
Parental Origin of the RB1 Gene Mutations in Families with Low Penetrance Hereditary Retinoblastoma

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Mutation Screening by NGS
2.3. Sanger Sequencing
2.4. Multiplex Ligation-Dependent Probe Amplification
2.5. Statistical Analysis
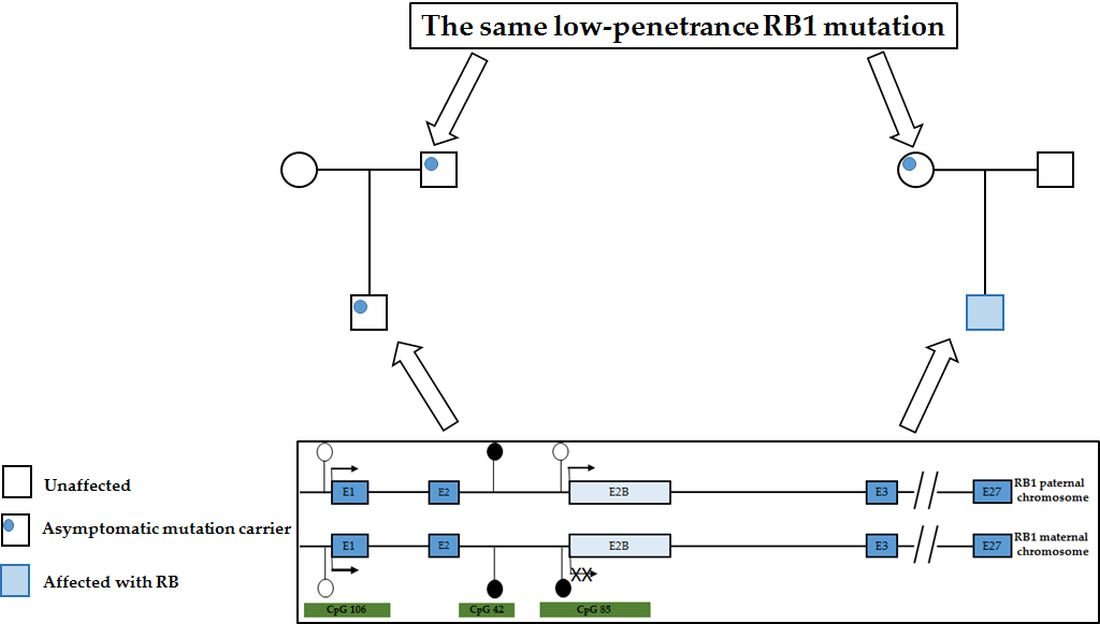
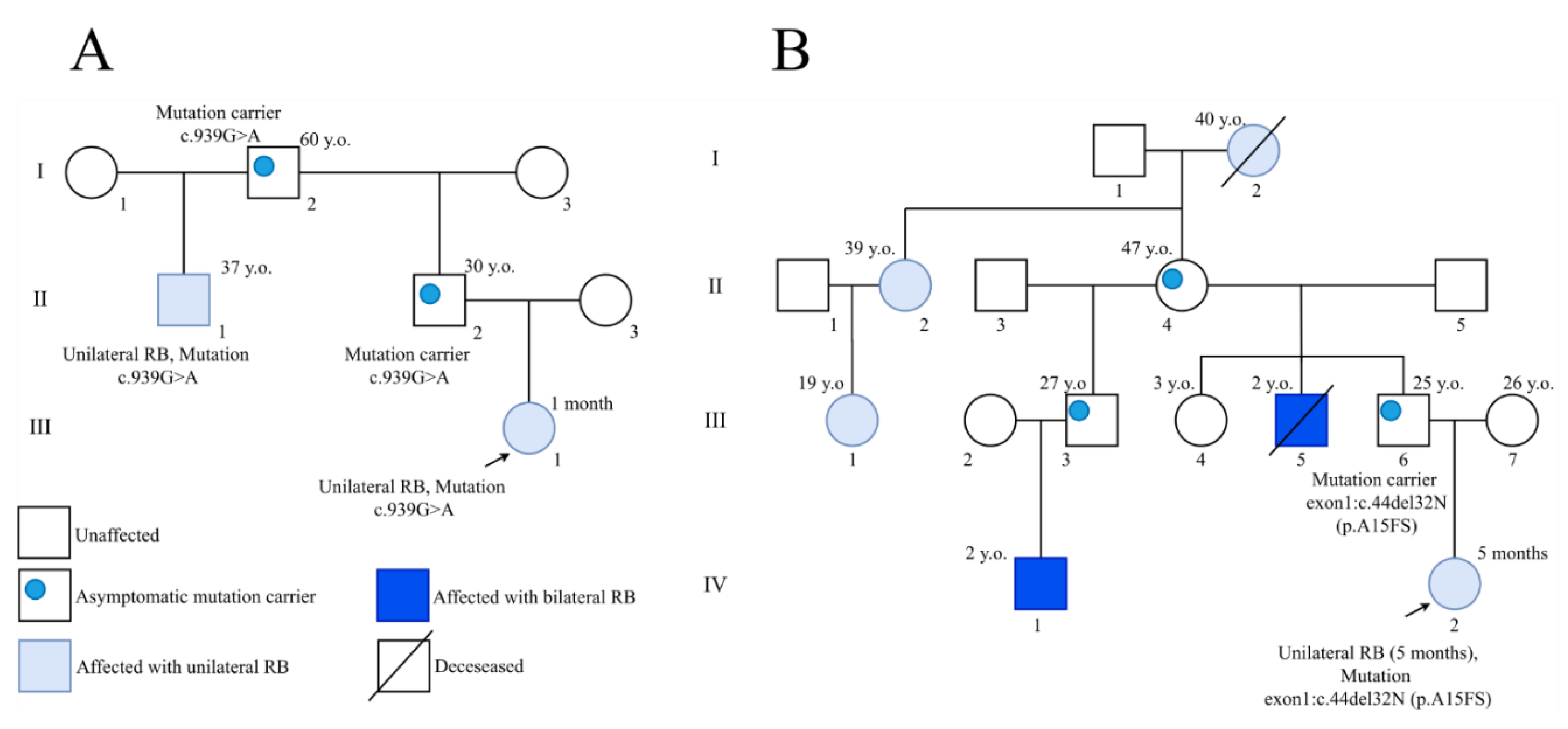
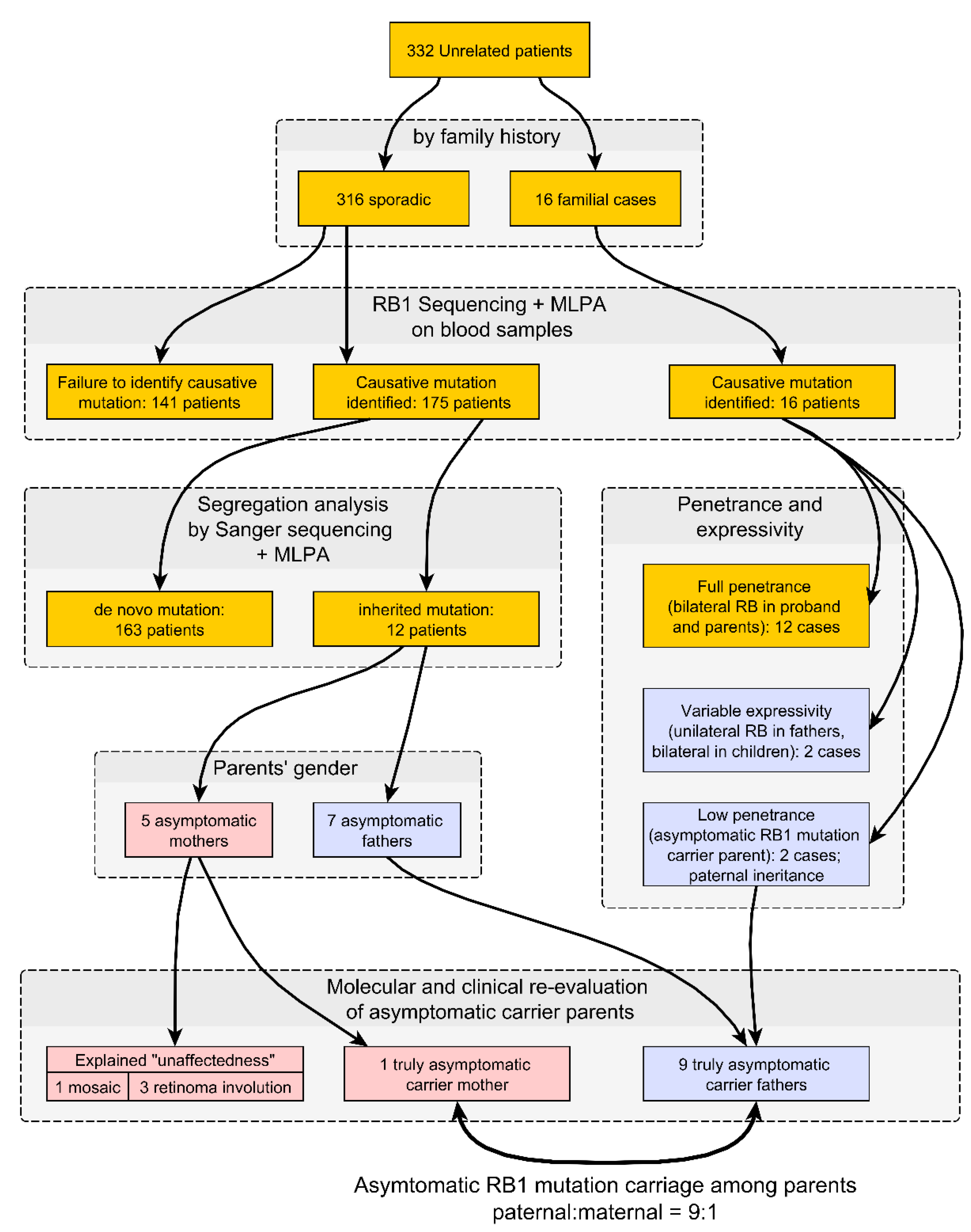
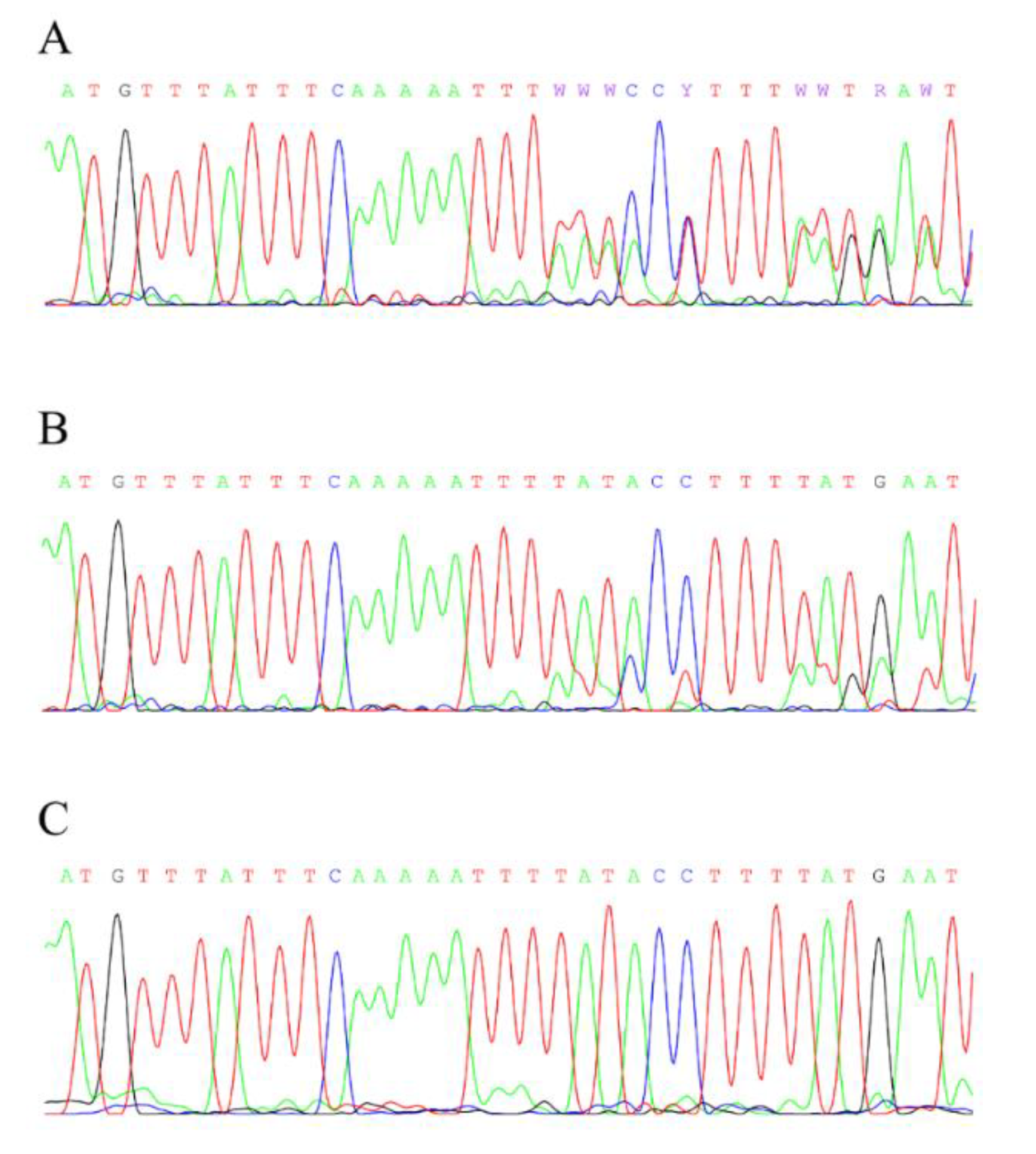
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, L.; Kashyap, S. Update on pathology of retinoblastoma. Int. J. Ophthalmol. 2018, 11, 2011–2016. [Google Scholar] [CrossRef] [PubMed]
- McEvoy, J.D.; Dyer, M.A. Genetic and Epigenetic Discoveries in Human Retinoblastoma. Crit. Rev. Oncog. 2015, 20, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Dimaras, H.; Corson, T.W.; Cobrinik, D.; White, A.; Zhao, J.; Munier, F.L.; Abramson, D.H.; Shields, C.L.; Chantada, G.L.; Njuguna, F.; et al. Retinoblastoma. Nat. Rev. Dis. Primers 2015, 1, 15021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R.; Honavar, S.G. Retinoblastoma. Indian J. Rediatr. 2017, 84, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Khodadoust, A.A.; Roozitalab, H.M.; Smith, R.E.; Green, W.R. Spontaneus regression of retinoblastoma. Surv. Ophthalmol. 1977, 21, 467–478. [Google Scholar] [CrossRef]
