
Epigenetic Silencing of miR-33b Promotes Peritoneal Metastases of Ovarian Cancer by Modulating the TAK1/FASN/CPT1A/NF-κB Axis

 , ,
, , 
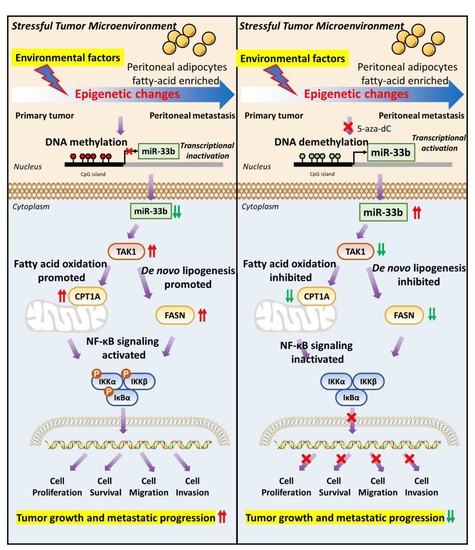
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Human Clinical Samples
2.2. Whole-Genome Methylation Profiling
2.3. Methylation-Specific PCR (MS-PCR) and Pyrosequencing Analysis
2.4. Construction of Cell Lines with Stable miR-33b Expression
2.5. Omental Conditioned Medium (OCM) and Commercial Kits
2.6. Real-Time Quantitative PCR
2.7. Western Blot Analysis
2.8. Cell Proliferation, Migration and Invasion Assays
2.9. CRISPR/Cas9-Mediated Gene Knockout
2.10. Dual-Luciferase Reporter Assay
2.11. In Situ Hybridization (ISH) and Immunohistochemistry (IHC)
2.12. Proteomics and Bioinformatics Analysis
2.13. Integrative Genomic Analyses for The Cancer Genome Atlas Ovarian Serous Cystadenocarcinoma (TCGA-OV) Data
2.14. In Vivo Intraperitoneal Dissemination Mouse Model
2.15. Statistical Analysis
3. Results
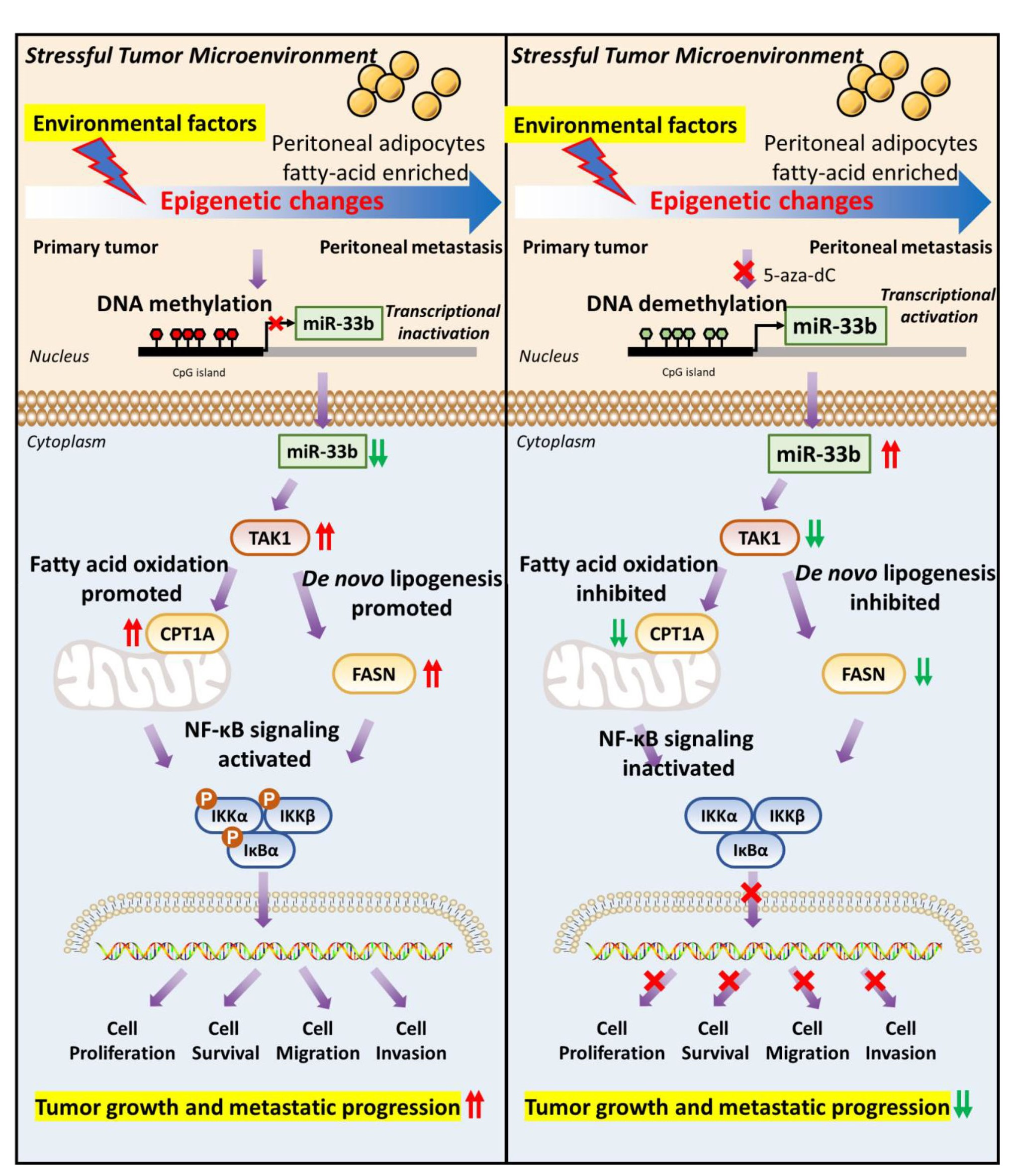
3.1. miR-33b Is Frequently Silenced by DNA Hypermethylation in Metastatic Ovarian Cancer Cells
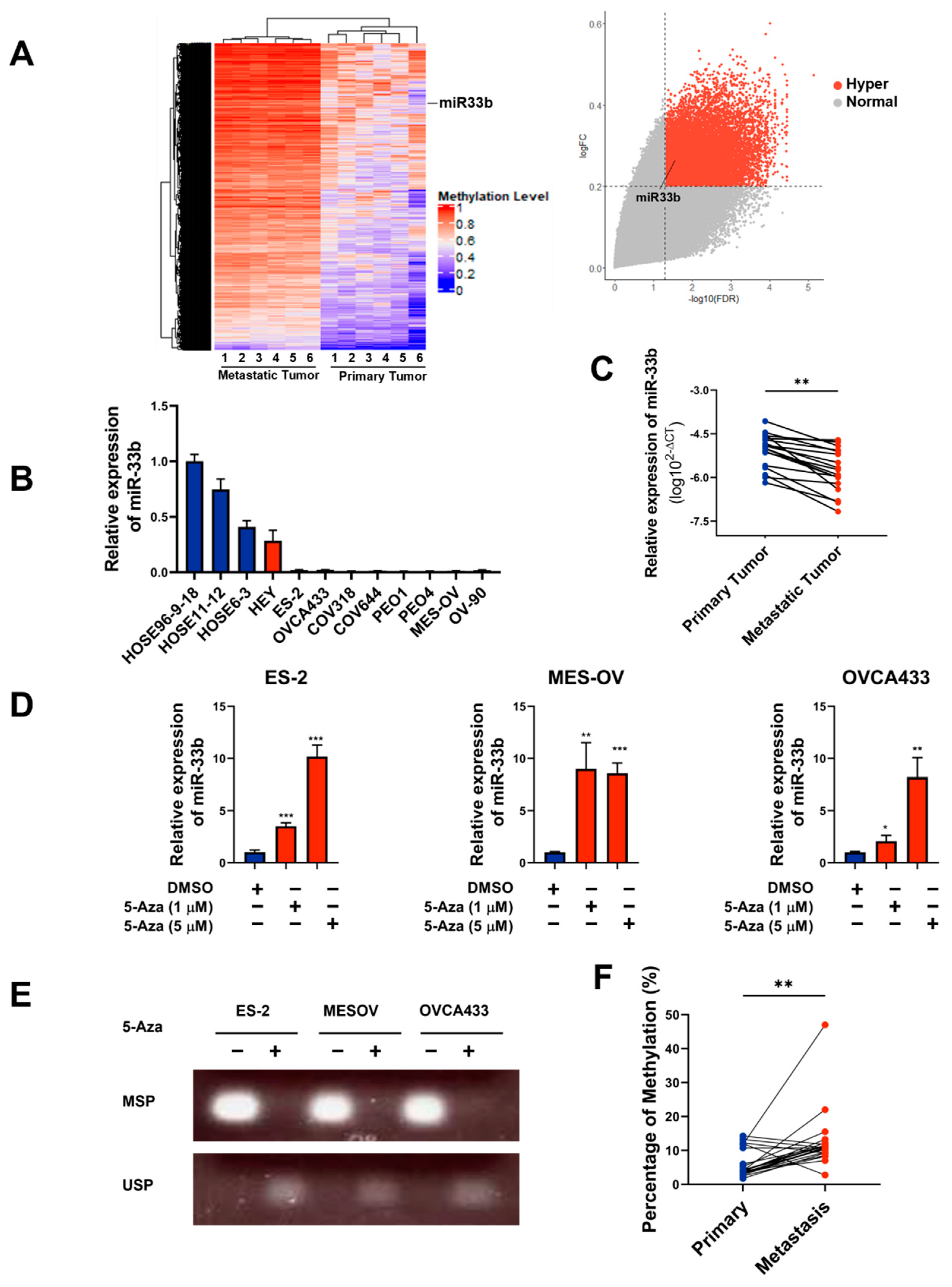
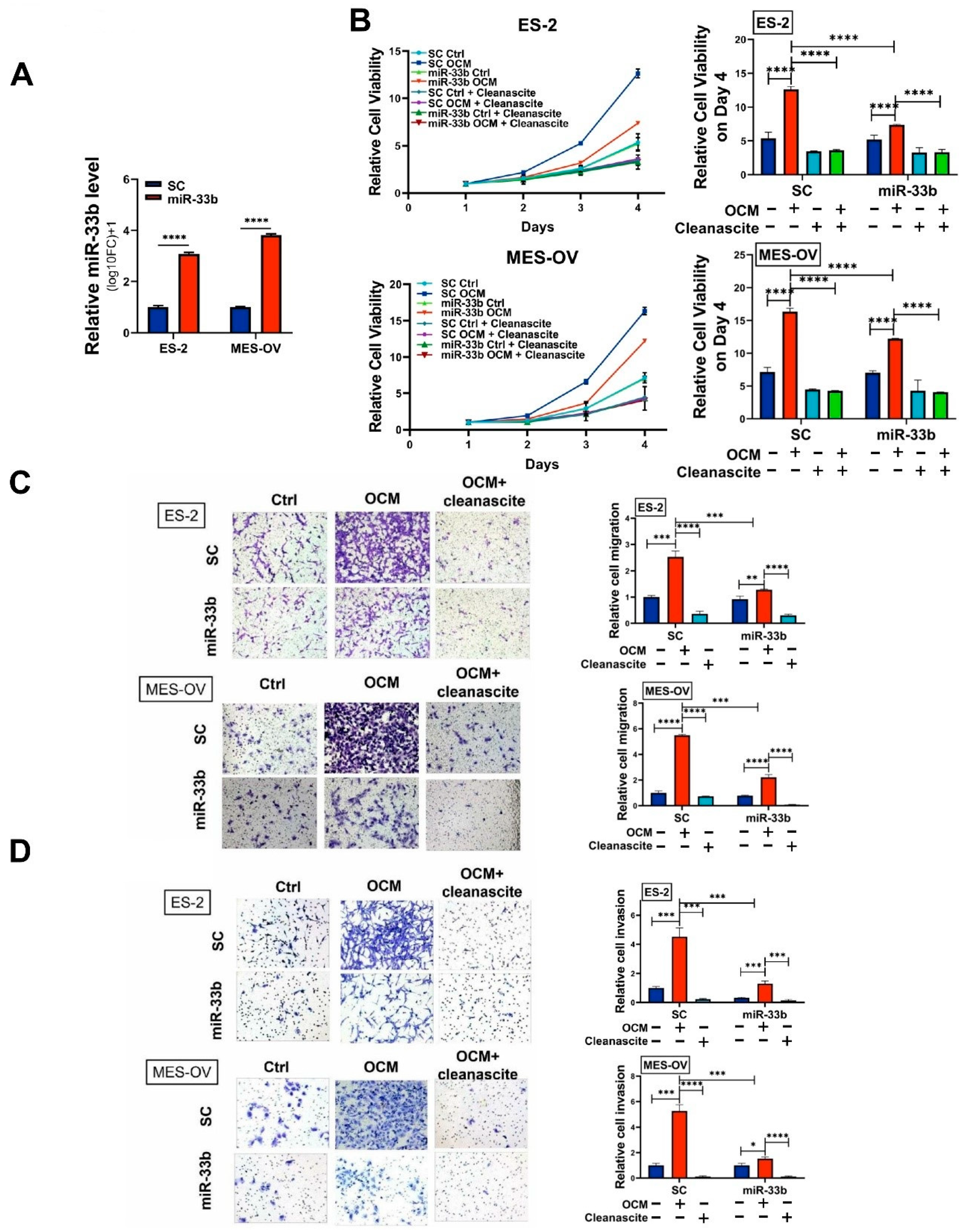
3.2. Overexpression of miR-33b Attenuates OCM-Mediated Oncogenic Properties of Ovarian Cancer Cells
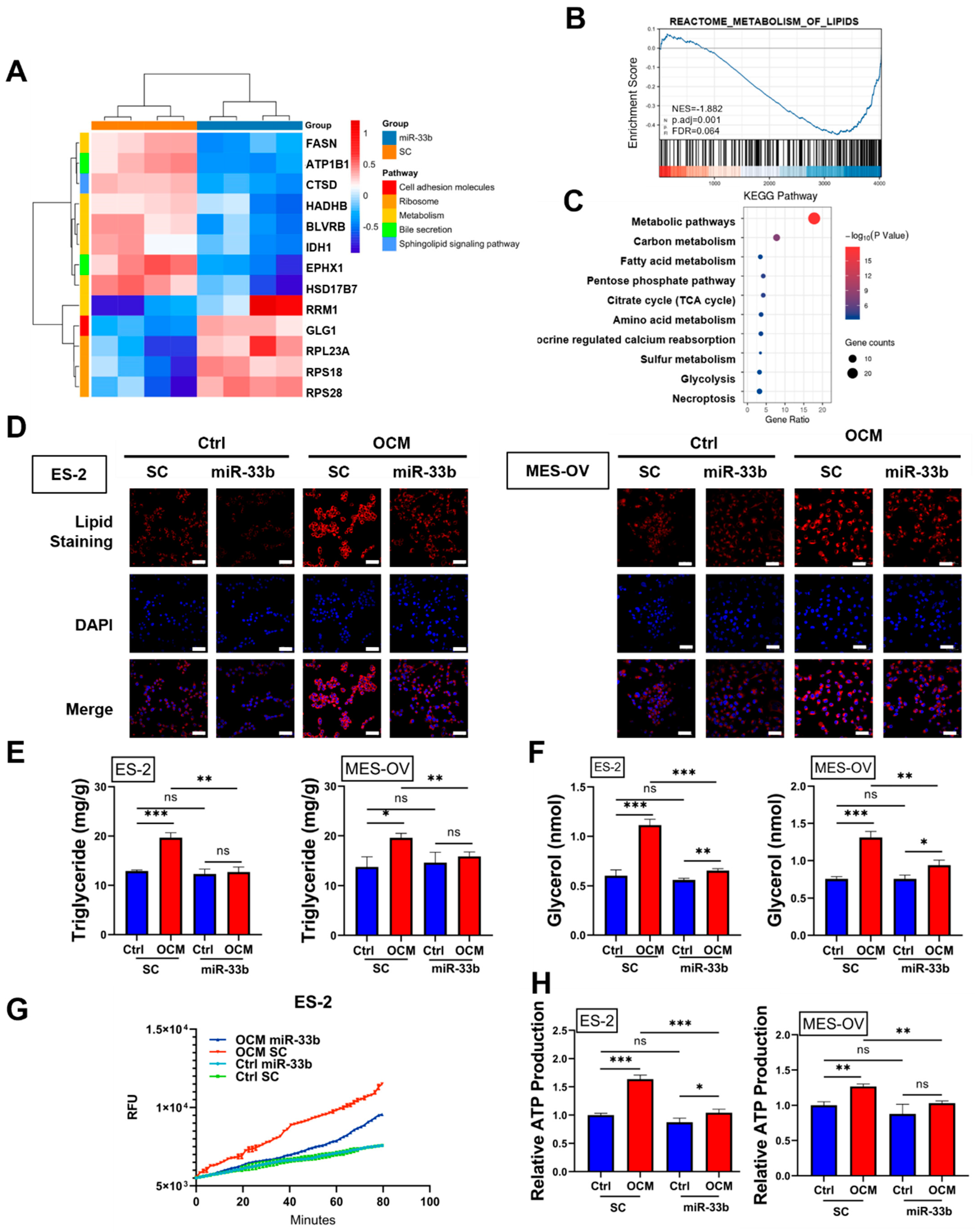
3.3. Enforced miR-33b Expression Inhibited OCM-Mediated High Lipid Metabolic Activities in Ovarian Cancer Cells
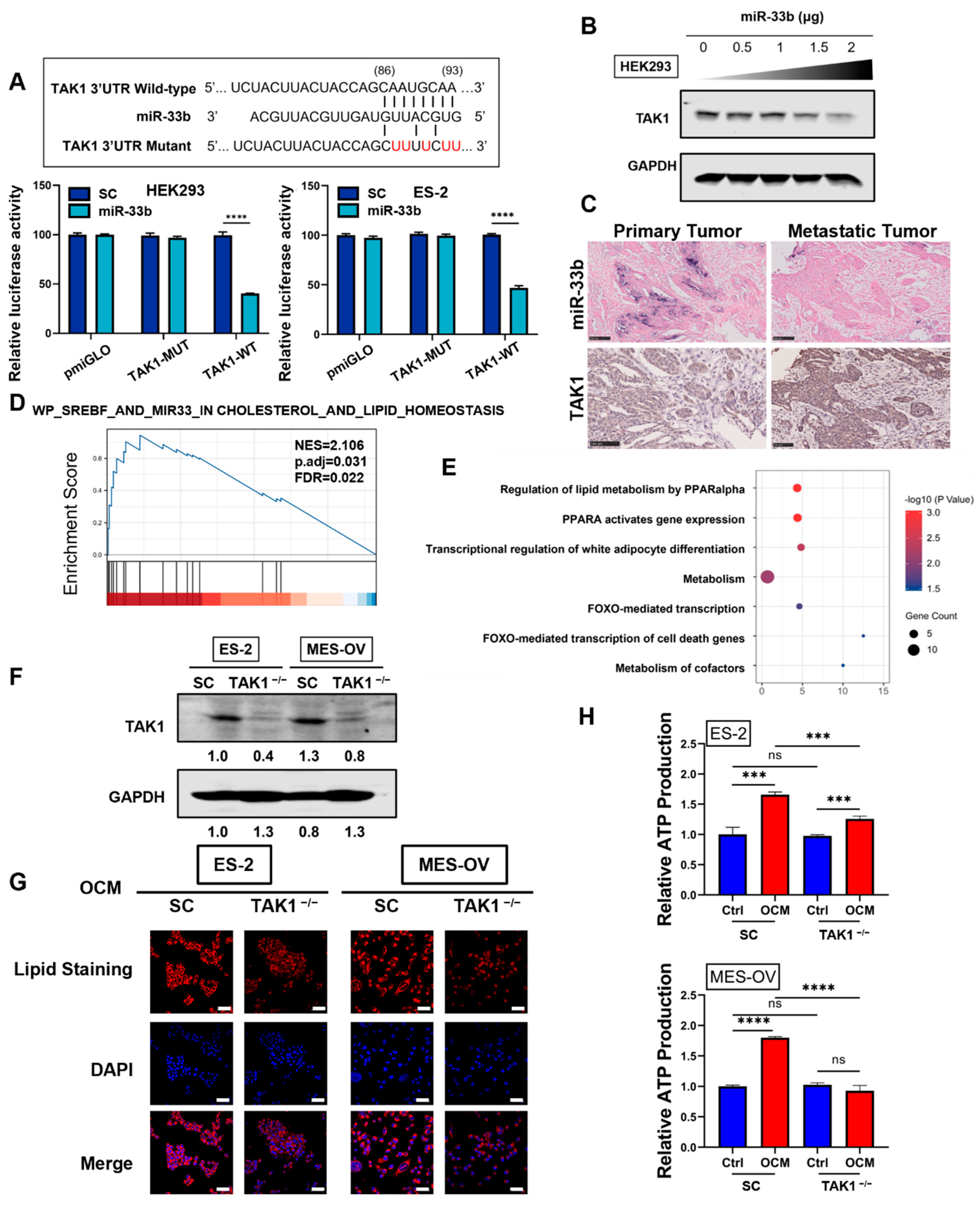
3.4. miR-33b Directly Targets TAK1, Which Is Involved in Lipid Metabolism Reprogramming of Ovarian Cancer
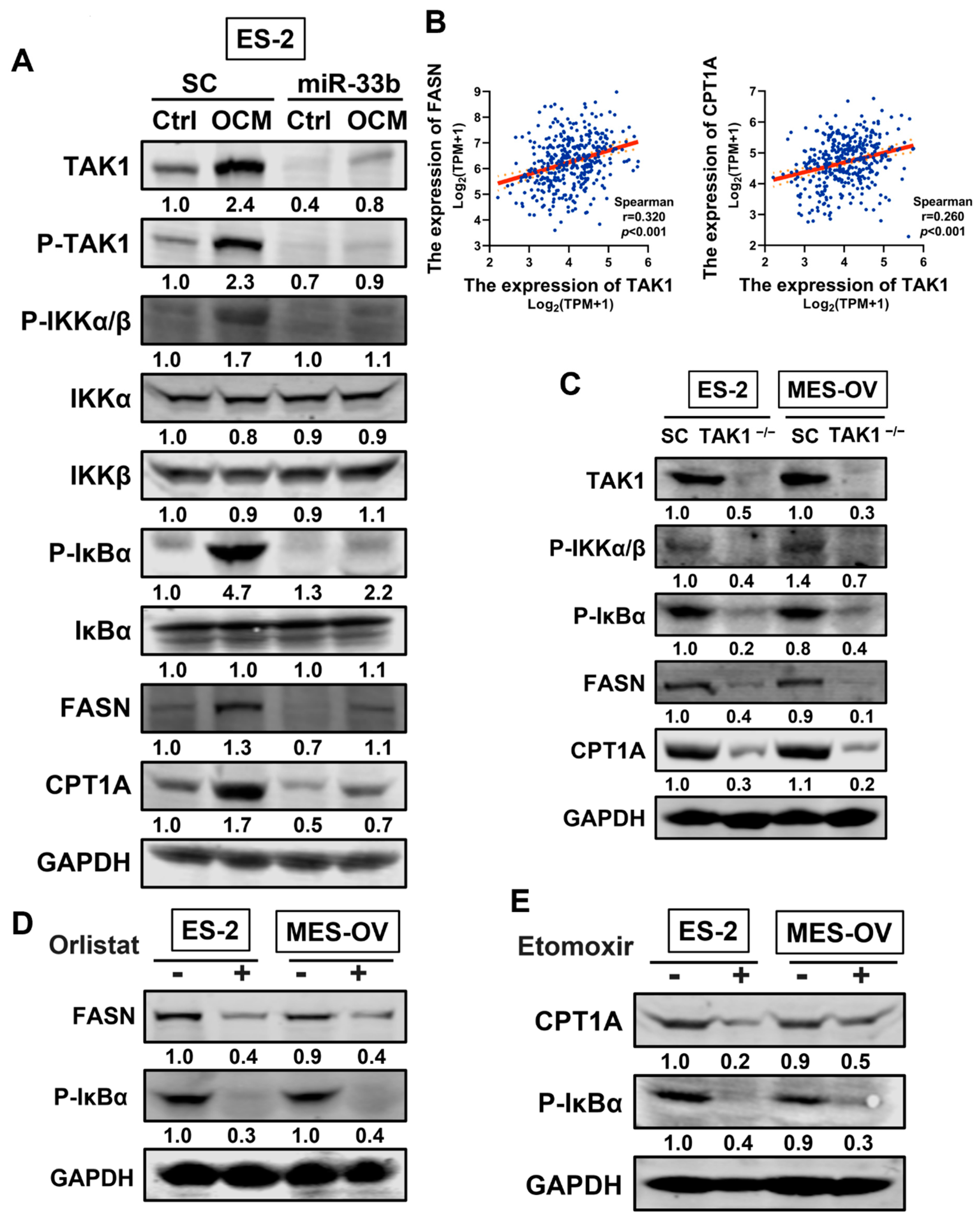
3.5. miR-33b Overexpression Suppresses TAK1/FASN/CPT1A/NF-κB Signaling in Ovarian Cancer
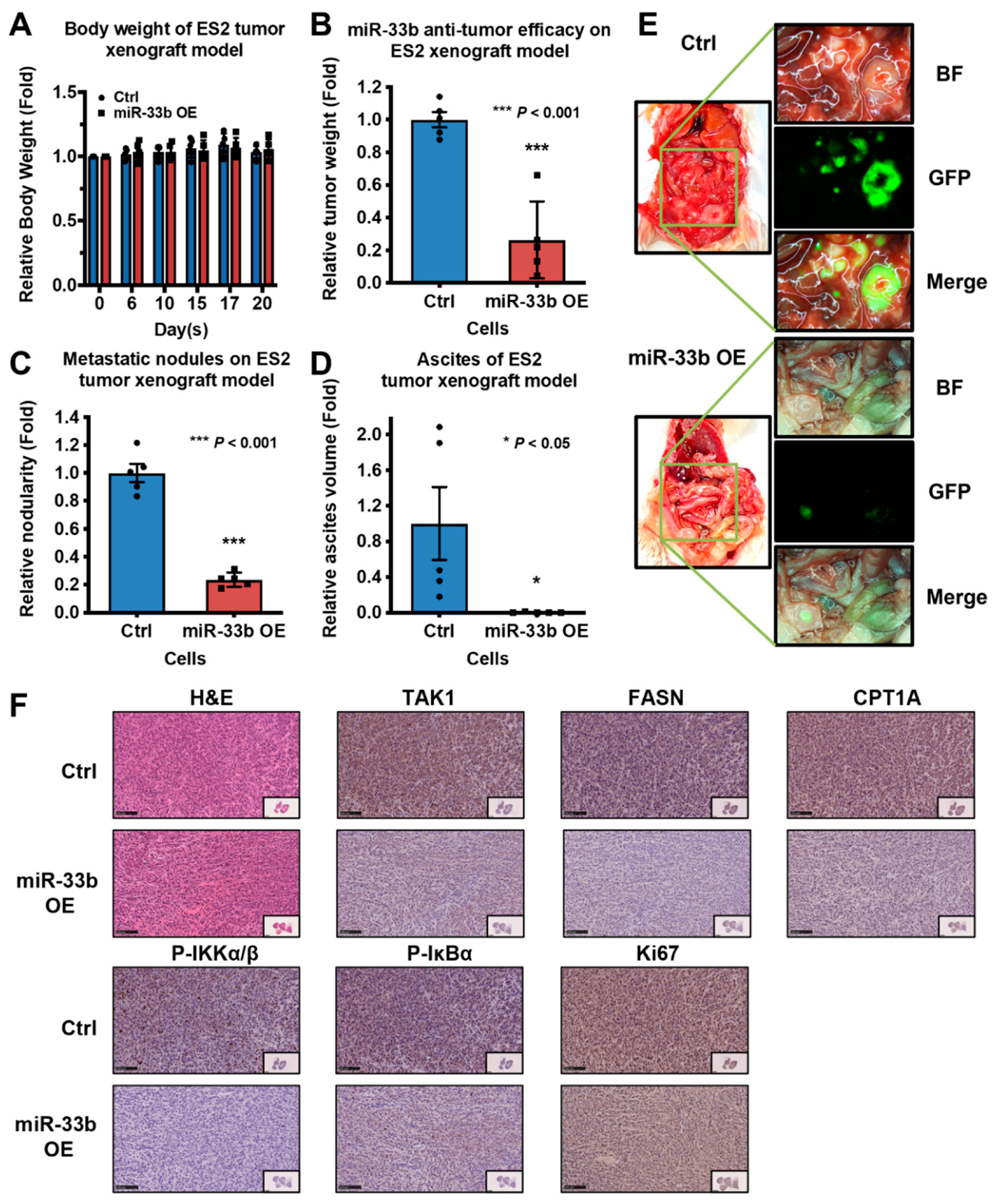
3.6. Induced Expression of miR-33b Suppresses Ovarian Cancer Growth In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CPT1A | Carnitine palmitoyltransferase 1A |
| EOC | Epithelial ovarian cancer |
| FASN | Fatty acid synthase |
| HGSOC | High-grade serous ovarian cancer |
| HOSE | Human ovarian surface epithelial |
| IHC | Immunohistochemical |
