1. Introduction
Neuroblastoma, a pediatric cancer of the peripheral nervous system, is one of the most common embryonal tumors outside the brain [
1]. It is thought to arise from tissues of the sympathoadrenergic system at early stages of embryonic development, specifically from neural crest progenitors that fail to differentiate during dorsolateral migration [
2]. From a histopathological perspective, neuroblastoma can be classified based on the grade of morphological differentiation into distinct categories, namely neuroblastoma, ganglioneuroblastoma, and ganglioneuroma, from less to more differentiated tumors. The last two categories are considered to be benign forms of neuroblastoma [
3]. The pathological category can be further classified according to the degree of cellular differentiation. For example, neuroblastoma can be further classified into undifferentiated, poorly differentiated, and differentiating tumors (reviewed in [
4]). The degree of differentiation is part of routine prognostic risk assessment. In fact, high-risk neuroblastomas, which represent up to ~60% of all diagnosed neuroblastomas, are usually undifferentiated or poorly differentiated tumors that present very aggressive behavior and have a five-year overall survival rate below 40% [
5]. Pro-differentiation therapies (i.e., with retinoic acid derivatives) are part of the standard of care in neuroblastoma, although they are restricted to the treatment of neuroblastoma minimal residual disease [
6]. However, not all patients respond to this treatment. Therefore, a better understanding of the molecular mechanisms that maintain these types of tumors in an undifferentiated state may reveal new opportunities for therapeutic intervention.
Epigenetic regulation (e.g., DNA methylation, histone post-translational modifications, non-coding RNA expression, etc.) is one of the mechanisms that controls the differentiation of neuroblasts (i.e., neuroblastoma precursors). The disruption of the homeostatic epigenetic balance contributes to the developmental arrest of sympathetic progenitors, thereby contributing to neuroblastoma oncogenesis [
7]. One of the genes that is determinant in the maintenance of neuronal progenitor identity is zuotin-related factor 1 (
