W.D.; Welch, D.R.; Steeg, P.S.; et al. Toward a drug development path that targets metastatic progression in osteosarcoma. Clin. Cancer Res. 2014, 20, 4200–4209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickel, K.; Fang, F.; Tao, J. Molecular genetics of osteosarcoma. Bone 2017, 102, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Collier, C.D.; Getty, P.J.; Greenfield, E.M. Targeting the Cancer Epigenome with Histone Deacetylase Inhibitors in Osteosarcoma. Adv. Exp. Med. Biol. 2020, 1258, 55–75. [Google Scholar] [CrossRef] [PubMed]
- Sayles, L.C.; Breese, M.R.; Koehne, A.L.; Leung, S.G.; Lee, A.G.; Liu, H.Y.; Spillinger, A.; Shah, A.T.; Tanasa, B.; Straessler, K.; et al. Genome-Informed Targeted Therapy for Osteosarcoma. Cancer Discov. 2019, 9, 46–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, J.; Jiang, M.M.; Jiang, L.; Salvo, J.S.; Zeng, H.C.; Dawson, B.; Bertin, T.K.; Rao, P.H.; Chen, R.; Donehower, L.A.; et al. Notch activation as a driver of osteogenic sarcoma. Cancer Cell 2014, 26, 390–401. [Google Scholar] [CrossRef] [Green Version]
- Liao, D.; Zhong, L.; Yin, J.; Zeng, C.; Wang, X.; Huang, X.; Chen, J.; Zhang, H.; Zhang, R.; Guan, X.Y.; et al. Chromosomal translocation-derived aberrant Rab22a drives metastasis of osteosarcoma. Nat. Cell Biol. 2020, 22, 868–881. [Google Scholar] [CrossRef]
- Smeester, B.A.; Slipek, N.J.; Pomeroy, E.J.; Laoharawee, K.; Osum, S.H.; Larsson, A.T.; Williams, K.B.; Stratton, N.; Yamamoto, K.; Peterson, J.J.; et al. PLX3397 treatment inhibits constitutive CSF1R-induced oncogenic ERK signaling, reduces tumor growth, and metastatic burden in osteosarcoma. Bone 2020, 136, 115353. [Google Scholar] [CrossRef]
- McGuire, J.J.; Nerlakanti, N.; Lo, C.H.; Tauro, M.; Utset-Ward, T.J.; Reed, D.R.; Lynch, C.C. Histone deacetylase inhibition prevents the growth of primary and metastatic osteosarcoma. Int. J. Cancer 2020, 147, 2811–2823. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Morrow, J.J.; Bayles, I.; Funnell, A.P.W.; Miller, T.E.; Saiakhova, A.; Lizardo, M.M.; Bartels, C.F.; Kapteijn, M.Y.; Hung, S.; Mendoza, A.; et al. Positively selected enhancer elements endow osteosarcoma cells with metastatic competence. Nat. Med. 2018, 24, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Gardner, H.L.; Sivaprakasam, K.; Briones, N.; Zismann, V.; Perdigones, N.; Drenner, K.; Facista, S.; Richholt, R.; Liang, W.; Aldrich, J.; et al. Canine osteosarcoma genome sequencing identifies recurrent mutations in DMD and the histone methyltransferase gene SETD2. Commun. Biol. 2019, 2, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiappetta, C.; Mancini, M.; Lessi, F.; Aretini, P.; De Gregorio, V.; Puggioni, C.; Carletti, R.; Petrozza, V.; Civita, P.; Franceschi, S.; et al. Whole-exome analysis in osteosarcoma to identify a personalized therapy. Oncotarget 2017, 8, 80416–80428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, E.L.; Westendorf, J.J. Histone Mutations and Bone Cancers. Adv. Exp. Med. Biol. 2021, 1283, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Liu, Y.; Liu, Y.; Alexandrov, L.B.; Edmonson, M.N.; Gawad, C.; Zhou, X.; Li, Y.; Rusch, M.C.; Easton, J.; et al. Pan-cancer genome and transcriptome analyses of 1699 paediatric leukaemias and solid tumours. Nature 2018, 555, 371–376. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [Green Version]
- Bradley, E.W.; McGee-Lawrence, M.E.; Westendorf, J.J. Hdac-mediated control of endochondral and intramembranous ossification. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Kelly, R.D.; Cowley, S.M. The physiological roles of histone deacetylase (HDAC) 1 and 2: Complex co-stars with multiple leading parts. Biochem. Soc. Trans. 2013, 41, 741–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A transcriptional activator of osteoblast differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Westendorf, J.J. Histone deacetylases in control of skeletogenesis. J. Cell Biochem. 2007, 102, 332–340. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug. Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Bae, Y.; Wang, L.; Lee, B. Chapter 85. Osteogenic Osteosarcoma. In Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, 8th ed.; Rosen, C.J., Ed.; Wiley: Ames, IA, USA, 2013; pp. 702–710. [Google Scholar] [CrossRef]