- Wu, S.; Zou, X.; Sun, Z.; Zhu, T.; Wei, X.; Sui, R. Unilateral retinocytoma associated with a variant in the RB1 gene. Mol. Genet. Genom. Med. 2020, 8, e1156. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.D.; Santos, M.C.M.; Shields, C.L.; Shields, J.A.; Eagle, R.C. Observations on 17 Patients With Retinocytoma. Arch. Ophthalmol. 2000, 2, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Babenko, O.V.; Zemlyakova, V.V.; Saakyan, S.V.; Brovkina, A.F.; Strelnikov, V.V.; Zaletaev, D.V.; Nemtsova, M.V. RB1 and CDKN2A functional defects resulting in retinoblastoma. Mol. Biol. 2002, 36, 625–630. [Google Scholar] [CrossRef]
- Babenko, O.V.; Saakyan, S.V.; Brovkina, A.F.; Kozlova, V.M.; Strelnikov, V.V.; Zaletaev, D.V.; Nemtsova, M.V. Spectrum and frequencies of RB1 structural defects in retinoblastoma. Mol. Biol. 2002, 36, 487–492. [Google Scholar] [CrossRef]
- Darwich, R.; Ghazawi, F.M.; Rahme, E.; Alghazawi, N.; Burnier, J.V.; Sasseville, D.; Burnier, M.N.; Litvinov, I.V. Retinoblastoma incidence trends in Canada: A national comprehensive population-based study. J. Pediatr. Ophthalmol. Strabismus 2019, 56, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.L.; Lally, S.E. Retinoblastoma. In Ocular Oncology; Springer: Singapore, 2019; pp. 91–99. [Google Scholar] [CrossRef]