ZRF1; also known as MPP11), which is encoded by the
DNAJC2 gene [
8,
9].
ZRF1 belongs to the M-phase phosphoprotein (MPP) family and was first discovered as a chaperone in the cytosol [
10,
11,
12]. However, later evidence has shown that it also acts as a chromatin regulator in the nucleus, where it is recruited to ubiquitinated histone H2A at ‘Lys-119’ (H2AK119ub), displacing the polycomb repressor complex 1 (PRC1) from chromatin and facilitating the transcription of neural progenitor-associated genes [
13].
In addition to its physiological role in the maintenance of the pluripotency of neural progenitor cells,
ZRF1 has already been functionally implicated in cancer.
ZRF1 was shown to be oncogenic in solid tumors such as breast [
14] and gastric [
15] cancers. Furthermore,
ZRF1 was shown to be overexpressed in acute myeloid leukemia (AML) acting as a negative regulator of differentiation. In the same study, Demajo and collaborators showed that
ZRF1 depletion cooperated with differentiating agents (i.e., retinoic acid) to suppress leukemia in vivo [
16].
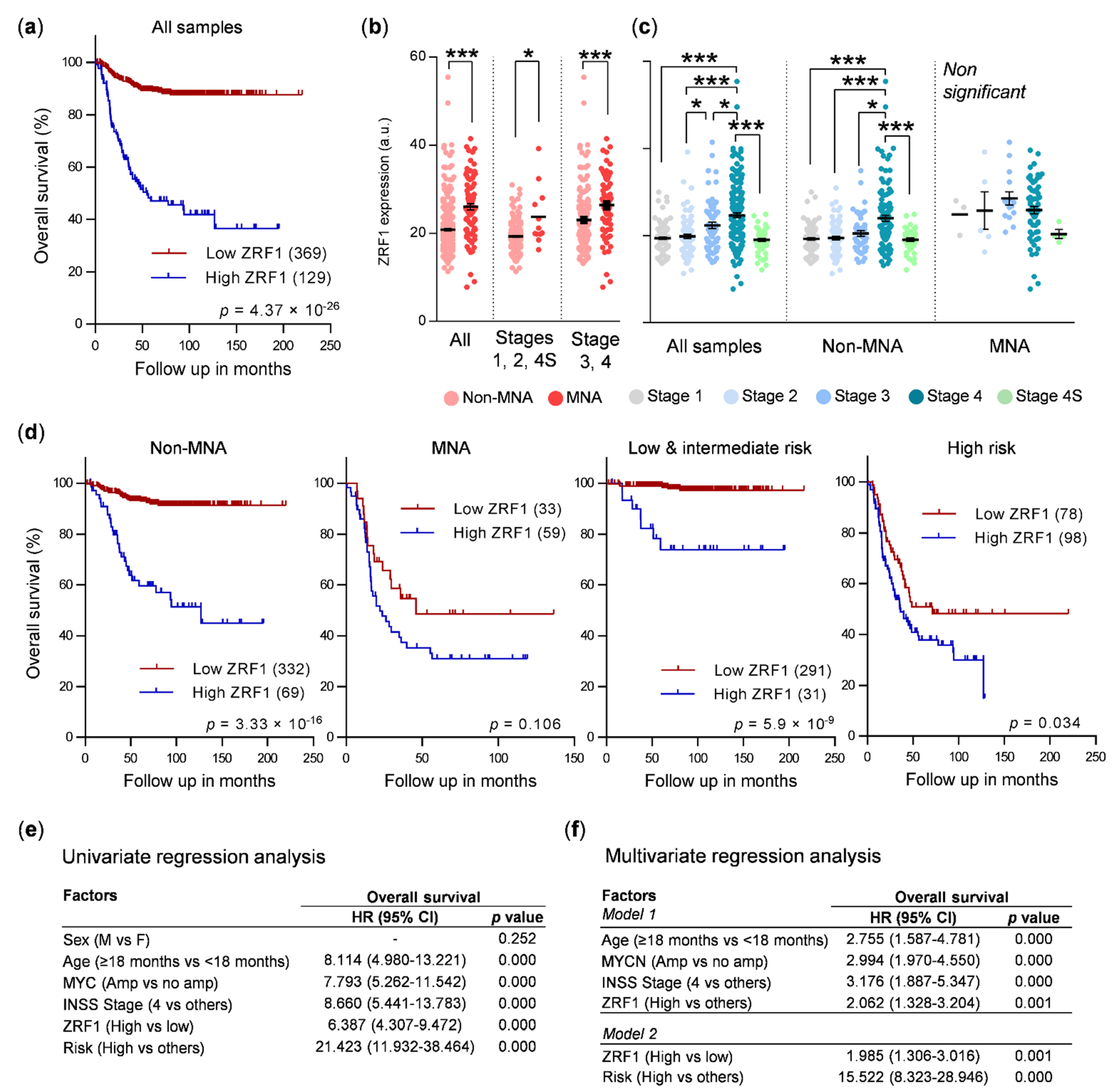
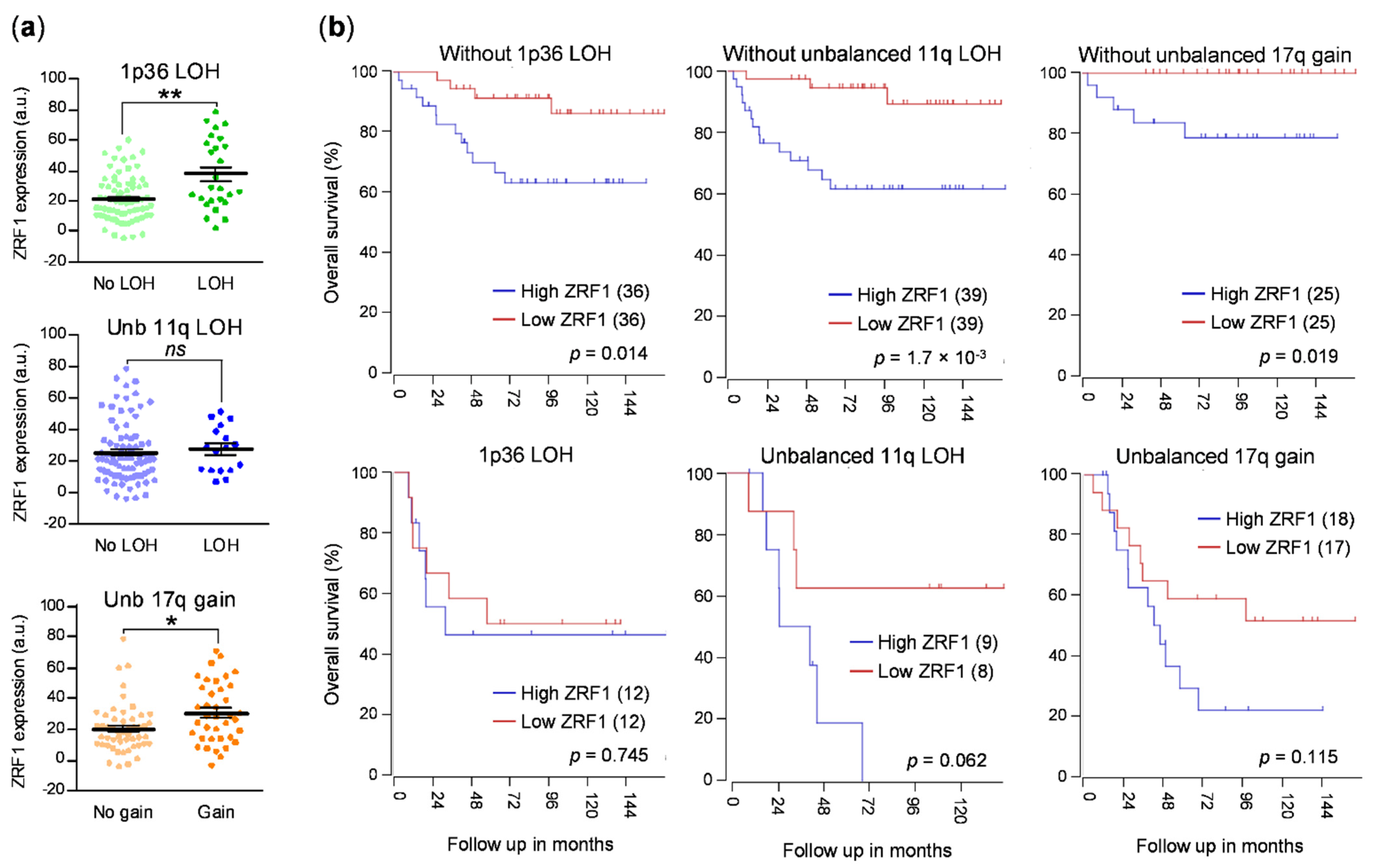
Thus, owing to the role of ZRF1 of maintaining the undifferentiated state of neural progenitor cells and its functional relevance in the differentiation of some tumors, we sought to determine whether ZRF1 plays a major role in neuroblastoma. Here, we found that the expression of ZRF1 mRNA is increased in advanced disease stages and in tumors, with the most common genetic alterations associated with poor prognosis in neuroblastoma, such as MYCN amplification, gain of chromosome 17q, and loss of 1p36. Moreover, the ZRF1 mRNA level was clearly associated with poor neuroblastoma survival in the absence of other poor-prognosis molecular alterations. However, our gain- and loss-of-function experiments suggest that ZRF1 is neither sufficient nor necessary to sustain the oncogenic properties of neuroblastoma cells, such as cell differentiation, proliferation, or migration. Our results validate ZRF1 as a potential prognostic marker, but discard it as a target for differentiation therapy.