- McGee-Lawrence, M.E.; Westendorf, J.J. Histone deacetylases in skeletal development and bone mass maintenance. Gene 2011, 474, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.W.; Suh, J.H.; Kim, A.Y.; Lee, Y.S.; Park, S.Y.; Kim, J.B. Histone Deacetylase 1-Mediated Histone Modification Regulates Osteoblast Differentiation. Mol. Endocrinol. 2006, 20, 2432–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, K.; Gan, G.; Ciarleglio, M.; Zhang, Y.; Tian, X.; Pedigo, C.E.; Cavanaugh, C.; Tate, J.; Wang, Y.; Cross, E.; et al. Podocyte histone deacetylase activity regulates murine and human glomerular diseases. J. Clin. Investig. 2019, 129, 1295–1313. [Google Scholar] [CrossRef] [Green Version]
- McGee-Lawrence, M.E.; Bradley, E.W.; Dudakovic, A.; Carlson, S.W.; Ryan, Z.C.; Kumar, R.; Dadsetan, M.; Yaszemski, M.J.; Chen, Q.; An, K.N.; et al. Histone deacetylase 3 is required for maintenance of bone mass during aging. Bone 2013, 52, 296–307. [Google Scholar] [CrossRef] [Green Version]
- Chaiyawat, P.; Pruksakorn, D.; Phanphaisarn, A.; Teeyakasem, P.; Klangjorhor, J.; Settakorn, J. Expression patterns of class I histone deacetylases in osteosarcoma: A novel prognostic marker with potential therapeutic implications. Mod. Pathol. 2018, 31, 264–274. [Google Scholar] [CrossRef] [Green Version]
- Deng, Z.; Liu, X.; Jin, J.; Xu, H.; Gao, Q.; Wang, Y.; Zhao, J. Histone Deacetylase Inhibitor Trichostatin a Promotes the Apoptosis of Osteosarcoma Cells through p53 Signaling Pathway Activation. Int. J. Biol. Sci. 2016, 12, 1298–1308. [Google Scholar] [CrossRef] [Green Version]
- Seto, E.; Yoshida, M. Erasers of histone acetylation: The histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscal, J.A.; Thompson, P.A.; Horton, T.M.; Ingle, A.M.; Ahern, C.H.; McGovern, R.M.; Reid, J.M.; Ames, M.M.; Espinoza-Delgado, I.; Weigel, B.J.; et al. A phase I trial of vorinostat and bortezomib in children with refractory or recurrent solid tumors: A Children’s Oncology Group phase I consortium study (ADVL0916). Pediatr. Blood Cancer 2013, 60, 390–395. [Google Scholar] [CrossRef] [Green Version]
- Keshelava, N.; Houghton, P.J.; Morton, C.L.; Lock, R.B.; Carol, H.; Keir, S.T.; Maris, J.M.; Reynolds, C.P.; Gorlick, R.; Kolb, E.A.; et al. Initial testing (stage 1) of vorinostat (SAHA) by the pediatric preclinical testing program. Pediatr. Blood Cancer 2009, 53, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Patatsos, K.; Shekhar, T.M.; Hawkins, C.J. Pre-clinical evaluation of proteasome inhibitors for canine and human osteosarcoma. Vet. Comp. Oncol. 2018, 16, 544–553. [Google Scholar] [CrossRef]
- Hou, M.; Huang, Z.; Chen, S.; Wang, H.; Feng, T.; Yan, S.; Su, Y.; Zuo, G. Synergistic antitumor effect of suberoylanilide hydroxamic acid and cisplatin in osteosarcoma cells. Oncol. Lett. 2018, 16, 4663–4670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Yang, C.; Feldman, M.J.; Wang, H.; Pang, Y.; Maggio, D.M.; Zhu, D.; Nesvick, C.L.; Dmitriev, P.; Bullova, P.; et al. Vorinostat suppresses hypoxia signaling by modulating nuclear translocation of hypoxia inducible factor 1 alpha. Oncotarget 2017, 8, 56110–56125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, M.C.; Sarver, A.L.; Tomiyasu, H.; Cornax, I.; Van Etten, J.; Varshney, J.; O’Sullivan, M.G.; Subramanian, S.; Modiano, J.F. Aberrant Retinoblastoma (RB)-E2F Transcriptional Regulation Defines Molecular Phenotypes of Osteosarcoma. J. Biol. Chem. 2015, 290, 28070–28083. [Google Scholar] [CrossRef] [Green Version]
- Foley, J.M.; Scholten, D.J., 2nd; Monks, N.R.; Cherba, D.; Monsma, D.J.; Davidson, P.; Dylewski, D.; Dykema, K.; Winn, M.E.; Steensma, M.R. Anoikis-resistant subpopulations of human osteosarcoma display significant chemoresistance and are sensitive to targeted epigenetic therapies predicted by expression profiling. J. Transl. Med. 2015, 13, 110. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Brynien, D.; Weiss, K.R. The HDAC inhibitor Vorinostat diminishes the in vitro metastatic behavior of Osteosarcoma cells. Biomed. Res. Int. 2015, 2015, 290368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Ma, C.; Shan, Z.; Ju, Y.; Li, S.; Zhao, Q. Histone deacetylase inhibitors suppress the growth of human osteosarcomas in vitro and in vivo. J. BUON 2013, 18, 1032–1037. [Google Scholar] [PubMed]
