
Brain and Breast Cancer Cells with PTEN Loss of Function Reveal Enhanced Durotaxis and RHOB Dependent Amoeboid Migration Utilizing 3D Scaffolds and Aligned Microfiber Tracts

 , ,
, ,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Melt Electrowriting Printing and Glass Slide Treatment
2.3. Melt Electrowriting Scaffold Functionalization
2.4. U87 Cells Stably Express the Farnesylated-tdTomato Fluorescent Protein
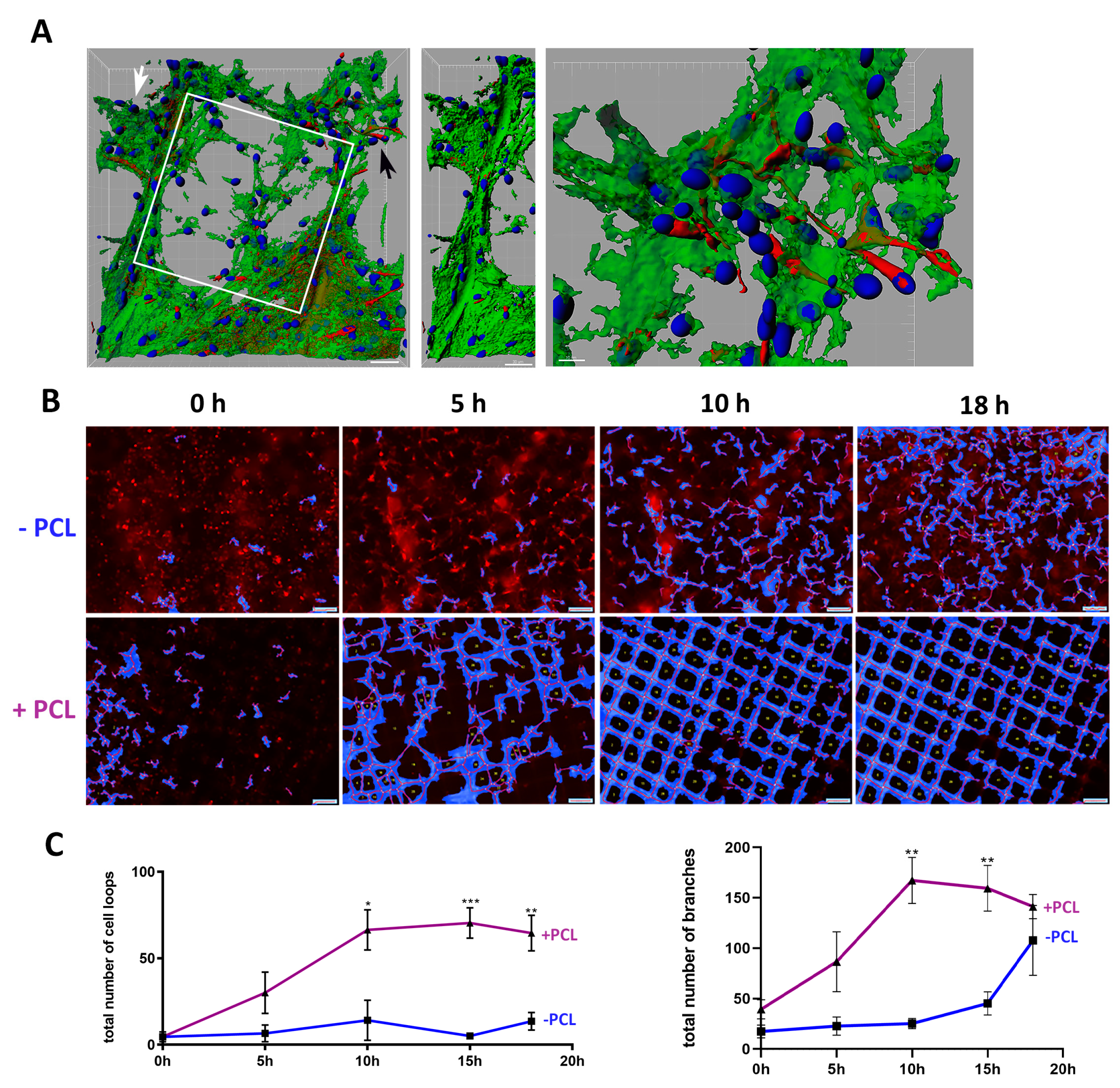
2.5. Cell Network Loop and Branch Formation Quantification