| ISH | In situ hybridization |
| MSP | Methylation specific primers |
| MS-PCR | Methylation-specific PCR |
| OCM | Omental conditioned medium |
| PVDF | Polyvinylidene difluoride |
| SDS-PAGE | Sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| TAK1 | Transforming growth factor beta-activated kinase 1 |
| TCGA | The Cancer Genome Atlas |
| TME | Tumor microenvironment |
| USP | Unmethylation specific primers |
References
- Zhao, Y.; Hong, X.; Chen, X.; Hu, C.; Lu, W.; Xie, B.; Zhong, L.; Zhang, W.; Cao, H.; Chen, B. Deregulation of Exo70 Facilitates Innate and Acquired Cisplatin Resistance in Epithelial Ovarian Cancer by Promoting Cisplatin Efflux. Cancers 2021, 13, 3467. [Google Scholar] [CrossRef]
- McCluggage, W.G. Morphological subtypes of ovarian carcinoma: A review with emphasis on new developments and pathogenesis. Pathology 2011, 43, 420–432. [Google Scholar] [CrossRef]
- Weidle, U.H.; Birzele, F.; Kollmorgen, G.; Rueger, R. Mechanisms and targets involved in dissemination of ovarian cancer. Cancer Genom. Proteom. 2016, 13, 407–423. [Google Scholar] [CrossRef] [Green Version]
- Alzamil, L.; Nikolakopoulou, K.; Turco, M.Y. Organoid systems to study the human female reproductive tract and pregnancy. Cell Death Differ. 2021, 28, 35–51. [Google Scholar] [CrossRef]
- Motohara, T.; Masuda, K.; Morotti, M.; Zheng, Y.; El-Sahhar, S.; Chong, K.Y.; Wietek, N.; Alsaadi, A.; Karaminejadranjbar, M.; Hu, Z. An evolving story of the metastatic voyage of ovarian cancer cells: Cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene 2019, 38, 2885–2898. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Nag, S.; Aggarwal, S.; Rauthan, A.; Warrier, N. Maintenance therapy for recurrent epithelial ovarian cancer: Current therapies and future perspectives—A review. J. Ovarian Res. 2019, 12, 1–15. [Google Scholar] [CrossRef]
- Nimmagadda, S.; Penet, M.-F. Ovarian cancer targeted theranostics. Front. Oncol. 2020, 9, 1537. [Google Scholar] [CrossRef]