- Shats, I.; Gatza, M.L.; Liu, B.; Angus, S.P.; You, L.; Nevins, J.R. FOXO transcription factors control E2F1 transcriptional specificity and apoptotic function. Cancer Res. 2013, 73, 6056–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blattmann, C.; Oertel, S.; Ehemann, V.; Thiemann, M.; Huber, P.E.; Bischof, M.; Witt, O.; Deubzer, H.E.; Kulozik, A.E.; Debus, J.; et al. Enhancement of radiation response in osteosarcoma and rhabdomyosarcoma cell lines by histone deacetylase inhibition. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 237–245. [Google Scholar] [CrossRef]
- Murahari, S.; Jalkanen, A.L.; Kulp, S.K.; Chen, C.S.; Modiano, J.F.; London, C.A.; Kisseberth, W.C. Sensitivity of osteosarcoma cells to HDAC inhibitor AR-42 mediated apoptosis. BMC Cancer 2017, 17, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.K.; Rue, S.W.; Lee, I.S.; Kim, H.C.; Lee, I.K.; Ahn, J.D.; Kim, H.S.; Yu, T.S.; Kwak, J.Y.; Heintz, N.H.; et al. Modulation of Sp1-dependent transcription by a cis-acting E2F element in dhfr promoter. Biochem. Biophys. Res. Commun. 2003, 306, 239–243. [Google Scholar] [CrossRef]
- Hirose, T.; Sowa, Y.; Takahashi, S.; Saito, S.; Yasuda, C.; Shindo, N.; Furuichi, K.; Sakai, T. p53-independent induction of Gadd45 by histone deacetylase inhibitor: Coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene 2003, 22, 7762–7773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, M.S.; Kim, C.W.; Park, B.S.; Kim, G.C.; Jeong, J.H.; Kwon, H.C.; Suh, D.J.; Cho, K.H.; Yee, S.B.; Yoo, Y.H. Mechanism of histone deacetylase inhibitor Trichostatin A induced apoptosis in human osteosarcoma cells. Apoptosis 2004, 9, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Li, Y.; Yang, L.; Xu, X.; Liang, F.; Liang, S.; Ba, G.; Xue, F.; Fu, Q. Upregulation of Polo-like kinase 2 gene expression by GATA-1 acetylation in human osteosarcoma MG-63 cells. Int. J. Biochem. Cell Biol. 2012, 44, 423–429. [Google Scholar] [CrossRef]
- Cheng, D.D.; Yang, Q.C.; Zhang, Z.C.; Yang, C.X.; Liu, Y.W. Antitumor activity of histone deacetylase inhibitor trichostatin A in osteosarcoma cells. Asian. Pac. J. Cancer Prev. 2012, 13, 1395–1399. [Google Scholar] [CrossRef] [Green Version]
- Yamanegi, K.; Kawabe, M.; Futani, H.; Nishiura, H.; Yamada, N.; Kato-Kogoe, N.; Kishimoto, H.; Yoshiya, S.; Nakasho, K. Sodium valproate, a histone deacetylase inhibitor, modulates the vascular endothelial growth inhibitor-mediated cell death in human osteosarcoma and vascular endothelial cells. Int. J. Oncol. 2015, 46, 1994–2002. [Google Scholar] [CrossRef]
- Zeng, H.; Zhang, J.M.; Du, Y.; Wang, J.; Ren, Y.; Li, M.; Li, H.; Cai, Z.; Chu, Q.; Yang, C. Crosstalk between ATF4 and MTA1/HDAC1 promotes osteosarcoma progression. Oncotarget 2016, 7, 7329–7342. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Chen, Y.; Chen, X.; Jiang, J.; Wang, X.; Wang, L.; Wang, J.; Zhang, J.; Gao, L. Trichostatin A activates FOXO1 and induces autophagy in osteosarcoma. Arch. Med. Sci. 2019, 15, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Capobianco, E.; Mora, A.; La Sala, D.; Roberti, A.; Zaki, N.; Badidi, E.; Taranta, M.; Cinti, C. Separate and combined effects of DNMT and HDAC inhibitors in treating human multi-drug resistant osteosarcoma HosDXR150 cell line. PLoS ONE 2014, 9, e95596. [Google Scholar] [CrossRef]
- Carol, H.; Gorlick, R.; Kolb, E.A.; Morton, C.L.; Manesh, D.M.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Maris, J.M.; Wozniak, A.; et al. Initial testing (stage 1) of the histone deacetylase inhibitor, quisinostat (JNJ-26481585), by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2014, 61, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cain, J.E.; McCaw, A.; Jayasekara, W.S.; Rossello, F.J.; Marini, K.D.; Irving, A.T.; Kansara, M.; Thomas, D.M.; Ashley, D.M.; Watkins, D.N. Sustained Low-Dose Treatment with the Histone Deacetylase Inhibitor LBH589 Induces Terminal Differentiation of Osteosarcoma Cells. Sarcoma 2013, 2013, 608964. [Google Scholar] [CrossRef]
- Yu, D.; Kahen, E.; Cubitt, C.L.; McGuire, J.; Kreahling, J.; Lee, J.; Altiok, S.; Lynch, C.C.; Sullivan, D.M.; Reed, D.R. Identification of Synergistic, Clinically Achievable, Combination Therapies for Osteosarcoma. Sci. Rep. 2015, 5, 16991. [Google Scholar] [CrossRef] [Green Version]
- Wirries, A.; Jabari, S.; Jansen, E.P.; Roth, S.; Figueroa-Juárez, E.; Wissniowski, T.T.; Neureiter, D.; Klieser, E.; Lechler, P.; Ruchholtz, S.; et al. Panobinostat mediated cell death: A novel therapeutic approach for osteosarcoma. Oncotarget 2018, 9, 32997–33010. [Google Scholar] [CrossRef] [Green Version]