2. Materials and Methods
2.1. Analysis of Neuroblastoma Gene Expression Datasets
ZRF1 mRNA expression levels were analyzed from neuroblastoma patient data from the GSE62564, GSE45547 and GSE3960 publicly available datasets. The GSE62564 dataset was used to construct receiver operating characteristic (ROC) curves to determine the prognostic value of
ZRF1 expression. The optimal cutoff value was defined according to the Youden index. Overall survival (OS) and the cumulative survival rate were estimated using the Kaplan–Meier method, and the log-rank test was performed to assess differences between groups. Univariate and multivariate Cox proportional hazard regression analyses were used to assess the prognostic significance of
ZRF1 on OS. These statistical analyses were performed using the IBM SPSS 21 software. For GSE45547 and GSE3960 datasets, gene expression data was extracted and Kaplan–Meier survival plots were generated using the R2 Genomics Analysis and Visualization Platform (
http://r2.amc.nl; accessed date: 21 January 2021).
ZRF1 mRNA expression levels between different patient groups were analyzed using GraphPad Prism Software (La Jolla, CA, USA), and statistical significance was assessed by Kruskal–Wallis test as a non-parametric ANOVA, and Dunn’s test for multiple comparisons.
2.2. Cell Lines
Neuroblastoma cell lines (SK-N-AS, SH-SY5Y, and IMR-32) and embryonic kidney cells (HEK293T) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), the CHLA-90 cell line was purchased from the Children’s Oncology Group Cell Culture and Xenograft Repository (Lubbock, TX, USA), and SK-N-BE(2), and LA1-5s were procured from the Public Health England Culture Collection (Salisbury, UK). Neuroblastoma cells were cultured and maintained in Iscove’s modified Dulbecco’s medium (Life Technologies, Waltham, MA, USA), supplemented with 20% heat-inactivated fetal bovine serum (South America Premium, Biowest, Nuaillé, France) and 1% insulin-transferrin-selenium supplement (Life Technologies). HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (Life Technologies), supplemented with 10% heat-inactivated fetal bovine serum. Media were supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin (Life Technologies), and 5 μg/mL plasmocin (InvivoGen, San Diego, CA, USA). Cultures were maintained at 37 °C in a 5% CO2 saturated atmosphere, and periodically tested for mycoplasma contamination.
2.3. Western Blot Analysis
Cells were lysed in RIPA buffer (Thermo Fisher Scientific, Waltham, MA, USA), supplemented with EDTA-free protease inhibitor cocktail (Roche, Basel, Switzerland). Cell lysates were quantified using a DC protein assay (Bio-Rad, Hercules, CA, USA) and 30 μg of protein was resolved on a 4–12% Tris-glycine sodium dodecyl sulfate polyacrylamide electrophoresis gel (Invitrogen, Carlsbad, CA, USA), then transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were blocked for 1 h with 5% non-fat milk or 5% bovine serum albumin (BSA) in Tris-buffered saline with 0.1% Tween and probed with primary antibodies overnight at 4 °C. Membranes were incubated with secondary antibodies for 1 h before developing with a chemiluminescent horseradish peroxidase substrate EZ-ECL Chemoluminiscence Detection Kit (Biological Industries, Kibbutz Beit-Haemek, Israel). Protein levels were quantified by densitometry using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Antibodies used for Western blot are listed in
Table S1. Original western blot images can be found in
Figure S7.