- Eloy, P.; Dehainault, C.; Sefta, M.; Aerts, I.; Doz, F.; Cassoux, N.; le Rouic, L.L.; Stoppa-Lyonnet, D.; Radvanyi, F.; Millot, G.A.; et al. A Parent-of-Origin Effect Impacts the Phenotype in Low Penetrance Retinoblastoma Families Segregating the c.1981C>T/p.Arg661Ttp Mutation of RB1. PLoS Genet. 2016, 12, e1005888. [Google Scholar] [CrossRef]
- Harbour, J.W. Molecular Basis of Low-Penetrance Retinoblastoma. Arch. Ophthalmol. 2001, 119, 1699–1704. [Google Scholar] [CrossRef] [Green Version]
- Kanber, D.; Berulava, T.; Ammerpohl, O.; Mitter, D.; Richter, J.; Siebert, R.; Horsthemke, B.; Lohmann, D.; Buiting, K. The Human Retinoblastoma Gene Is Imprinted. PLoS Genet. 2009, 5, e1000790. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variations from next-generation sequencing data. Nuclear Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Biesecker, L.G.; Spinner, N.B. A genomic view of mosaicism and human disease. Nat. Rev. Genet. 2013, 14, 307–320. [Google Scholar] [CrossRef]
- Dimaras, H.; Khetan, V.; Halliday, W.; Orlic, M.; Prigoda, N.L.; Piovesan, B.; Marrano, P.; Corson, T.W.; Eagle, R.C., Jr.; Squire, J.A.; et al. Loss of RB1 induces non-proliferative retinoma: Increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet. 2008, 17, 1363–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theriault, B.; Dimaras, H.; Gallie, B.L.; Corson, T.W. The genomic landscape of retinoblastoma: A review. Clin. Exp. Ophthalmol. 