- Clark, R.; Krishnan, V.; Schoof, M.; Rodriguez, I.; Theriault, B.; Chekmareva, M.; Rinker-Schaeffer, C. Milky spots promote ovarian cancer metastatic colonization of peritoneal adipose in experimental models. Am. J. Pathol. 2013, 183, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Valkenburg, K.C.; de Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Patel, S.A.; Vanharanta, S. Epigenetic determinants of metastasis. Mol. Oncol. 2017, 11, 79–96. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Zhan, X.; Dong, C.; Liu, L. Genomics alterations of metastatic and primary tissues across 15 cancer types. Sci. Rep. 2017, 7, 13262. [Google Scholar] [CrossRef] [Green Version]
- Bertucci, F.; Finetti, P.; Guille, A.; Adelaide, J.; Garnier, S.; Carbuccia, N.; Monneur, A.; Charafe-Jauffret, E.; Goncalves, A.; Viens, P.; et al. Comparative genomic analysis of primary tumors and metastases in breast cancer. Oncotarget 2016, 7, 27208–27219. [Google Scholar] [CrossRef] [Green Version]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic drivers of tumourigenesis and cancer metastasis. Semin. Cancer Biol. 2018, 51, 149–159. [Google Scholar] [CrossRef]
- Reyes, H.D.; Devor, E.J.; Warrier, A.; Newtson, A.M.; Mattson, J.; Wagner, V.; Duncan, G.N.; Leslie, K.K.; Gonzalez-Bosquet, J. Differential DNA methylation in high-grade serous ovarian cancer (HGSOC) is associated with tumor behavior. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Moufarrij, S.; Dandapani, M.; Arthofer, E.; Gomez, S.; Srivastava, A.; Lopez-Acevedo, M.; Villagra, A.; Chiappinelli, K.B. Epigenetic therapy for ovarian cancer: Promise and progress. Clin. Epigenet. 2019, 11, 1–11. [Google Scholar] [CrossRef]
- Hentze, J.L.; Høgdall, C.K.; Høgdall, E.V. Methylation and ovarian cancer: Can DNA methylation be of diagnostic use? Mol. Clin. Oncol. 2019, 10, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Menon, D.R.; Hammerlindl, H.; Torrano, J.; Schaider, H.; Fujita, M. Epigenetics and metabolism at the crossroads of stress-induced plasticity, stemness and therapeutic resistance in cancer. Theranostics 2020, 10, 6261. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, W.; Zhang, Y.; Sun, W.; Yung, M.M.; Sun, J.; Li, J.; Chen, C.-W.; Li, Z.; Meng, Y. ERK regulates HIF1α-mediated platinum resistance by directly targeting PHD2 in ovarian cancer. Clin. Cancer Res. 2019, 25, 5947–5960. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Gupta, S.; Varma, K.; Sachan, M. MicroRNA as biomarker in ovarian cancer management: Advantages and challenges. DNA Cell Biol. 2020, 39, 2103–2124. [Google Scholar] [CrossRef]