2.4. Proliferation and Colony Formation Assays
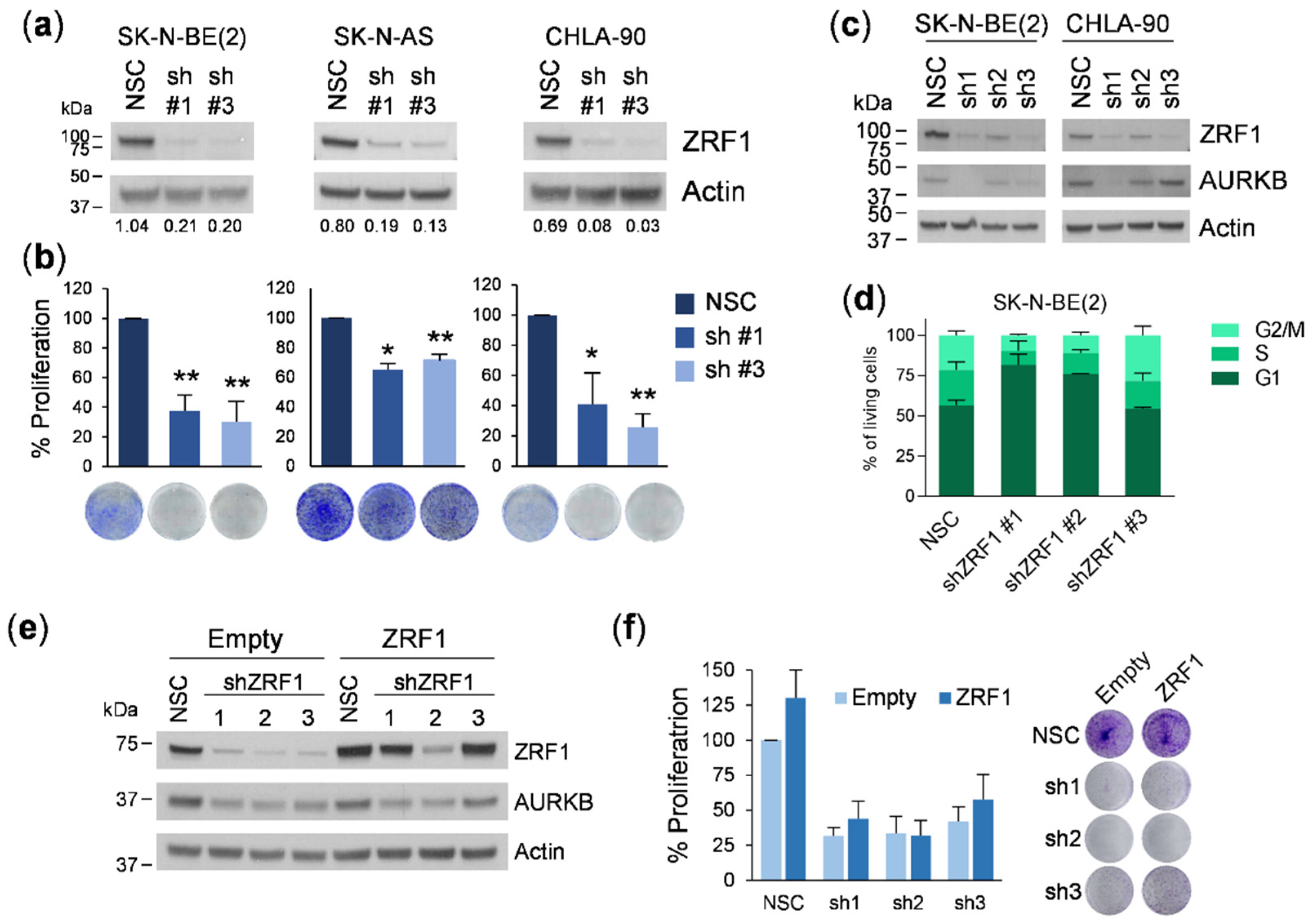
For the proliferation experiments, transduced or transfected cells were plated at a density of 2–8 × 104 cells /well in 6-well plates and allowed to grow for 7 days with a medium change on day 4. Cells were fixed with 1% glutaraldehyde and stained with 0.5% crystal violet. Stained cells were treated in 15% acetic acid, and the absorbance was read at 590 nm. For the colony formation experiments, cells were plated at a very low density (5–10 × 102 cells/well in 6-well plates) and the medium was changed every 3–4 days. The plates were fixed in glutaraldehyde and stained with crystal violet at day 10 or when colonies were visible to the naked eye. Colonies were photographed and counted using ImageJ software.