2014, 42, 33–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dommering, C.J.; Mol, B.M.; Moll, A.C.; Burton, M.; Cloos, J.; Dorsman, J.C.; Meijers-Heijboer, H.; van der Hout, A.H. RB1 mutation spectrum in a comprehensive nationwide cohort of retinoblastoma patients. J. Med. Genet. 2014, 51, 366–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomar, S.; Sethi, R.; Sundar, G.; Quah, T.C.; Quah, B.L.; Lai, P.S. Mutation spectrum of RB1 mutations in retinoblastoma cases from Singapore with implications for genetic management and counselling. PLoS ONE 2017, 12, e0178776. [Google Scholar] [CrossRef] [Green Version]
- Tomar, A.S.; Finger, P.T.; Gallie, B.; Mallipatna, A.; Kivelä, T.T.; Zhang, C.; Zhao, J.; Wilson, M.W.; Brenna, R.C.; Burges, M. A Multicenter, International Collaborative Study for AJCC-Staging of Retinoblastoma: Treatment Success and Globe Salvage. Ophthalmology 2020. [Google Scholar] [CrossRef]
- Ushakova, T.L.; Trofimov, I.A.; Gorovtsova, O.V.; Yarovoy, A.A.; Saakyan, S.V.; Letyagin, I.A.; Matinyan, N.V.; Kukushkin, A.V.; Martynov, L.A.; Pogrebnyakov, I.V. New Era of Organ-Preserving Treatment in Pediatric Intraocular Retinoblastoma in Russia: A Multicentre Study. Onkopediatria 2018, 5, 51–69. [Google Scholar] [CrossRef]
- Buiting, K.; Kanber, D.; Horsthemke, B.; Lohmann, D. Imprinting of RB1 (the new kid on the block). Brief Funct. Genom. 