- Loh, A.H.P.; Stewart, E.; Bradley, C.L.; Chen, X.; Daryani, V.; Stewart, C.F.; Calabrese, C.; Funk, A.; Miller, G.; Karlstrom, A.; et al. Combinatorial screening using orthotopic patient derived xenograft-expanded early phase cultures of osteosarcoma identify novel therapeutic drug combinations. Cancer Lett. 2019, 442, 262–270. [Google Scholar] [CrossRef]
- Yang, C.; Choy, E.; Hornicek, F.J.; Wood, K.B.; Schwab, J.H.; Liu, X.; Mankin, H.; Duan, Z. Histone deacetylase inhibitor (HDACI) PCI-24781 potentiates cytotoxic effects of doxorubicin in bone sarcoma cells. Cancer Chemother. Pharmacol. 2011, 67, 439–446. [Google Scholar] [CrossRef]
- Di Pompo, G.; Salerno, M.; Rotili, D.; Valente, S.; Zwergel, C.; Avnet, S.; Lattanzi, G.; Baldini, N.; Mai, A. Novel histone deacetylase inhibitors induce growth arrest, apoptosis, and differentiation in sarcoma cancer stem cells. J. Med. Chem. 2015, 58, 4073–4079. [Google Scholar] [CrossRef]
- Ozaki, T.; Wu, D.; Sugimoto, H.; Nagase, H.; Nakagawara, A. Runt-related transcription factor 2 (RUNX2) inhibits p53-dependent apoptosis through the collaboration with HDAC6 in response to DNA damage. Cell Death Dis. 2013, 4, e610. [Google Scholar] [CrossRef]
- Castillo-Juárez, P.; Sanchez, S.C.; Chávez-Blanco, A.D.; Mendoza-Figueroa, H.; Correa-Basurto, J. Apoptotic Effects of N-(2-hydroxyphenyl)-2-propylpentanamide on U87-MG and U-2 OS Cells and Antiangiogenic Properties. Anticancer. Agents Med. Chem. 2020. [Google Scholar] [CrossRef]
- La Noce, M.; Paino, F.; Mele, L.; Papaccio, G.; Regad, T.; Lombardi, A.; Papaccio, F.; Desiderio, V.; Tirino, V. HDAC2 depletion promotes osteosarcoma’s stemness both in vitro and in vivo: A study on a putative new target for CSCs directed therapy. J. Exp. Clin. Cancer Res. 2018, 37, 296. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Ye, C.; Lin, P.; Li, B.; Zhang, W.; Sun, L.; Wang, Z.; Xue, D.; Teng, W.; et al. Valproic Acid Combined with Zoledronate Enhance γδ T Cell-Mediated Cytotoxicity against Osteosarcoma Cells via the Accumulation of Mevalonate Pathway Intermediates. Front. Immunol. 2018, 9, 377. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Wang, H.; Zhang, F.; Tian, Y.; Tian, Z.; Cai, Z.; Lim, D.; Feng, Z. The Effect of VPA on Increasing Radiosensitivity in Osteosarcoma Cells and Primary-Culture Cells from Chemical Carcinogen-Induced Breast Cancer in Rats. Int. J. Mol. Sci. 2017, 18, 1027. [Google Scholar] [CrossRef] [Green Version]
- Wittenburg, L.A.; Ptitsyn, A.A.; Thamm, D.H. A systems biology approach to identify molecular pathways altered by HDAC inhibition in osteosarcoma. J. Cell Biochem. 2012, 113, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Khalil, M.A.; Hrabeta, J.; Cipro, S.; Stiborova, M.; Vicha, A.; Eckschlager, T. Neuroblastoma stem cells-mechanisms of chemoresistance and histone deacetylase inhibitors. Neoplasma 2012, 59, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.P. How the NOTCH pathway contributes to the ability of osteosarcoma cells to metastasize. Cancer Treat. Res. 2009, 152, 479–496. [Google Scholar] [CrossRef]
- Yamanegi, K.; Yamane, J.; Hata, M.; Ohyama, H.; Yamada, N.; Kato-Kogoe, N.; Futani, H.; Nakasho, K.; Okamura, H.; Terada, N. Sodium valproate, a histone deacetylase inhibitor, decreases the secretion of soluble Fas by human osteosarcoma cells and increases their sensitivity to Fas-mediated cell death. J. Cancer Res. Clin. Oncol. 2009, 135, 879–889. [Google Scholar] [CrossRef]
- Perego, S.; Sansoni, V.; Banfi, G.; Lombardi, G. Sodium butyrate has anti-proliferative, pro-differentiating, and immunomodulatory effects in osteosarcoma cells and counteracts the TNFα-induced low-grade inflammation. Int. J. Immunopathol. Pharmacol. 2018, 31, 1–14. [Google Scholar] [CrossRef]
- Xie, C.; Wu, B.; Chen, B.; Shi, Q.; Guo, J.; Fan, Z.; Huang, Y. Histone deacetylase inhibitor sodium butyrate suppresses proliferation and promotes apoptosis in osteosarcoma cells by regulation of the MDM2-p53 signaling. Onco Targets Ther. 2016, 9, 4005–4013. [Google Scholar] [CrossRef]
- El-Naggar, A.M.; Somasekharan, S.P.; Wang, Y.; Cheng, H.; Negri, G.L.; Pan, M.; Wang, X.Q.; Delaidelli, A.; Rafn, B.; Cran, J.; et al. Class I HDAC inhibitors enhance YB-1 acetylation and oxidative stress to block sarcoma metastasis. EMBO Rep. 2019, 20, e48375. [Google Scholar] [CrossRef]