- Ma, X. The omentum, a niche for premetastatic ovarian cancer. J. Exp. Med. 2020, 217, e20192312. [Google Scholar] [CrossRef] [Green Version]
- Klymenko, Y.; Nephew, K.P. Epigenetic crosstalk between the tumor microenvironment and ovarian cancer cells: A therapeutic road less traveled. Cancers 2018, 10, 295. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.R.; Yung, M.M.; Xuan, Y.; Zhan, S.; Leung, L.L.; Liang, R.R.; Leung, T.H.; Yang, H.; Xu, D.; Sharma, R. Targeting of lipid metabolism with a metabolic inhibitor cocktail eradicates peritoneal metastases in ovarian cancer cells. Commun. Biol. 2019, 2, 1–15. [Google Scholar] [CrossRef]
- Chen, K.; Liu, M.X.; Mak, C.S.-L.; Yung, M.M.-H.; Leung, T.H.-Y.; Xu, D.; Ngu, S.-F.; Chan, K.K.-L.; Yang, H.; Ngan, H.Y.-S. Methylation-associated silencing of miR-193a-3p promotes ovarian cancer aggressiveness by targeting GRB7 and MAPK/ERK pathways. Theranostics 2018, 8, 423. [Google Scholar] [CrossRef]
- Chan, D.W.; Yung, M.M.; Chan, Y.-S.; Xuan, Y.; Yang, H.; Xu, D.; Zhan, J.-B.; Chan, K.K.; Ng, T.-B.; Ngan, H.Y. MAP30 protein from Momordica charantia is therapeutic and has synergic activity with cisplatin against ovarian cancer in vivo by altering metabolism and inducing ferroptosis. Pharmacol. Res. 2020, 161, 105157. [Google Scholar] [CrossRef]
- Matei, D.E.; Nephew, K.P. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol. Oncol. 2010, 116, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.W.; Lam, W.Y.; Chen, F.; Yung, M.M.H.; Chan, Y.S.; Chan, W.S.; He, F.; Liu, S.S.; Chan, K.K.L.; Li, B.; et al. Genome-wide DNA methylome analysis identifies methylation signatures associated with survival and drug resistance of ovarian cancers. Clin. Epigenet. 2021, 13, 142. [Google Scholar] [CrossRef]