2.5. Migration and Invasion Assays
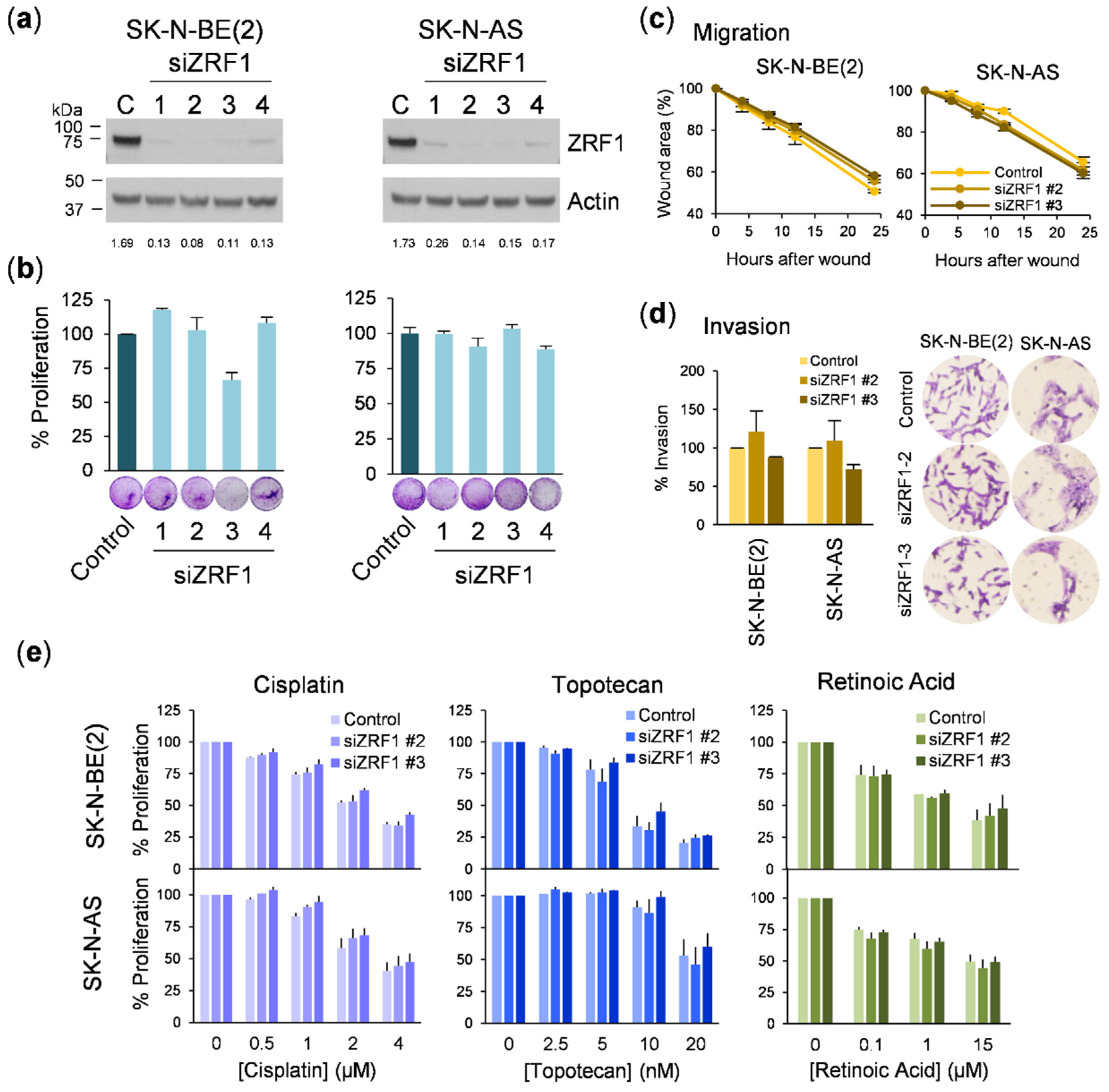
For the wound-healing assays, neuroblastoma cells were plated at a density of 3 × 106 cells/well in a 6-well plate. The next day, an artificial wound was created in the confluent cell monolayer. Six predefined fields per condition were photographed under contrast phase microscopy at the indicated time points, and the wound area was measured using Image J software. The migration rate was calculated by normalizing the wound area to time 0. For the transwell invasion assays, 2 × 105 cells were seeded in serum-free media in the upper chamber of 8.0 µm pore size transwells (Corning Life Sciences, Corning, NY, USA) previously coated with a barrier of rat tail collagen I (Corning). The lower chamber was filled with media supplemented with fetal bovine serum. After 16 h, remaining cells were removed from the upper chamber and the cells that migrated to the lower surface of the membrane were fixed with 4% paraformaldehyde, and stained with crystal violet. Invading cells were imaged by bright field microscopy, quantified by diluting crystals in acetic acid, and read at 590 nm.
2.6. Differentiation Assays
Neuroblastoma cells were plated at low density (1–1.2 × 10
5 cells) in collagen-coated 60 mm plates. One day later, cells were treated with 10 µM 13-
cis-retinoic acid (Selleckchem, Munich, Germany). Cells were collected at day 5 post-treatment for Western blot analysis. For
RARβ gene expression analysis, RNA was extracted from cell lysates using a miRNeasy Mini Kit (Qiagen, Germantown, MA, USA) and retrotranscribed with a high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific). Real-time PCR was performed with PerfeCTa SYBR Green Fastmix (Quantabio, Beverly, MA, USA) using L27 as the internal standard. Primers are listed in
Table S2. Relative quantification of gene expression was calculated using the 2
−DDCt method [
17]. For neurite length analyses, 1× 10
4 cells per well were seeded in collagen-coated glass covers in 24-well plates and treated with retinoic acid for 5 days before fixation with 4% paraformaldehyde. Cells were stained with phalloidin-iFluor 594 (Abcam, Cambridge, UK), following manufacturer’s instructions, and DAPI 10 µ/mL (Invitrogen). Slides were visualized with a FV1000 confocal microscope (Olympus, Shinjuku, Tokyo, Japan). Ten fields were acquired for each biological replicate and processed using ImageJ software. Actin prolongations longer than twice the length of the nucleus (~30 µm) were considered as neurites.