- Jaboin, J.; Wild, J.; Hamidi, H.; Khanna, C.; Kim, C.J.; Robey, R.; Bates, S.E.; Thiele, C.J. MS-27-275, an inhibitor of histone deacetylase, has marked in vitro and in vivo antitumor activity against pediatric solid tumors. Cancer Res. 2002, 62, 6108–6115. [Google Scholar]
- Koshkina, N.V.; Rao-Bindal, K.; Kleinerman, E.S. Effect of the histone deacetylase inhibitor SNDX-275 on Fas signaling in osteosarcoma cells and the feasibility of its topical application for the treatment of osteosarcoma lung metastases. Cancer 2011, 117, 3457–3467. [Google Scholar] [CrossRef]
- Kiany, S.; Huang, G.; Kleinerman, E.S. Effect of entinostat on NK cell-mediated cytotoxicity against osteosarcoma cells and osteosarcoma lung metastasis. Oncoimmunology 2017, 6, e1333214. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Okamoto, K.; Yonehara, S. Sensitization of osteosarcoma cells to death receptor-mediated apoptosis by HDAC inhibitors through downregulation of cellular FLIP. Cell Death Differ. 2005, 12, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Ouchida, M.; Morimoto, Y.; Yoshida, A.; Jitsumori, Y.; Ozaki, T.; Sonobe, H.; Inoue, H.; Shimizu, K. Significant growth suppression of synovial sarcomas by the histone deacetylase inhibitor FK228 in vitro and in vivo. Cancer Lett. 2005, 224, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Okada, T.; Tanaka, K.; Nakatani, F.; Sakimura, R.; Matsunobu, T.; Li, X.; Hanada, M.; Nakamura, T.; Oda, Y.; Tsuneyoshi, M.; et al. Involvement of P-glycoprotein and MRP1 in resistance to cyclic tetrapeptide subfamily of histone deacetylase inhibitors in the drug-resistant osteosarcoma and Ewing’s sarcoma cells. Int. J. Cancer 2006, 118, 90–97. [Google Scholar] [CrossRef]
- Graham, C.; Tucker, C.; Creech, J.; Favours, E.; Billups, C.A.; Liu, T.; Fouladi, M.; Freeman, B.B., 3rd; Stewart, C.F.; Houghton, P.J. Evaluation of the antitumor efficacy, pharmacokinetics, and pharmacodynamics of the histone deacetylase inhibitor depsipeptide in childhood cancer models in vivo. Clin. Cancer Res. 2006, 12, 223–234. [Google Scholar] [CrossRef] [Green Version]
- Matsubara, H.; Watanabe, M.; Imai, T.; Yui, Y.; Mizushima, Y.; Hiraumi, Y.; Kamitsuji, Y.; Watanabe, K.; Nishijo, K.; Toguchida, J.; et al. Involvement of extracellular signal-regulated kinase activation in human osteosarcoma cell resistance to the histone deacetylase inhibitor FK228 [(1S,4S,7Z,10S,16E,21R)-7-ethylidene-4,21-bis(propan-2-yl)-2-oxa-12,13-dithia-5,8,20,23-tetraazabicyclo[8.7.6]tricos-16-ene-3,6,9,19,22-pentone]. J. Pharmacol. Exp. Ther. 2009, 328, 839–848. [Google Scholar] [CrossRef]
- Wobser, M.; Weber, A.; Glunz, A.; Tauch, S.; Seitz, K.; Butelmann, T.; Hesbacher, S.; Goebeler, M.; Bartz, R.; Kohlhof, H.; et al. Elucidating the mechanism of action of domatinostat (4SC-202) in cutaneous T cell lymphoma cells. J. Hematol. Oncol. 2019, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhang, Z.; Kuang, X.; Ma, D.; Xiong, J.; Lu, T.; Zhang, Y.; Yu, K.; Zhang, S.; Wang, J.; et al. 4SC-202 induces apoptosis in myelodysplastic syndromes and the underlying mechanism. Am. J. Transl. Res. 2020, 12, 2968–2983. [Google Scholar]
- Pinkerneil, M.; Hoffmann, M.J.; Kohlhof, H.; Schulz, W.A.; Niegisch, G. Evaluation of the Therapeutic Potential of the Novel Isotype Specific HDAC Inhibitor 4SC-202 in Urothelial Carcinoma Cell Lines. Target Oncol. 2016, 11, 783–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Yin, Y.; Dorfman, R.G.; Zou, T.; Pan, Y.; Li, Y.; Wang, Y.; Zhou, Q.; Zhou, L.; Kong, B.; et al. Down-regulation of HDAC3 inhibits growth of cholangiocarcinoma by inducing apoptosis. Oncotarget 2017, 8, 99402–99413. [Google Scholar] [CrossRef] [Green Version]
- Zhijun, H.; Shusheng, W.; Han, M.; Jianping, L.; Li-Sen, Q.; Dechun, L. Pre-clinical characterization of 4SC-202, a novel class I HDAC inhibitor, against colorectal cancer cells. Tumour. Biol. 2016, 37, 10257–10267. [Google Scholar] [CrossRef] [PubMed]
- Mishra, V.K.; Wegwitz, F.; Kosinsky, R.L.; Sen, M.; Baumgartner, R.; Wulff, T.; Siveke, J.T.; Schildhaus, H.U.; Najafova, Z.; Kari, V.; et al. Histone deacetylase class-I inhibition promotes epithelial gene expression in pancreatic cancer cells in a BRD4- and MYC-dependent manner. Nucleic. Acids. Res. 2017, 45, 6334–6349. [Google Scholar] [CrossRef]