- Dávalos, A.; Goedeke, L.; Smibert, P.; Ramírez, C.M.; Warrier, N.P.; Andreo, U.; Cirera-Salinas, D.; Rayner, K.; Suresh, U.; Pastor-Pareja, J.C. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 9232–9237. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Hagymási, K.; Tulassay, Z. Helicobacter pylori infection: New pathogenetic and clinical aspects. World J. Gastroenterol. 2014, 20, 6386. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, B.T.; Qamar, L.; Yamamoto, T.M.; McMellen, A.; Watson, Z.L.; Richer, J.K.; Behbakht, K.; Schlaepfer, I.R.; Bitler, B.G. Targeting fatty acid oxidation to promote anoikis and inhibit ovarian cancer progression. Mol. Cancer Res. 2020, 18, 1088–1098. [Google Scholar] [CrossRef] [Green Version]
- Cai, P.C.; Shi, L.; Liu, V.W.; Tang, H.W.; Liu, I.J.; Leung, T.H.; Chan, K.K.; Yam, J.W.; Yao, K.-M.; Ngan, H.Y. Elevated TAK1 augments tumor growth and metastatic capacities of ovarian cancer cells through activation of NF-κB signaling. Oncotarget 2014, 5, 7549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, M.M.-H.; Tang, H.W.-M.; Cai, P.C.-H.; Leung, T.H.-Y.; Ngu, S.-F.; Chan, K.K.-L.; Xu, D.; Yang, H.; Ngan, H.Y.-S.; Chan, D.W. GRO-α and IL-8 enhance ovarian cancer metastatic potential via the CXCR2-mediated TAK1/NFκB signaling cascade. Theranostics 2018, 8, 1270. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Shen, Y.; Feng, X.; Kong, Y.; Shao, Y.; Meng, J.; Zhang, X.; Yang, G. Deregulation of lipid metabolism: The critical factors in ovarian cancer. Front. Oncol. 2020, 10, 2288. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, F.; Xu, Q.; Han, L.; Xu, J.; Gao, L.; Sun, X.; Li, Y.; Li, Y.; Qian, M. Revisiting ovarian cancer microenvironment: A friend or a foe? Protein Cell 2018, 9, 674–692. [Google Scholar] [CrossRef] [Green Version]
- Cagan, R.L.; Zon, L.I.; White, R.M. Modeling cancer with flies and fish. Dev. Cell 2019, 49, 317–324. [Google Scholar] [CrossRef]
- Azmi, A.S.; Li, Y.; Aboukameel, A.; Muqbil, I.; Philip, P.A.; Mohammad, R.M. DNA-methylation-caused downregulation of miR-30 contributes to the high expression of XPO1 and the aggressive growth of tumors in pancreatic ductal adenocarcinoma. Cancers 2019, 11, 1101. [Google Scholar] [CrossRef] [Green Version]
