
The Unfolded Protein Response Is a Major Driver of LCN2 Expression in BCR–ABL- and JAK2V617F-Positive MPN

 , , , ,
, , , ,  , and add
Show full author list
, and add
Show full author list
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Primary Patient Samples
2.2. Cell Culture
2.3. Cytokines and Chemical Compounds
2.4. Multiplex Immunoassay for Serum Samples
2.5. RT-qPCR
2.6. SDS Page and Western Blotting
2.7. Propidium Iodide Staining
2.8. Generation of Promoter Constructs
2.9. Site Directed Mutagenesis
2.10. Luciferase Assay
2.11. Statistical Analysis
3. Results
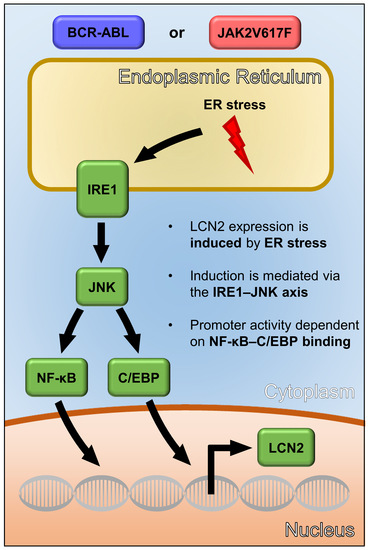
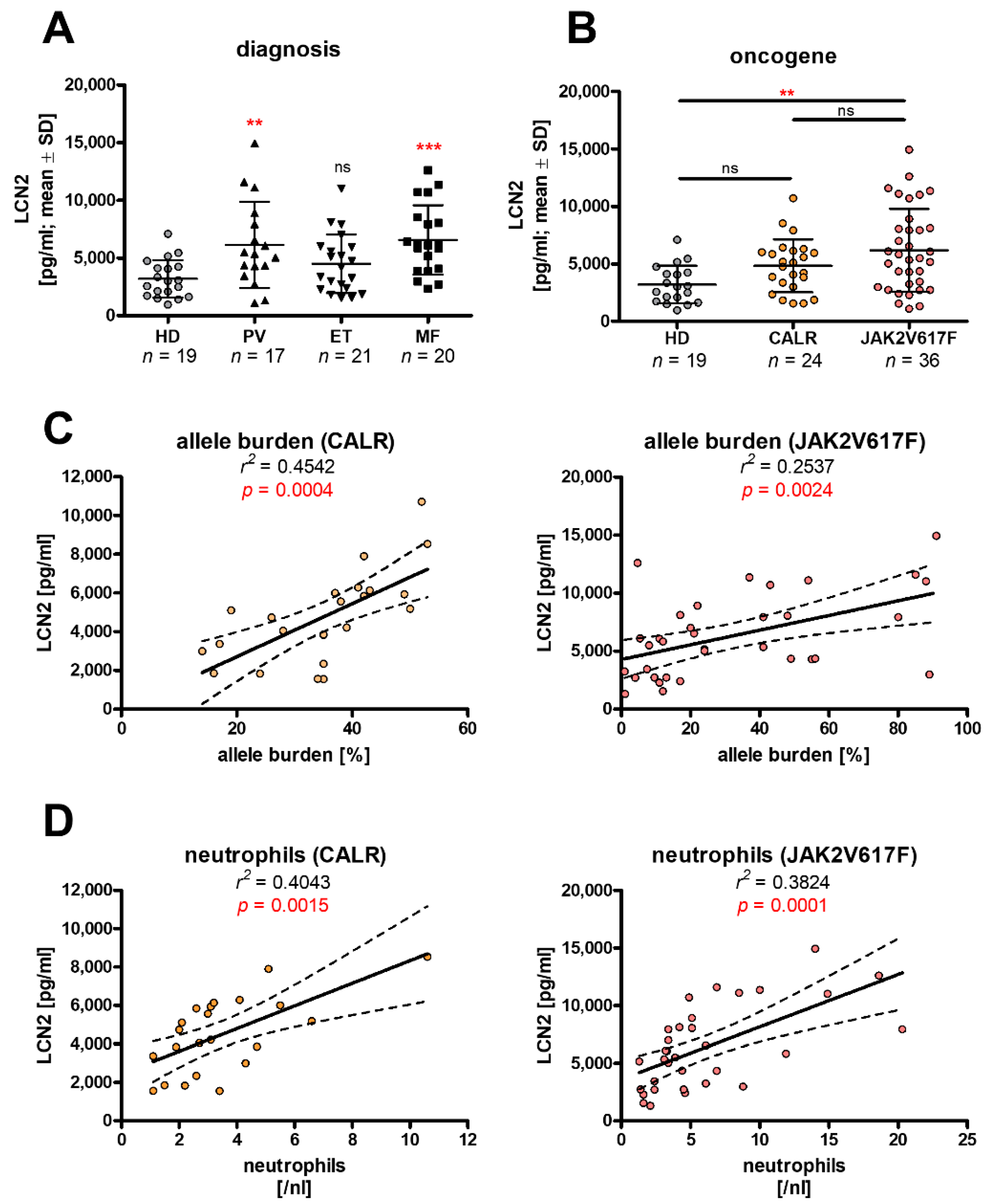
3.1. Differences in LCN2 Expression Are Linked Both to Oncogenes and Cellular Phenotype
3.2. ER Stress Induces Lcn2 mRNA Expression Independent of Oncogene Activity
3.3. UPR-Induced Lcn2 Expression Is Regulated by the IRE1α—JNK Axis
3.4. LCN2 Promoter Activity upon ER Stress Is Dependent on C/EBP and NF-κB Promoter Binding Sites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koschmieder, S.; Chatain, N. Role of inflammation in the biology of myeloproliferative neoplasms. Blood Rev. 2020, 42, 100711. [Google Scholar] [CrossRef]
- Sawyers, C.L. Chronic myeloid leukemia. N. Engl. J. Med. 1999, 340, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Klampfl, T.; Gisslinger, H.; Harutyunyan, A.S.; Nivarthi, H.; Rumi, E.; Milosevic, J.D.; Them, N.C.; Berg, T.; Gisslinger, B.; Pietra, D.; et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N. Engl. J. Med. 2013, 369, 2379–2390. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Massie, C.E.; Baxter, E.J.; Nice, F.L.; Gundem, G.; Wedge, D.C.; Avezov, E.; Li, J.; Kollmann, K.; Kent, D.G.; et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N. Engl. J. Med. 2013, 369, 2391–2405. [Google Scholar] [CrossRef] [Green Version]
- Lussana, F.; Rambaldi, A. Inflammation and myeloproliferative neoplasms. J. Autoimmun. 2017, 85, 58–63. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Hasselbalch, H.C.; Bjorn, M.E. MPNs as Inflammatory Diseases: The Evidence, Consequences, and Perspectives. Mediators Inflamm. 2015, 2015, 102476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, C.H.; Cha, J. Analysis of neutrophil gelatinase-associated lipocalin, vascular endothelial growth factor, and soluble receptor for advanced glycation end-products in bone marrow supernatant in hematologic malignancies. Clin. Biochem. 2020, 80, 19–24. [Google Scholar] [CrossRef]
- Kjeldsen, L.; Johnsen, A.H.; Sengelov, H.; Borregaard, N. Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J. Biol. Chem. 1993, 268, 10425–10432. [Google Scholar] [CrossRef]
- Villalva, C.; Sorel, N.; Bonnet, M.L.; Guilhot, J.; Mayeur-Rousse, C.; Guilhot, F.; Chomel, J.C.; Turhan, A.G. Neutrophil gelatinase-associated lipocalin expression in chronic myeloid leukemia. Leuk. Lymphoma 2008, 49, 984–988. [Google Scholar] [CrossRef] [PubMed]