2010, 9, 347–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, D.N.; Krawczak, M.; Polychronakos, C.; Tyler-Smith, C.; Kehrer-Sawatzki, H. Where genotype is not predictive of phenotype: Towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum. Genet. 2013, 132, 1077–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, K.E.; Parker, R. Nonsense-mediated mRNA decay: Terminating erroneous gene expression. Curt. Opin. Cell Biol. 2004, 16, 292–299. [Google Scholar] [CrossRef]
- Sánchez-Sánchez, F.; Ramírez-Castillejo, C.; Weekes, D.B.; Beneyto, M.; Prieto, F.; Nájera, C.; Mittnacht, S. Attenuation of disease phenotype through alternative translation initiation in low-penetrance retinoblastoma. Human Mutat. 2007, 28, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Klutz, M.; Brockmann, D.; Lohmann, D.R. A parent-of-origin effect in two families with retinoblastoma is associated with a distinct splice mutatin in the RB1 gene. Am. J. Hum. Genet. 2002, 7, 174–179. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.; Dehainault, C.; Desjardins, L.; Doz, F.; Levy, C.; Sastre, X.; Couturier, J.; Stoppa-Lyonnet, D.; Gauthier, C.H.; Villars, M. Genotype-phenotype correlations in hereditary familial retinoblastoma. Hum. Mutat. 2007, 28, 284–293. [Google Scholar] [CrossRef]
- Zhang, K.; Nowak, I.; Rushlow, D.; Gallie, B.L.; Lohmann, D.R. Patterns of missplicing caused by RB1 gene mutations in patients with retinoblastoma and association with phenotypic expression. Human Mutat. 2008, 29, 475–484. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