- Gruber, W.; Peer, E.; Elmer, D.P.; Sternberg, C.; Tesanovic, S.; Del Burgo, P.; Coni, S.; Canettieri, G.; Neureiter, D.; Bartz, R.; et al. Targeting class I histone deacetylases by the novel small molecule inhibitor 4SC-202 blocks oncogenic hedgehog-GLI signaling and overcomes smoothened inhibitor resistance. Int. J. Cancer 2018, 142, 968–975. [Google Scholar] [CrossRef]
- Hoffman, M.M.; Zylla, J.S.; Bhattacharya, S.; Calar, K.; Hartman, T.W.; Bhardwaj, R.D.; Miskimins, W.K.; de la Puente, P.; Gnimpieba, E.Z.; Messerli, S.M. Analysis of Dual Class I Histone Deacetylase and Lysine Demethylase Inhibitor Domatinostat (4SC-202) on Growth and Cellular and Genomic Landscape of Atypical Teratoid/Rhabdoid. Cancers 2020, 12, 756. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Tai, S.; Deng, M.; Fan, Z.; Ping, F.; He, L.; Zhang, C.; Huang, Y.; Cheng, B.; Xia, J. Metformin and 4SC-202 synergistically promote intrinsic cell apoptosis by accelerating ΔNp63 ubiquitination and degradation in oral squamous cell carcinoma. Cancer Med. 2019, 8, 3479–3490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Wan, F.; Li, Z.; Zhang, F. 4SC-202 activates ASK1-dependent mitochondrial apoptosis pathway to inhibit hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2016, 471, 267–273. [Google Scholar] [CrossRef]
- Song, L.; Bretz, A.C.; Gravemeyer, J.; Spassova, I.; Muminova, S.; Gambichler, T.; Sriram, A.; Ferrone, S.; Becker, J.C. The HDAC Inhibitor Domatinostat Promotes Cell-Cycle Arrest, Induces Apoptosis, and Increases Immunogenicity of Merkel Cell Carcinoma Cells. J. Investig. Dermatol. 2021, 141, 903–912.e904. [Google Scholar] [CrossRef]
- Von Tresckow, B.; Sayehli, C.; Aulitzky, W.E.; Goebeler, M.E.; Schwab, M.; Braz, E.; Krauss, B.; Krauss, R.; Hermann, F.; Bartz, R.; et al. Phase I study of domatinostat (4SC-202), a class I histone deacetylase inhibitor in patients with advanced hematological malignancies. Eur. J. Haematol. 2019, 102, 163–173. [Google Scholar] [CrossRef]
- Roberts, W.M.; Douglass, E.C.; Peiper, S.C.; Houghton, P.J.; Look, A.T. Amplification of the gli gene in childhood sarcomas. Cancer Res. 1989, 49, 5407–5413. [Google Scholar]
- Khalid, U.; Simovic, M.; Iskar, M.; Wong, J.K.; Kumar, R.; Jugold, M.; Sill, M.; Bolkestein, M.; Kolb, T.; Hergt, M.; et al. A synergistic interaction between HDAC- and PARP inhibitors in childhood tumors with chromothripsis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Harris, S.A.; Enger, R.J.; Riggs, B.L.; Spelsberg, T.C. Development and characterization of a conditionally immortalized human fetal osteoblastic cell line. J. Bone. Miner. Res. 1995, 10, 178–186. [Google Scholar] [CrossRef]
- Blattmann, C.; Oertel, S.; Thiemann, M.; Weber, K.J.; Schmezer, P.; Zelezny, O.; Lopez Perez, R.; Kulozik, A.E.; Debus, J.; Ehemann, V. Suberoylanilide hydroxamic acid affects γH2AX expression in osteosarcoma, atypical teratoid rhabdoid tumor and normal tissue cell lines after irradiation. Strahlenther. Onkol. 2012, 188, 168–176. [Google Scholar] [CrossRef]
- Fang, F.; VanCleave, A.; Helmuth, R.; Torres, H.; Rickel, K.; Wollenzien, H.; Sun, H.; Zeng, E.; Zhao, J.; Tao, J. Targeting the Wnt/beta-catenin pathway in human osteosarcoma cells. Oncotarget 2018, 9, 36780–36792. [Google Scholar] [CrossRef] [Green Version]
- VanCleave, A.; Palmer, M.; Fang, F.; Torres, H.; Rodezno, T.; Li, Q.; Fuglsby, K.; Evans, C.; Afeworki, Y.; Ross, A.; et al. Development and characterization of the novel human osteosarcoma cell line COS-33 with sustained activation of the mTOR pathway. Oncotarget 2020, 11, 2597–2610. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Gregory, C.A.; Gunn, W.G.; Peister, A.; Prockop, D.J. An Alizarin red-based assay of mineralization by adherent cells in culture: Comparison with cetylpyridinium chloride extraction. Anal. Biochem. 2004, 329, 77–84. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2020. Available online: http://www.R-project.org/ (accessed on 3 February 2020).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome. Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Society. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kolde, R.; Pheatmap: Pretty Heatmaps 2019. R Package Version 1.0.12. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 3 February 2020).