- Leng, X.; Lin, H.; Ding, T.; Wang, Y.; Wu, Y.; Klumpp, S.; Sun, T.; Zhou, Y.; Monaco, P.; Belmont, J.; et al. Lipocalin 2 is required for BCR-ABL-induced tumorigenesis. Oncogene 2008, 27, 6110–6119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Xia, L.; Liu, Y.C.; Hochman, T.; Bizzari, L.; Aruch, D.; Lew, J.; Weinberg, R.; Goldberg, J.D.; Hoffman, R. Lipocalin produced by myelofibrosis cells affects the fate of both hematopoietic and marrow microenvironmental cells. Blood 2015, 126, 972–982. [Google Scholar] [CrossRef] [Green Version]
- Kagoya, Y.; Yoshimi, A.; Tsuruta-Kishino, T.; Arai, S.; Satoh, T.; Akira, S.; Kurokawa, M. JAK2V617F+ myeloproliferative neoplasm clones evoke paracrine DNA damage to adjacent normal cells through secretion of lipocalin-2. Blood 2014, 124, 2996–3006. [Google Scholar] [CrossRef] [Green Version]
- Borkham-Kamphorst, E.; Drews, F.; Weiskirchen, R. Induction of lipocalin-2 expression in acute and chronic experimental liver injury moderated by pro-inflammatory cytokines interleukin-1beta through nuclear factor-kappaB activation. Liver Int. 2011, 31, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Sunil, V.R.; Patel, K.J.; Nilsen-Hamilton, M.; Heck, D.E.; Laskin, J.D.; Laskin, D.L. Acute endotoxemia is associated with upregulation of lipocalin 24p3/Lcn2 in lung and liver. Exp. Mol. Pathol. 2007, 83, 177–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Karoui, K.; Viau, A.; Dellis, O.; Bagattin, A.; Nguyen, C.; Baron, W.; Burtin, M.; Broueilh, M.; Heidet, L.; Mollet, G.; et al. Endoplasmic reticulum stress drives proteinuria-induced kidney lesions via Lipocalin 2. Nat. Commun. 2016, 7, 10330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from Endoplasmic Reticulum and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Khateb, A.; Ronai, Z.A. Unfolded Protein Response in Leukemia: From Basic Understanding to Therapeutic Opportunities. Trends Cancer 2020, 6, 960–973. [Google Scholar] [CrossRef]
- Kusio-Kobialka, M.; Podszywalow-Bartnicka, P.; Peidis, P.; Glodkowska-Mrowka, E.; Wolanin, K.; Leszak, G.; Seferynska, I.; Stoklosa, T.; Koromilas, A.E.; Piwocka, K. The PERK-eIF2alpha phosphorylation arm is a pro-survival pathway of BCR-ABL signaling and confers resistance to imatinib treatment in chronic myeloid leukemia cells. Cell Cycle 2012, 11, 4069–4078. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Lin, D.C.; Guo, X.; Kharabi Masouleh, B.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1alpha-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef]
- Han, L.; Schubert, C.; Kohler, J.; Schemionek, M.; Isfort, S.; Brummendorf, T.H.; Koschmieder, S.; Chatain, N. Calreticulin-mutant proteins induce megakaryocytic signaling to transform hematopoietic cells and undergo accelerated degradation and Golgi-mediated secretion. J. Hematol. Oncol. 2016, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Kaur, S.; Guha, S.; Batra, S.K. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim. Biophys Acta 2012, 1826, 129–169. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A. Endoplasmic reticulum proteostasis: A key checkpoint in cancer. Am. J. Physiol. Cell Physiol. 2017, 312, C93–C102. [Google Scholar] [CrossRef] [Green Version]