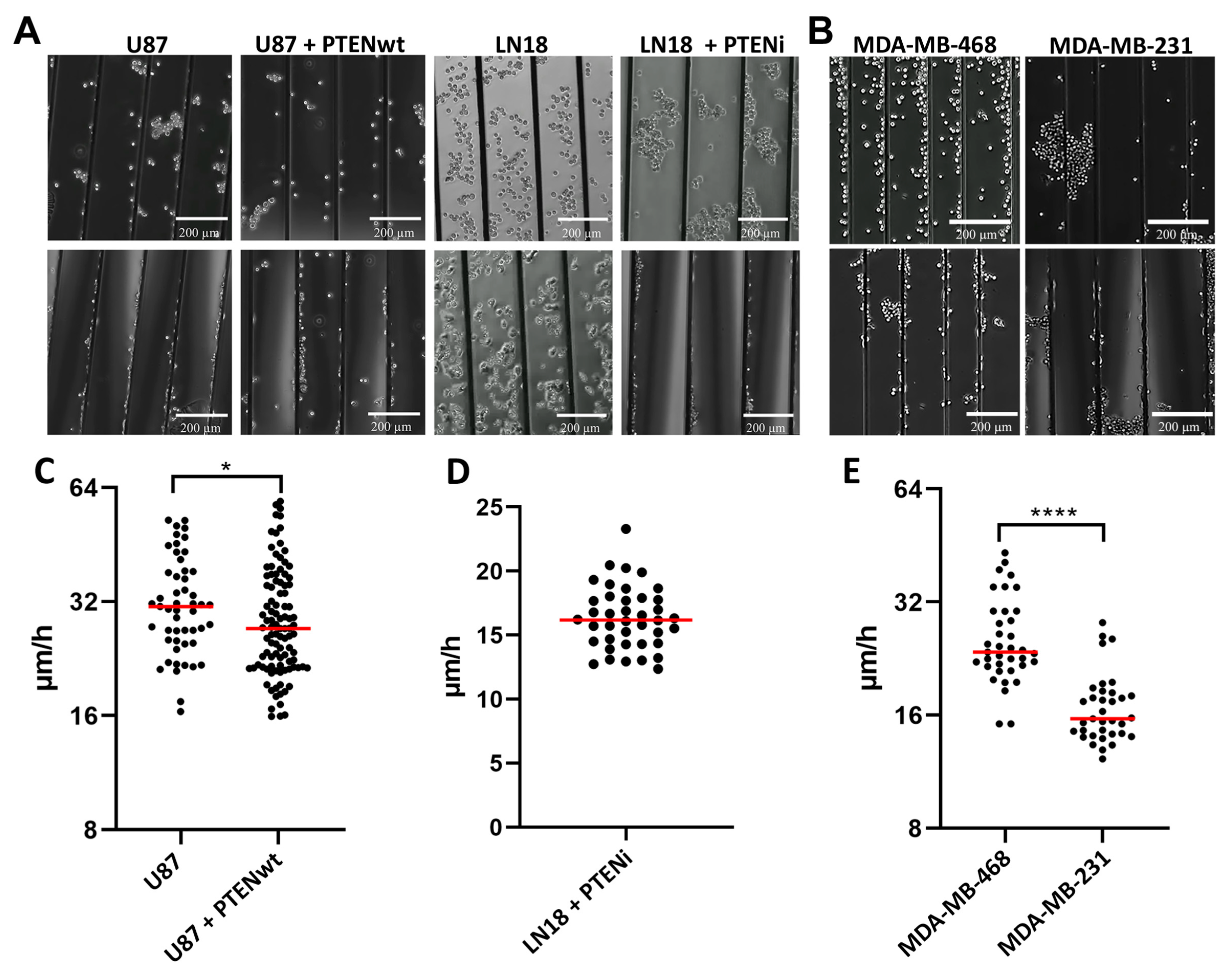
2.6. Cell Migration Assessment Following Treatment with PTEN, RHO and ROCK Inhibitors or Rescuing PTEN Function with Live Cell Imaging
2.7. Cell Viability
2.8. Immunocytochemistry and F-Actin Staining