- Oronsky, B.; Oronsky, N.; Scicinski, J.; Fanger, G.; Lybeck, M.; Reid, T. Rewriting the epigenetic code for tumor resensitization: A review. Transl. Oncol. 2014, 7, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Sookram, J.; Zheng, A.; Linden, K.M.; Morgan, A.B.; Brown, S.A.; Ostrovsky, O. Epigenetic therapy can inhibit growth of ovarian cancer cells and reverse chemoresistant properties acquired from metastatic omentum. Int. J. Gynecol. Obstet. 2019, 145, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Su, H.Y.; Lai, H.C.; Lin, Y.W.; Liu, C.Y.; Chen, C.K.; Chou, Y.C.; Lin, S.P.; Lin, W.C.; Lee, H.Y.; Yu, M.H. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through Wnt signaling pathway. Int. J. Cancer 2010, 127, 555–567. [Google Scholar] [CrossRef]
- Vang, S.; Wu, H.-T.; Fischer, A.; Miller, D.H.; MacLaughlan, S.; Douglass, E.; Steinhoff, M.; Collins, C.; Smith, P.J.; Brard, L. Identification of ovarian cancer metastatic miRNAs. PLoS ONE 2013, 8, e58226. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.X.; Siu, M.K.; Liu, S.S.; Yam, J.W.; Ngan, H.Y.; Chan, D.W. Epigenetic silencing of microRNA-199b-5p is associated with acquired chemoresistance via activation of JAG1-Notch1 signaling in ovarian cancer. Oncotarget 2014, 5, 944. [Google Scholar] [CrossRef] [Green Version]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef]
- Tao, Y.; Xu, S.; Wang, J.; Xu, L.; Zhang, C.; Chen, K.; Lian, Z.; Zhou, J.; Xie, H.; Zheng, S. Delivery of microRNA-33 antagomirs by mesoporous silica nanoparticles to ameliorate lipid metabolic disorders. Front. Pharmacol. 2020, 11, 921. [Google Scholar] [CrossRef]
- Yang, C.; Xia, B.-R.; Zhang, Z.-C.; Zhang, Y.-J.; Lou, G.; Jin, W.-L. Immunotherapy for Ovarian Cancer: Adjuvant, Combination, and Neoadjuvant. Front. Immunol. 2020, 11, 2595. [Google Scholar] [CrossRef]
- Lyssiotis, C.A.; Kimmelman, A.C. Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 2017, 27, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Wolpaw, A.J.; Dang, C.V. Exploiting metabolic vulnerabilities of cancer with precision and accuracy. Trends Cell Biol. 2018, 28, 201–212. [Google Scholar] [CrossRef]