- Le Cabec, V.; Cowland, J.B.; Calafat, J.; Borregaard, N. Targeting of proteins to granule subsets is determined by timing and not by sorting: The specific granule protein NGAL is localized to azurophil granules when expressed in HL-60 cells. Proc. Natl. Acad. Sci. USA 1996, 93, 6454–6457. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.H.; Yoon, J.; Kim, D.S.; Kim, S.J.; Sung, H.J.; Lee, S.R. Association of peripheral blood neutrophil gelatinase-associated lipocalin levels with bone marrow neutrophil gelatinase-associated lipocalin levels and neutrophil count in hematologic malignancy. J. Clin. Lab. Anal. 2019, 33, e22920. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.H.; Cha, J.; Chang, E.A.; Nam, M.H.; Park, S.J.; Sung, H.J.; Lee, S.R. Analysis of bone marrow supernatant neutrophil gelatinase-associated lipocalin and hematological parameters in hematological malignancy. J. Clin. Lab. Anal. 2020, 34, e23253. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.Q.; Wu, Y.; Wang, F.; Cui, W. Prognostic value of CALR vs. JAK2V617F mutations on splenomegaly, leukemic transformation, thrombosis, and overall survival in patients with primary fibrosis: A meta-analysis. Ann. Hematol. 2016, 95, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Sorensen, O.E.; Sehested, M.; Borregaard, N. Neutrophil gelatinase-associated lipocalin is up-regulated in human epithelial cells by IL-1 beta, but not by TNF-alpha. J. Immunol. 2003, 171, 6630–6639. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Vaidya, R.; Caramazza, D.; Finke, C.; Lasho, T.; Pardanani, A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: A comprehensive cytokine profiling study. J Clin. Oncol. 2011, 29, 1356–1363. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H.; Wei, S.; Wang, Z.; Ji, G. Inhibition of ER stress-related IRE1alpha/CREB/NLRP1 pathway promotes the apoptosis of human chronic myelogenous leukemia cell. Mol. Immunol. 2018, 101, 377–385. [Google Scholar] [CrossRef]
- Mahadevan, N.R.; Rodvold, J.; Almanza, G.; Perez, A.F.; Wheeler, M.C.; Zanetti, M. ER stress drives Lipocalin 2 upregulation in prostate cancer cells in an NF-kappaB-dependent manner. BMC Cancer 2011, 11, 229. [Google Scholar] [CrossRef] [Green Version]
- Schardt, J.A.; Mueller, B.U.; Pabst, T. Activation of the unfolded protein response in human acute myeloid leukemia. Methods Enzymol. 2011, 489, 227–243. [Google Scholar] [CrossRef]
- Zhou, C.; Martinez, E.; Di Marcantonio, D.; Solanki-Patel, N.; Aghayev, T.; Peri, S.; Ferraro, F.; Skorski, T.; Scholl, C.; Frohling, S.; et al. JUN is a key transcriptional regulator of the unfolded protein response in acute myeloid leukemia. Leukemia 2017, 31, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Kharabi Masouleh, B.; Geng, H.; Hurtz, C.; Chan, L.N.; Logan, A.C.; Chang, M.S.; Huang, C.; Swaminathan, S.; Sun, H.; Paietta, E.; et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E2219–E2228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borkham-Kamphorst, E.; Van