2.7. Vectors and Lentiviral Infection
pEV-
ZRF1, pCAG-
ZRF1, and pLKO with different sh
ZRF1 vectors were kindly provided by Luciano DiCroce. Lentiviruses were generated in HEK293T cells using previously described methods [
18,
19]. Silent mutations were introduced into the
ZRF1 overexpression vector using three sequential site-directed mutagenesis reactions in pCAG-
ZRF1, using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent, Santa Clara, CA, USA) and checked by Sanger sequencing. Primers used for site-directed mutagenesis and sequencing are listed in
Table S2, and the shRNA target sequences are listed in
Table S3. After mutagenesis, the
ZRF1 sequence was excised from the pCAG by XhoI digestion and ligated into the FG12 lentiviral overexpression vector.
2.8. ZRF1 Overexpression Experiments
Neuroblastoma cells plated at a density of 5 × 105 cells in 60 mm plates were transduced with viral supernatant. pEV-transduced cells were selected by separating green fluorescence protein (GFP)-positive cells by fluorescence-assisted cell sorting (FACSAria, BD Biosciences, San Jose, CA, USA) at the Flow Cytometry facility of VHIR.
2.9. ZRF1 Knockdown Experiments
For shRNA transduction, 2–8 × 10
5 cells were plated in 60 mm plates with viral supernatant containing either pLKO-non-silencing control (NSC) or shZRF #1, #2, or #3. After 16 h, the supernatant was replaced with fresh medium, and 24 h later, transduced cells were selected by puromycin resistance (1 µg/mL). Three days after transduction, the cells were detached and used for proliferation experiments. For siRNA knockdown, a set of four pre-designed ON-TARGETplus siRNAs against
ZRF1 were purchased from Dharmacon (Lafayette, CO, USA). The siRNA target sequences are listed in
Table S3. Neuroblastoma cell lines at a concentration of 1.67 × 10
5 cells/mL were transfected with siRNA at 25 nM using Lipofectamine 2000 (Life Technologies) following the manufacturer’s instructions. After incubation for 16 h, the medium was replaced.
4. Discussion
Neuroblastoma is thought to originate from cells of the neural crest that are transformed during differentiation and migration toward tissues of the sympathoadrenergic lineage. The occurrence of a transformative event during tissue differentiation dictates the aggressiveness of the tumor. In general, patients with poor prognosis have histologically undifferentiated tumors, whereas those with better prognosis have tumors with histological evidence of cellular differentiation [
23]. This differentiation program is tightly regulated by a complex set of signals, including external signaling, activation of specific transcriptional programs, and/or epigenetic events (reviewed in [
24]). Experimental results in transgenic mouse models have identified activating ALK mutations and MYCN overexpression as the main oncogenic drivers of neuroblastoma [
25,
26]. These molecular alterations often converge on mechanisms that block differentiation and confer sustained proliferation capabilities. In particular, genes associated with the maintenance of embryonic and adult stem cells, such as the components of PRC1 or PRC2, have been linked to the initiation and progression of neuroblastoma [
27]. For example, BMI1, a core component of the PRC1 complex, has been shown to cooperate in MYCN-driven neuroblastomas by inhibiting cell death and differentiation [
28,
29,
30]. Focusing on the role of PRC1 in neuronal differentiation, Aloia et al. identified
ZRF1 as a transcriptional regulator of neural fates in embryonic stem cells [
9]. Furthermore,
ZRF1 expression has been associated with poor outcomes in other tumors, such as breast [
14,
31] or gastric cancer [
15], thus suggesting an oncogenic role in cancer. Thus, we sought to determine whether
ZRF1 plays a functional role in aggressive neuroblastomas. Data mining of multiple neuroblastoma gene expression datasets confirmed that
ZRF1 mRNA expression was elevated in subsets of patients with the most common genetic alterations associated with poor outcomes, thereby supporting our initial hypothesis, although this correlation could not be confirmed at the protein level in neuroblastoma patient samples.