- Kim, J.; DeBerardinis, R.J. Mechanisms and implications of metabolic heterogeneity in cancer. Cell Metab. 2019, 30, 434–446. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 1–16. [Google Scholar] [CrossRef]
- Sassmann-Schweda, A.; Singh, P.; Tang, C.; Wietelmann, A.; Wettschureck, N.; Offermanns, S. Increased apoptosis and browning of TAK1-deficient adipocytes protects against obesity. JCI Insight 2016, 1, e81175. [Google Scholar] [CrossRef] [Green Version]
- Gallot, Y.S.; McMillan, J.D.; Xiong, G.; Bohnert, K.R.; Straughn, A.R.; Hill, B.G.; Kumar, A. Distinct roles of TRAF6 and TAK1 in the regulation of adipocyte survival, thermogenesis program, and high-fat diet-induced obesity. Oncotarget 2017, 8, 112565. [Google Scholar] [CrossRef] [Green Version]
- Inokuchi-Shimizu, S.; Park, E.J.; Roh, Y.S.; Yang, L.; Zhang, B.; Song, J.; Liang, S.; Pimienta, M.; Taniguchi, K.; Wu, X. TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J. Clin. Investig. 2014, 124, 3566–3578. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zhang, P.; Zhou, J.; Liao, R.; Che, Y.; Gao, M.-M.; Sun, J.; Cai, J.; Cheng, X.; Huang, Y. TNFAIP3 interacting protein 3 overexpression suppresses nonalcoholic steatohepatitis by blocking TAK1 activation. Cell Metab. 2020, 31, 726–740. [Google Scholar] [CrossRef]
- Rayet, B.; Gelinas, C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene 1999, 18, 6938–6947. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, C.R.; Woo, J.-H.; Tully, E.; Wilsbach, K.; Gabrielson, E. Nuclear Factor-κB (NF-κB) Mediates a Protective Response in Cancer Cells Treated with Inhibitors of Fatty Acid Synthase. J. Biol. Chem. 2011, 286, 31457–31465. [Google Scholar] [CrossRef] [Green Version]
- Senga, S.; Kobayashi, N.; Kawaguchi, K.; Ando, A.; Fujii, H. Fatty acid-binding protein 5 (FABP5) promotes lipolysis of lipid droplets, de novo fatty acid (FA) synthesis and activation of nuclear factor-kappa B (NF-κB) signaling in cancer cells. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018, 1863, 1057–1067. [Google Scholar] [CrossRef]