de Leur, E.; Haas, U.; Weiskirchen, R. Liver parenchymal cells lacking Lipocalin 2 (LCN2) are prone to endoplasmic reticulum stress and unfolded protein response. Cell Signal 2019, 55, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, S.K.; Asimakopoulou, A.; Tillmann, S.; Koschmieder, S.; Weiskirchen, R. TNF-alpha controls Lipocalin-2 expression in PC-3 prostate cancer cells. Cytokine 2020, 135, 155214. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Stephens, J.M. STAT1, NF-kappaB and ERKs play a role in the induction of lipocalin-2 expression in adipocytes. Mol. Metab. 2013, 2, 161–170. [Google Scholar] [CrossRef]
- Winston, J.T.; Strack, P.; Beer-Romero, P.; Chu, C.Y.; Elledge, S.J.; Harper, J.W. The SCFbeta-TRCP-ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IkappaBalpha and beta-catenin and stimulates IkappaBalpha ubiquitination in vitro. Genes Dev. 1999, 13, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Spiegelman, V.S.; Stavropoulos, P.; Latres, E.; Pagano, M.; Ronai, Z.; Slaga, T.J.; Fuchs, S.Y. Induction of beta-transducin repeat-containing protein by JNK signaling and its role in the activation of NF-kappaB. J. Biol. Chem. 2001, 276, 27152–27158. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Hu, G.K. Pink1 protects cortical neurons from thapsigargin-induced oxidative stress and neuronal apoptosis. Biosci. Rep. 2015, 35. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.K.; Tseng, C.K.; Liaw, C.C.; Huang, C.Y.; Wei, C.K.; Sheu, J.H.; Lee, J.C. Lobohedleolide suppresses hepatitis C virus replication via JNK/c-Jun-C/EBP-mediated down-regulation of cyclooxygenase-2 expression. Sci. Rep. 2018, 8, 8676. [Google Scholar] [CrossRef] [Green Version]
- Glaros, T.; Fu, Y.; Xing, J.; Li, L. Molecular mechanism underlying persistent induction of LCN2 by lipopolysaccharide in kidney fibroblasts. PLoS ONE 2012, 7, e34633. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tillmann, S.; Olschok, K.; Schröder, S.K.; Bütow, M.; Baumeister, J.; Kalmer, M.; Preußger, V.; Weinbergerova, B.; Kricheldorf, K.; Mayer, J.; et al. The Unfolded Protein Response Is a Major Driver of LCN2 Expression in BCR–ABL- and JAK2V617F-Positive MPN. Cancers 2021, 13, 4210. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13164210
Tillmann S, Olschok K, Schröder SK, Bütow M, Baumeister J, Kalmer M, Preußger V, Weinbergerova B, Kricheldorf K, Mayer J, et al. The Unfolded Protein Response Is a Major Driver of LCN2 Expression in BCR–ABL- and JAK2V617F-Positive MPN. Cancers. 2021; 13(16):4210. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13164210
Chicago/Turabian StyleTillmann, Stefan, Kathrin Olschok, Sarah K. Schröder, Marlena Bütow, Julian Baumeister, Milena Kalmer, Vera Preußger, Barbora Weinbergerova, Kim Kricheldorf, Jiri Mayer, and et al. 2021. "The Unfolded Protein Response Is a Major Driver of LCN2 Expression in BCR–ABL- and JAK2V617F-Positive MPN" Cancers 13, no. 16: 4210. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13164210