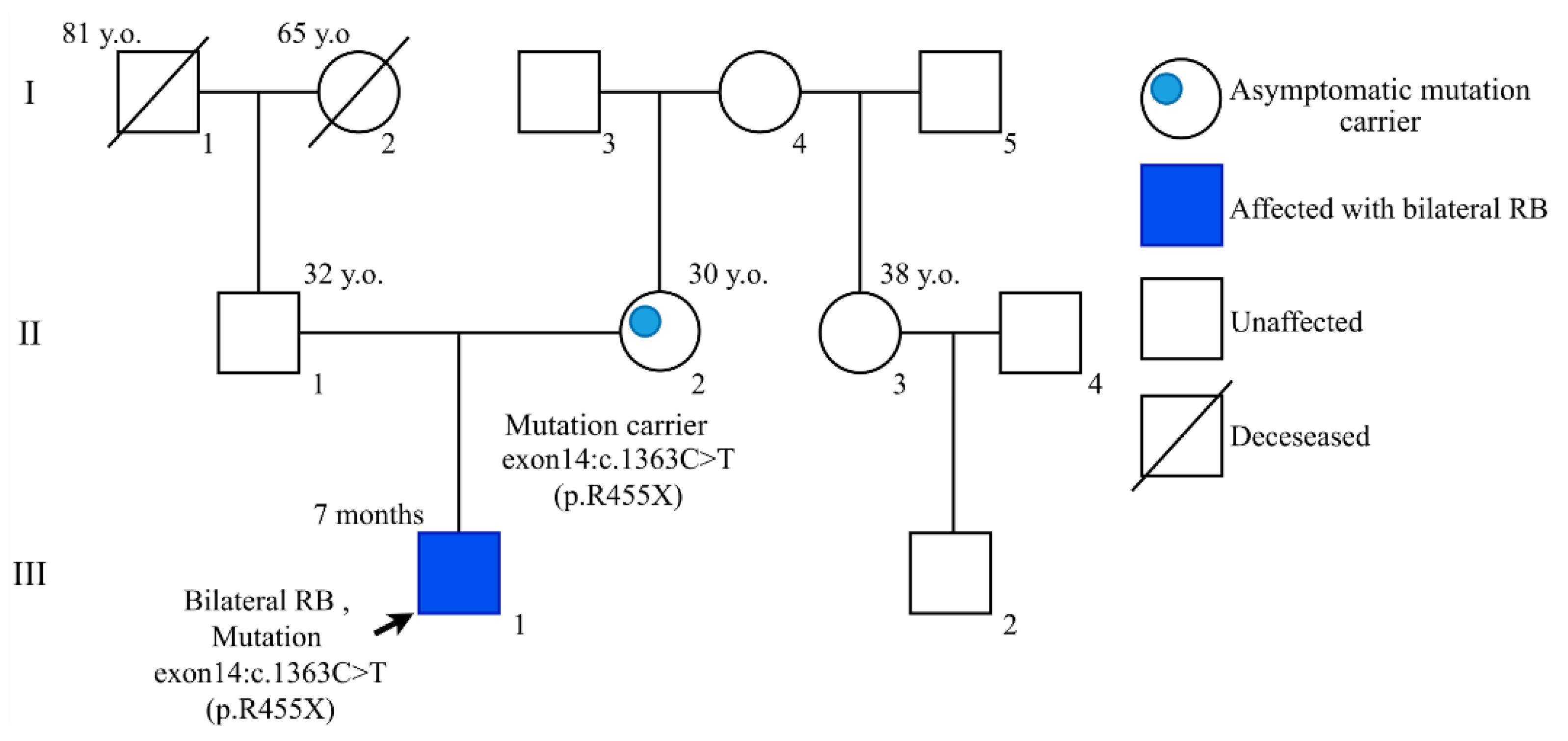{kind=link}
{kind=link}
| Family # | Mutation | Mutation Type | Location | RB Clinical form in the Proband | Other Mutation Carriers in the Family/Retinoblastoma Form |
|---|---|---|---|---|---|
| 164 | c.32_63del | Frameshift | Exon 1 | Bilateral | Mother/bilateral |
| 261 | c.939G > A | Splice site (missense splice) | Exon 9 | Unilateral |
|
| 286 | c.380+1G > A | Splice site | Exon 3 | Bilateral | Father/unilateral |
| 306 | c.1072C > T | Nonsense | Exon 11 | Bilateral | Father/bilateral |
| 319 | c.45_76del | Frameshift | Exon 1 | Unilateral |
|
| 323 | c.958C > T | Nonsense | Exon 10 | Bilateral | Father/bilateral |
| 327 | c.958C > T | Nonsense | Exon 10 | Bilateral | Mother/bilateral |
| 347 | c.54_76del | Frameshift | Exon 1 | Bilateral | Father/bilateral |
| 360 | c.1696-2A > G | Splice site | Intron 17 | Bilateral |
|
| 372 | c.1735C > T | Nonsense | Exon 18 | Bilateral | Mother/bilateral |
| 388 | NC_000013.11:g. (43412928_48258929)_ (48381391_48453040)del | Gross deletion | Exons 1–27 | Bilateral | Mother/bilateral |
| 394 | c.1654C > T | Nonsense | Exon 17 | Bilateral | Father/bilateral |
| 398 | c.1724del | Frameshift | Exon 18 | Bilateral |
|
| 409 | c.608-12T > G | Splice site | Intron 6 | Bilateral | Mother/bilateral |