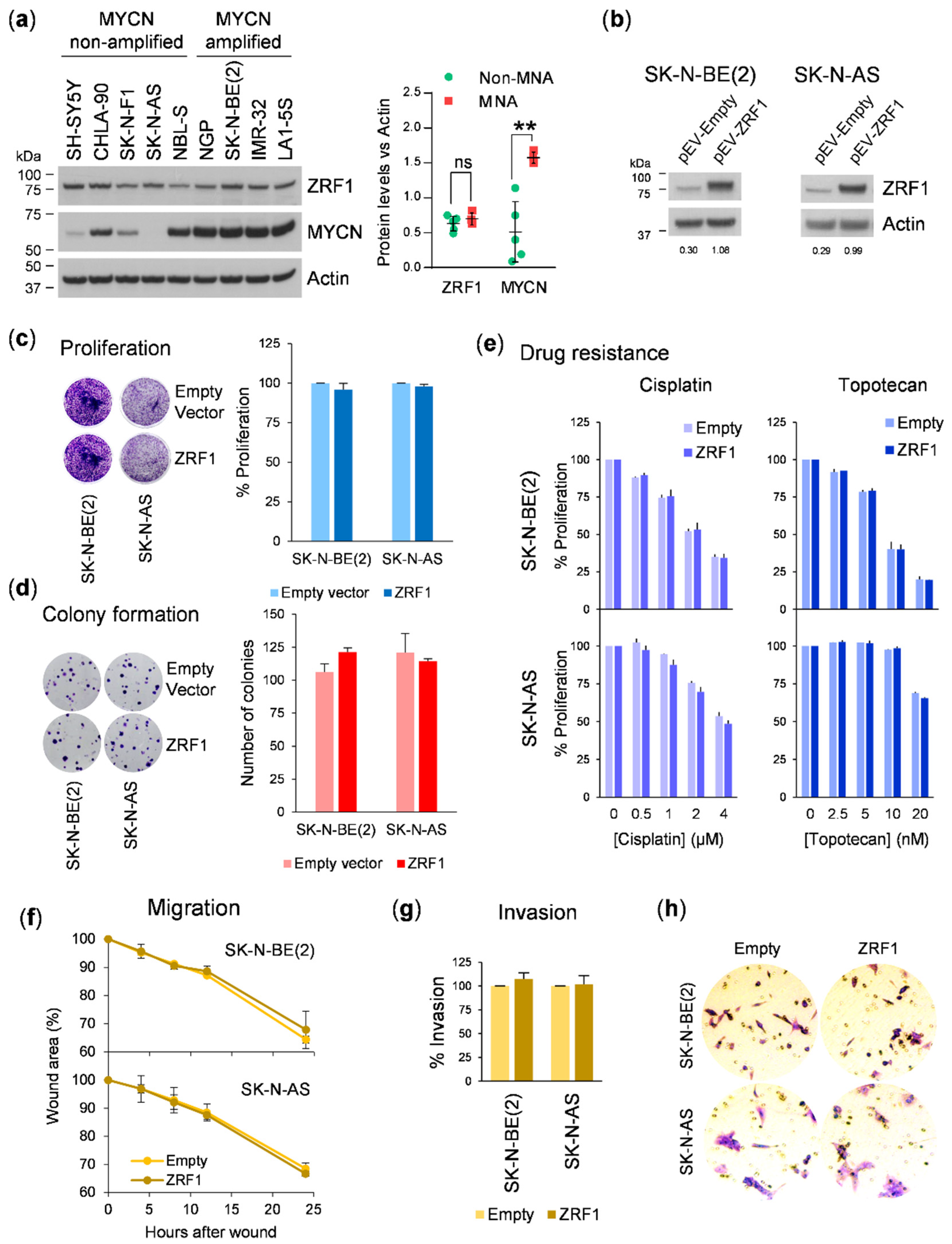
Nevertheless, our functional data do not support a relevant contribution of
ZRF1 in the oncogenesis of neuroblastoma. Ectopic expression of
ZRF1 did not offer proliferation, colony formation, or migratory or invasive advantages to neuroblastoma cells. In acute myeloid leukemia,
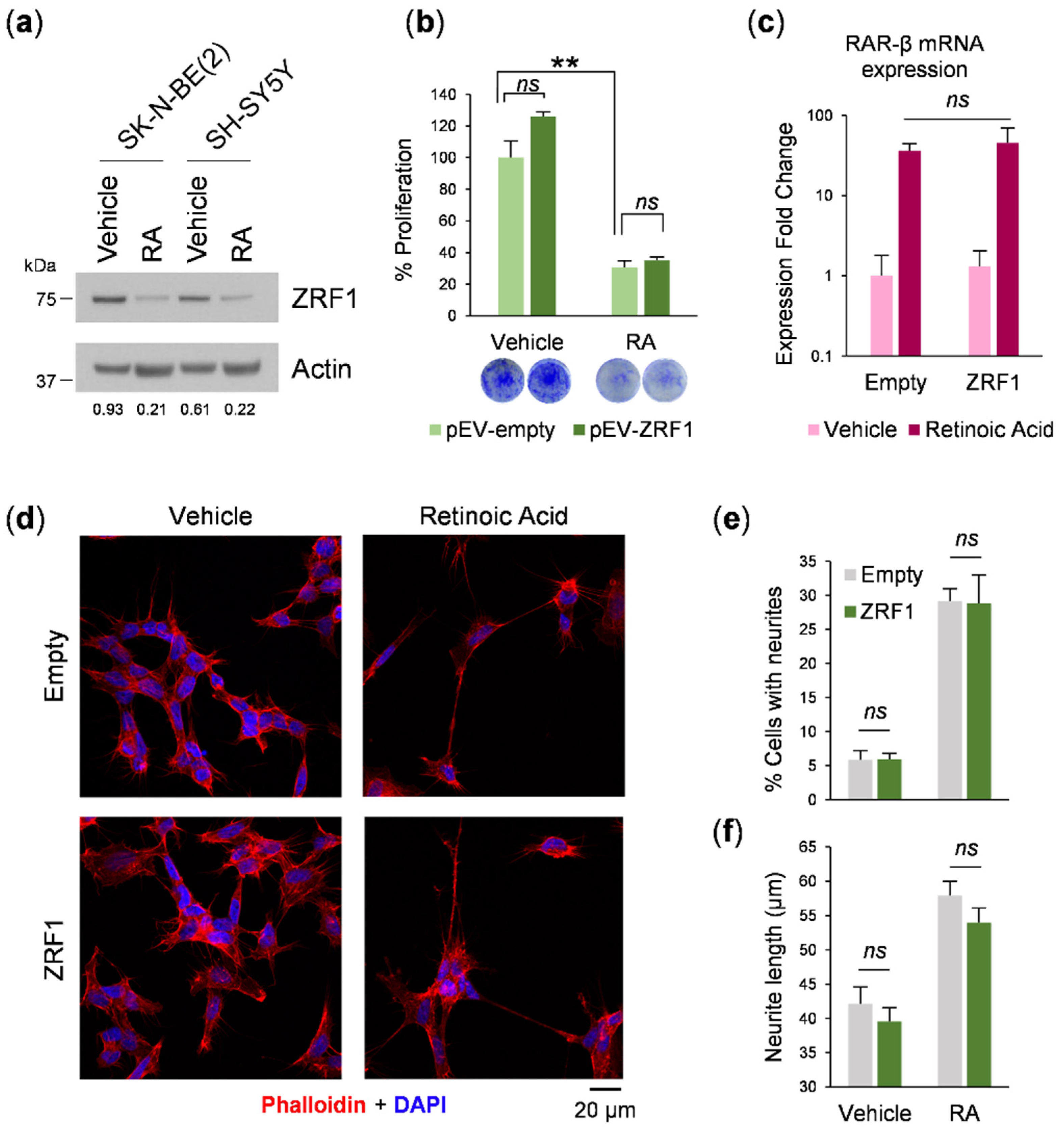
ZRF1 is a regulator of RA-induced differentiation [
16], and because of the relevance of retinoids in neuroblastoma therapy [
22], we investigated whether
ZRF1 interferes with the response of neuroblastoma cells to RA. When neuroblastoma cells were exposed to 13-
cis-RA, a clear reduction in the
ZRF1 level was observed, concomitant with an expected reduction in cell proliferation and morphological changes such as neurite outgrowth. However, maintaining high
ZRF1 expression ectopically was not sufficient to reverse the RA effect. These results suggest that the downregulation of
ZRF1 is a consequence of the RA-induced differentiation process.
Although
ZRF1 is not sufficient to provide oncogenic advantages, it could still be necessary to maintain the undifferentiated and highly proliferative state of neuroblastoma cells. Previous reports demonstrated that depletion of
ZRF1 resulted in a reduction of cell proliferation and the induction of apoptosis in gastric [
15] or breast cancer models [
14]. Conversely, Kaymak et al. also showed that the reduction in cell proliferation mediated by
ZRF1 depletion was accompanied by an increase in the migration and invasion properties of breast cancer cells [
31]. Our first set of experiments silencing
ZRF1 using two different shRNAs resulted in a marked reduction in cell proliferation in six neuroblastoma cell lines. However, these effects were not rescued by overexpressing the shRNA-insensitive form of
ZRF1, thereby indicating that the depletion of
ZRF1 was not the causal factor for the reduction in cell proliferation. Consistent with this previous finding, siRNA-mediated depletion of
ZRF1 did not alter the proliferative, migratory, or invasive capacities or the sensitivity to RA in neuroblastoma cells, thus confirming that