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug. Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Engin, F.; Bertin, T.; Ma, O.; Jiang, M.M.; Wang, L.; Sutton, R.E.; Donehower, L.A.; Lee, B. Notch signaling contributes to the pathogenesis of human osteosarcomas. Hum. Mol. Genet. 2009, 18, 1464–1470. [Google Scholar] [CrossRef] [Green Version]
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 2018, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yan, X.; Tang, J.; Wang, Y.; Tang, J.; Wu, W.; Liu, M. HDAC2-mediated upregulation of IL-6 triggers the migration of osteosarcoma cells. Cell Biol. Toxicol. 2019, 35, 423–433. [Google Scholar] [CrossRef]
- Kopljar, I.; Gallacher, D.J.; De Bondt, A.; Cougnaud, L.; Vlaminckx, E.; Van den Wyngaert, I.; Lu, H.R. Functional and Transcriptional Characterization of Histone Deacetylase Inhibitor-Mediated Cardiac Adverse Effects in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem. Cells Transl. Med. 2016, 5, 602–612. [Google Scholar] [CrossRef] [Green Version]
- McGee-Lawrence, M.E.; Carpio, L.R.; Schulze, R.J.; Pierce, J.L.; McNiven, M.A.; Farr, J.N.; Khosla, S.; Oursler, M.J.; Westendorf, J.J. Hdac3 Deficiency Increases Marrow Adiposity and Induces Lipid Storage and Glucocorticoid Metabolism in Osteochondroprogenitor Cells. J. bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2016, 31, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Nusbaum, O.; Chen, X.; Zhu, Y. Valeric Acid Suppresses Liver Cancer Development by Acting as a Novel HDAC Inhibitor. Mol. Ther. Oncolytics 2020, 19, 8–18. [Google Scholar] [CrossRef]
- Molyneux, S.D.; Di Grappa, M.A.; Beristain, A.G.; McKee, T.D.; Wai, D.H.; Paderova, J.; Kashyap, M.; Hu, P.; Maiuri, T.; Narala, S.R.; et al. Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclass in mice. J. Clin. Investig. 2010, 120, 3310–3325. [Google Scholar] [CrossRef] [Green Version]
- Yen, M.L.; Chien, C.C.; Chiu, I.M.; Huang, H.I.; Chen, Y.C.; Hu, H.I.; Yen, B.L. Multilineage differentiation and characterization of the human fetal osteoblastic 1.19 cell line: A possible in vitro model of human mesenchymal progenitors. Stem. Cells 2007, 25, 125–131. [Google Scholar] [CrossRef]
- Messerli, S.M.; Hoffman, M.M.; Gnimpieba, E.Z.; Kohlhof, H.; Bhardwaj, R.D. 4SC-202 as a Potential Treatment for the Pediatric Brain Tumor Medulloblastoma. Brain Sci. 2017, 7, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Setoguchi, T.; Hirotsu, M.; Gao, H.; Sasaki, H.; Matsunoshita, Y.; Komiya, S. Inhibition of Notch pathway prevents osteosarcoma growth by cell cycle regulation. Br. J. Cancer 2009, 100, 1957–1965. [Google Scholar] [CrossRef] [Green Version]
- Singla, A.; Wang, J.; Yang, R.; Geller, D.S.; Loeb, D.M.; Hoang, B.H. Wnt Signaling in Osteosarcoma. Adv. Exp. Med. Biol. 2020, 1258, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Kansara, M.; Tsang, M.; Kodjabachian, L.; Sims, N.A.; Trivett, M.K.; Ehrich, M.; Dobrovic, A.; Slavin, J.; Choong, P.F.; Simmons, P.J.; et al. Wnt inhibitory factor 1 is epigenetically silenced in human osteosarcoma, and targeted disruption accelerates osteosarcomagenesis in mice. J. Clin. Investig. 2009, 119, 837–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, M.; Rainusso, N.; Lee, Y.C.; Dawson, B.; Coarfa, C.; Han, R.; Larson, J.L.; Shuck, R.; Kurenbekova, L.; Yustein, J.T. Tegavivint and the β-Catenin/ALDH Axis in Chemotherapy-Resistant and Metastatic Osteosarcoma. J. Natl. Cancer Inst. 2019, 111, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Jianjun, H.; Xue, Y.; Shuwei, Y.; Liyuan, Z.; Jie, W.; Lixian, C. MicroRNA-133b Inhibits Cell Proliferation and Invasion in Osteosarcoma by Targeting Sirt1. Oncol. Res. 2017, 25, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Schofield, A.V.; Gamell, C.; Bernard, O. Tubulin polymerization promoting protein 1 (TPPP1) increases β-catenin expression through inhibition of HDAC6 activity in U2OS osteosarcoma cells. Biochem. Biophys. Res. Commun. 2013, 436, 571–577. [Google Scholar] [CrossRef]