| 538 | NC_000013.11:g. (48463554_48465087)_ (48465224_48473094)del | Intragenic deletion | Exons 22–23 | Bilateral | Mother/bilateral |
| 539 | c.1233_1254 ins TAAAGAACTGC ACAGTGAATCC | Frameshift | Exon 13 | Bilateral | Father/bilateral |
| Family # | Mutation | Mutation Type | Location | Retinoblastoma Form in the Proband | Parent—Asymptomatic Mutation Carrier |
|---|---|---|---|---|---|
| Families with Purely Asymptomatic Mutation Carrier Parents | |||||
| 319 | c.45_76del | Frameshift | Exon 1 | Unilateral | Father |
| 485 | c.83del | Frameshift | Exon 1 | Unilateral | Father |
| 393 | c.607+1G > T | Splice site | Intron 6 | Unilateral | Father |
| 533 | c.607+1G > T | Splice site | Intron 6 | Unilateral | Father |
| 261 | c.939G > A | Splice site (missense splice) | Exon 9 | Unilateral | Father |
| 255 | c. 1364G > C | Missense | Exon 14 | Unilateral | Father |
| 566 | c.1573G > A | Missense | Exon 17 | Unilateral | Father |
| 437 | c.1695+5G > T | Splice site | Intron 17 | Bilateral | Father |
| 424 | c.1981C > T | Missense | Exon 20 | Unilateral | Father |
| 522 | c.861G > C | Splice site (missense splice) | Exon 8 | Bilateral | Mother |
| Families with asymptomatic mutation carriage in parents explained by either incomplete penetrance or mosaic mutation carriage | |||||
| 482 | c.887del | Frameshift | Exon 9 | Bilateral | Mother (mosaic) |