ZRF1 is dispensable for the progression of this type of tumor. It is important to highlight that inconsistencies between different gene silencing methods could have misled the conclusion of this study. However, our use of independent silencing tools and rescue experiments with target protein ectopic expression leaves no room for doubt.
Imamura et al. demonstrated that while the effects of
ZRF1 on cell proliferation were p53-dependent, those related to migration and invasion were p53-independent [
15]. In our work, we covered this aspect by using cell lines with non-functional (CHLA-90, SK-N-BE(2), and SK-N-AS) and functional p53 (SH-SY5Y and IMR-32), and we did not find any differential response in the gain-of-function or loss-of-function experiments.
The paradoxical discrepancy between the strong correlation between
ZRF1 expression and poor patient outcome, and its dispensable function in tumor cells could be due to one or more of the following reasons: (i) one key characteristic of cancer is uncontrolled transcription. Thus, many genes are likely to be differentially expressed incidentally, rather than reflecting a gene that is driving a biologically significant outcome [
32]; (ii) the
ZRF1-PRC1 axis may regulate different sets of genes in a lineage-dependent manner; (iii) molecular alterations present in neuroblastoma (i.e., mutation burden, chromosomal copy number variations, etc.) deactivate the physiological regulation of
ZRF1-PRC1 on cell proliferation/differentiation programs and become
ZRF1 independent; and (iv) the ambivalent molecular function of
ZRF1 in neuroblastoma cells may be inclined to a non-essential chaperone role. This hypothesis is supported by our subcellular fractionation analysis in different neuroblastoma cell lines, where
ZRF1 was found to be predominantly enriched in the cytosolic fraction (
Figure S6).
In summary, our data suggest the potential use of ZRF1 expression as an independent prognostic factor, particularly in cases without any other associated molecular prognostic factors. However, ZRF1 does not seem to be a promising target candidate for differentiation therapy for neuroblastoma.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