- Zhang, A.; He, S.; Sun, X.; Ding, L.; Bao, X.; Wang, N. Wnt5a promotes migration of human osteosarcoma cells by triggering a phosphatidylinositol-3 kinase/Akt signals. Cancer Cell. Int. 2014, 14, 15. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, K.; Li, X.; Ikegaki, S.; Oneyama, C.; Okada, M.; Nishita, M.; Minami, Y. Dissection of Wnt5a-Ror2 signaling leading to matrix metalloproteinase (MMP-13) expression. J. Biol. Chem. 2012, 287, 1588–1599. [Google Scholar] [CrossRef] [Green Version]
- Hoang, B.H.; Kubo, T.; Healey, J.H.; Sowers, R.; Mazza, B.; Yang, R.; Huvos, A.G.; Meyers, P.A.; Gorlick, R. Expression of LDL receptor-related protein 5 (LRP5) as a novel marker for disease progression in high-grade osteosarcoma. Int. J. Cancer 2004, 109, 106–111. [Google Scholar] [CrossRef]
- Enomoto, M.; Hayakawa, S.; Itsukushima, S.; Ren, D.Y.; Matsuo, M.; Tamada, K.; Oneyama, C.; Okada, M.; Takumi, T.; Nishita, M.; et al. Autonomous regulation of osteosarcoma cell invasiveness by Wnt5a/Ror2 signaling. Oncogene 2009, 28, 3197–3208. [Google Scholar] [CrossRef] [Green Version]
- Yustein, J.T.; Liu, Y.C.; Gao, P.; Jie, C.; Le, A.; Vuica-Ross, M.; Chng, W.J.; Eberhart, C.G.; Bergsagel, P.L.; Dang, C.V. Induction of ectopic Myc target gene JAG2 augments hypoxic growth and tumorigenesis in a human B-cell model. Proc. Natl. Acad. Sci. USA 2010, 107, 3534–3539. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Erez, A.; Lee, B. One NOTCH Further: Jagged 1 in Bone Metastasis. Cancer Cell 2011, 19, 159–161. [Google Scholar] [CrossRef] [Green Version]
- Palermo, R.; Checquolo, S.; Giovenco, A.; Grazioli, P.; Kumar, V.; Campese, A.F.; Giorgi, A.; Napolitano, M.; Canettieri, G.; Ferrara, G.; et al. Acetylation controls Notch3 stability and function in T-cell leukemia. Oncogene 2012, 31, 3807–3817. [Google Scholar] [CrossRef]
- Torres, H.M.; Van Cleave, A.; Palmer, M.; Callahan, D.; Smithback, A.; May, D.; Roux, K.; Tao, J. Abstract 2917: Antitumor effects of an epigenetic inhibitor, 4SC-202, on human osteosarcoma cells. Cancer Res. 2020, 80, 2917. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Yamanegi, K.; Yamane, J.; Kobayashi, K.; Kato-Kogoe, N.; Ohyama, H.; Nakasho, K.; Yamada, N.; Hata, M.; Nishioka, T.; Fukunaga, S.; et al. Sodium valproate, a histone deacetylase inhibitor, augments the expression of cell-surface NKG2D ligands, MICA/B, without increasing their soluble forms to enhance susceptibility of human osteosarcoma cells to NK cell-mediated cytotoxicity. Oncol. Rep. 2010, 24, 1621–1627. [Google Scholar] [CrossRef] [Green Version]
- Schott, C.; Shah, A.T.; Sweet-Cordero, E.A. Genomic Complexity of Osteosarcoma and Its Implication for Preclinical and Clinical Targeted Therapies. Adv. Exp. Med. Biol. 2020, 1258, 1–19. [Google Scholar] [CrossRef]
- Castillo-Tandazo, W.; Mutsaers, A.J.; Walkley, C.R. Osteosarcoma in the Post Genome Era: Preclinical Models and Approaches to Identify Tractable Therapeutic Targets. Curr. Osteoporos Rep. 2019, 17, 343–352. [Google Scholar] [CrossRef]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; Pfeil, J.; Egolf, L.E.; Way, G.P.; Farrel, A.; et al. Genomic Profiling of Childhood Tumor Patient-Derived Xenograft Models to Enable Rational Clinical Trial Design. Cell Rep. 2019, 29, 1675–1689.e1679. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, H.M.; VanCleave, A.M.; Vollmer, M.; Callahan, D.L.; Smithback, A.; Conn, J.M.; Rodezno-Antunes, T.; Gao, Z.; Cao, Y.; Afeworki, Y.; et al. Selective Targeting of Class I Histone Deacetylases in a Model of Human Osteosarcoma. Cancers 2021, 13, 4199. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13164199
Torres HM, VanCleave AM, Vollmer M, Callahan DL, Smithback A, Conn JM, Rodezno-Antunes T, Gao Z, Cao Y, Afeworki Y, et al. Selective Targeting of Class I Histone Deacetylases in a Model of Human Osteosarcoma. Cancers. 2021; 13(16):4199. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13164199
Chicago/Turabian StyleTorres, Haydee M., Ashley M. VanCleave, Mykayla Vollmer, Dakota L. Callahan, Austyn Smithback, Josephine M. Conn, Tania Rodezno-Antunes, Zili Gao, Yuxia Cao, Yohannes Afeworki, and et al. 2021. "Selective Targeting of Class I Histone Deacetylases in a Model of Human Osteosarcoma" Cancers 13, no. 16: 4199. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13164199