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.-X. Lipid desaturation is a metabolic marker and therapeutic target of ovarian cancer stem cells. Cell Stem Cell 2017, 20, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Li, J.; Chen, S.; Cai, J.; Ban, Y.; Peng, Q.; Zhou, Y.; Zeng, Z.; Peng, S.; Li, X. Emerging role of lipid metabolism alterations in Cancer stem cells. J. Exp. Clin. Cancer Res. 2018, 37, 1–18. [Google Scholar]
- Kobayashi, M.; Sawada, K.; Miyamoto, M.; Shimizu, A.; Yamamoto, M.; Kinose, Y.; Nakamura, K.; Kawano, M.; Kodama, M.; Hashimoto, K. Exploring the potential of engineered exosomes as delivery systems for tumor-suppressor microRNA replacement therapy in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 527, 153–161. [Google Scholar] [CrossRef]
- Wang, X.; Holgado, B.L.; Ramaswamy, V.; Mack, S.; Zayne, K.; Remke, M.; Wu, X.; Garzia, L.; Daniels, C.; Kenney, A.M. miR miR on the wall, who’s the most malignant medulloblastoma miR of them all? Neuro-oncology 2018, 20, 313–323. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Yung, M.M.H.; Sharma, R.; Chen, F.; Poon, Y.-T.; Lam, W.-Y.; Li, B.; Ngan, H.Y.S.; Chan, K.K.L.; Chan, D.W. Epigenetic Silencing of miR-33b Promotes Peritoneal Metastases of Ovarian Cancer by Modulating the TAK1/FASN/CPT1A/NF-κB Axis. Cancers 2021, 13, 4795. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13194795
Wang X, Yung MMH, Sharma R, Chen F, Poon Y-T, Lam W-Y, Li B, Ngan HYS, Chan KKL, Chan DW. Epigenetic Silencing of miR-33b Promotes Peritoneal Metastases of Ovarian Cancer by Modulating the TAK1/FASN/CPT1A/NF-κB Axis. Cancers. 2021; 13(19):4795. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13194795
Chicago/Turabian StyleWang, Xueyu, Mingo M. H. Yung, Rakesh Sharma, Fushun Chen, Ying-Tung Poon, Wai-Yip Lam, Benjamin Li, Hextan Y. S. Ngan, Karen K. L. Chan, and David W. Chan. 2021. "Epigenetic Silencing of miR-33b Promotes Peritoneal Metastases of Ovarian Cancer by Modulating the TAK1/FASN/CPT1A/NF-κB Axis" Cancers 13, no. 19: 4795. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13194795