| 472 | c.1345G > T | Nonsense | Exon 14 | Bilateral | Mother (retinoma involution) |
| 359 | c.1363C > T | Nonsense | Exon 14 | Bilateral | Mother (retinoma involution) |
| 594 | c.2293_2297 del | Frameshift | Exon 22 | Bilateral | Mother (retinoma involution) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alekseeva, E.A.; Babenko, O.V.; Kozlova, V.M.; Ushakova, T.L.; Kazubskaya, T.P.; Nemtsova, M.V.; Chesnokova, G.G.; Mikhaylenko, D.S.; Bure, I.V.; Kalinkin, A.I.; et al. Parental Origin of the RB1 Gene Mutations in Families with Low Penetrance Hereditary Retinoblastoma. Cancers 2021, 13, 5068. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205068
Alekseeva EA, Babenko OV, Kozlova VM, Ushakova TL, Kazubskaya TP, Nemtsova MV, Chesnokova GG, Mikhaylenko DS, Bure IV, Kalinkin AI, et al. Parental Origin of the RB1 Gene Mutations in Families with Low Penetrance Hereditary Retinoblastoma. Cancers. 2021; 13(20):5068. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205068
Chicago/Turabian StyleAlekseeva, Ekaterina A., Olga V. Babenko, Valentina M. Kozlova, Tatiana L. Ushakova, Tatiana P. Kazubskaya, Marina V. Nemtsova, Galina G. Chesnokova, Dmitry S. Mikhaylenko, Irina V. Bure, Alexey I. Kalinkin, and et al. 2021. "Parental Origin of the RB1 Gene Mutations in Families with Low Penetrance Hereditary Retinoblastoma" Cancers 13, no. 20: 5068. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205068