2.9. Confocal and Scanning Electron Microscopy
2.10. RNA Extraction, qPCR and RNA-Sequencing
2.11. Western Blot
2.12. Statistical Analysis and Data Presentation
3. Results
3.1. Design and Fabrication of 3D Scaffolds and Aligned Microfibers for Cell Functional Studies
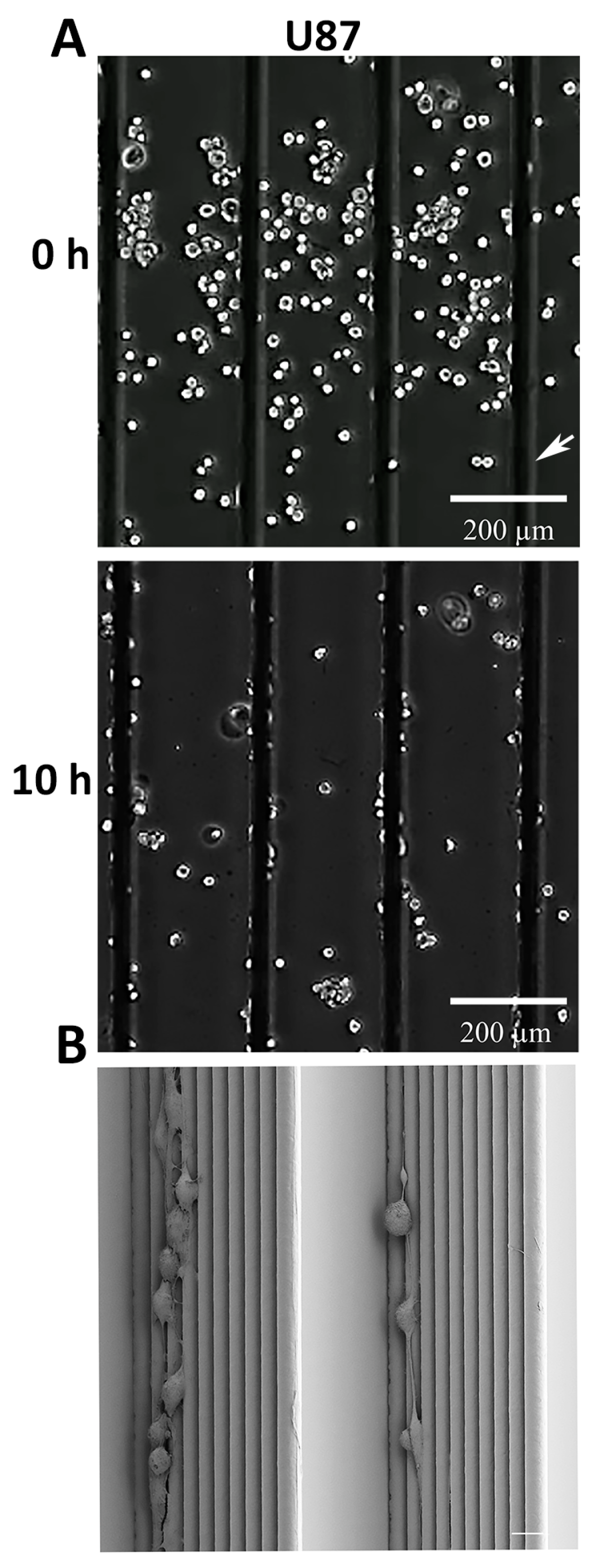
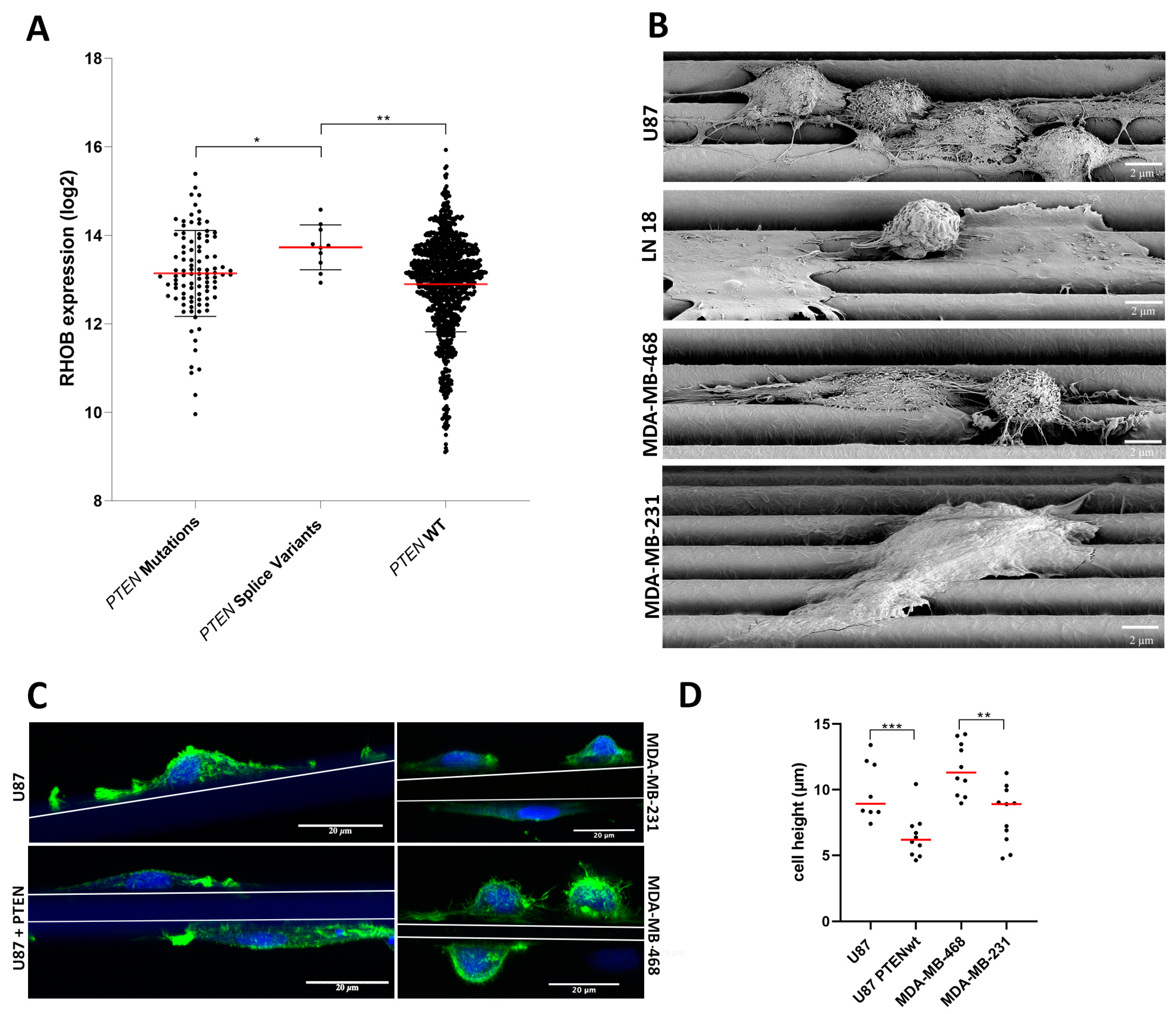
3.2. U87 Cells Show an Enhanced Scaffold Directed Durotaxis, Adhesion and Network Formation
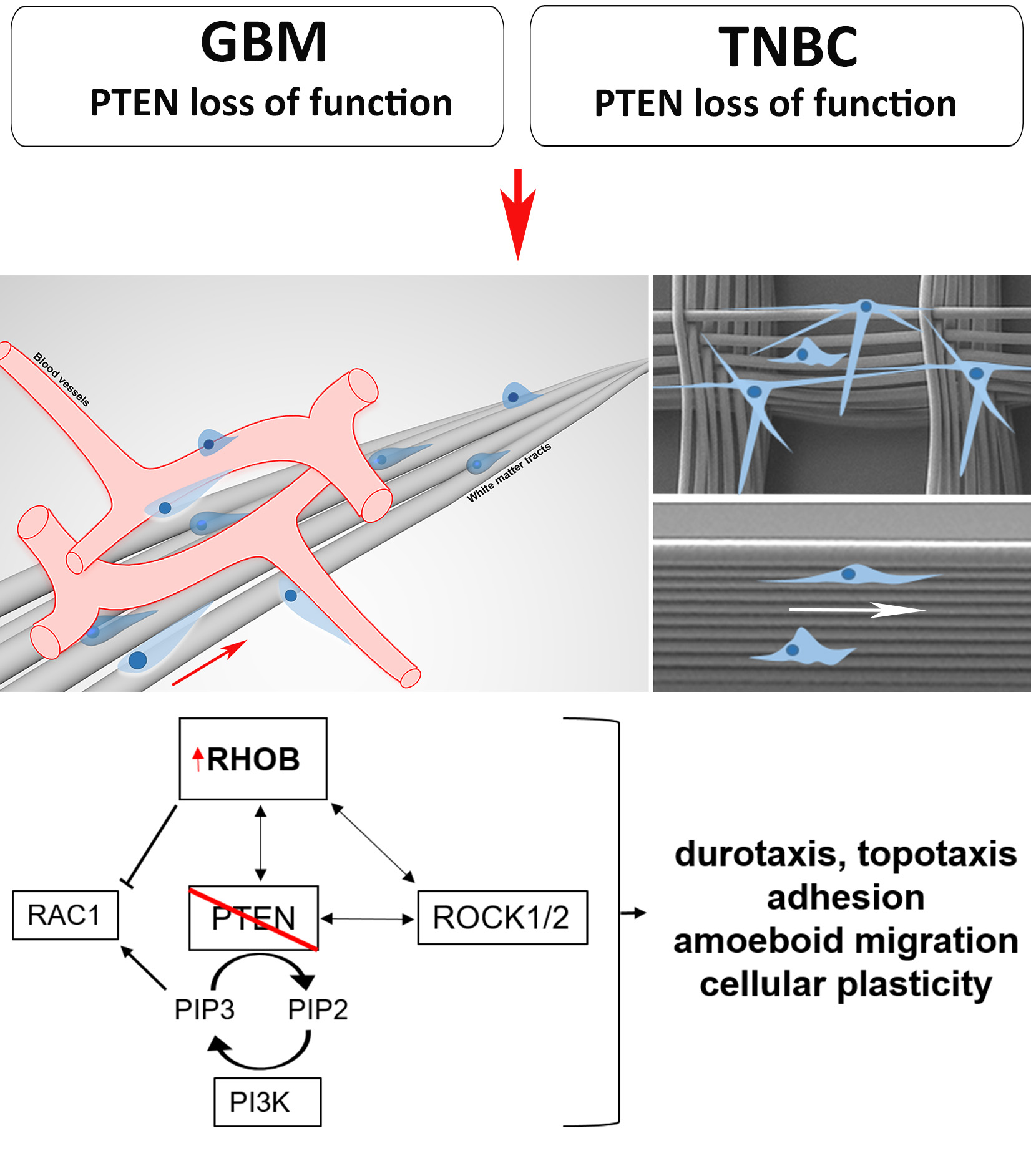
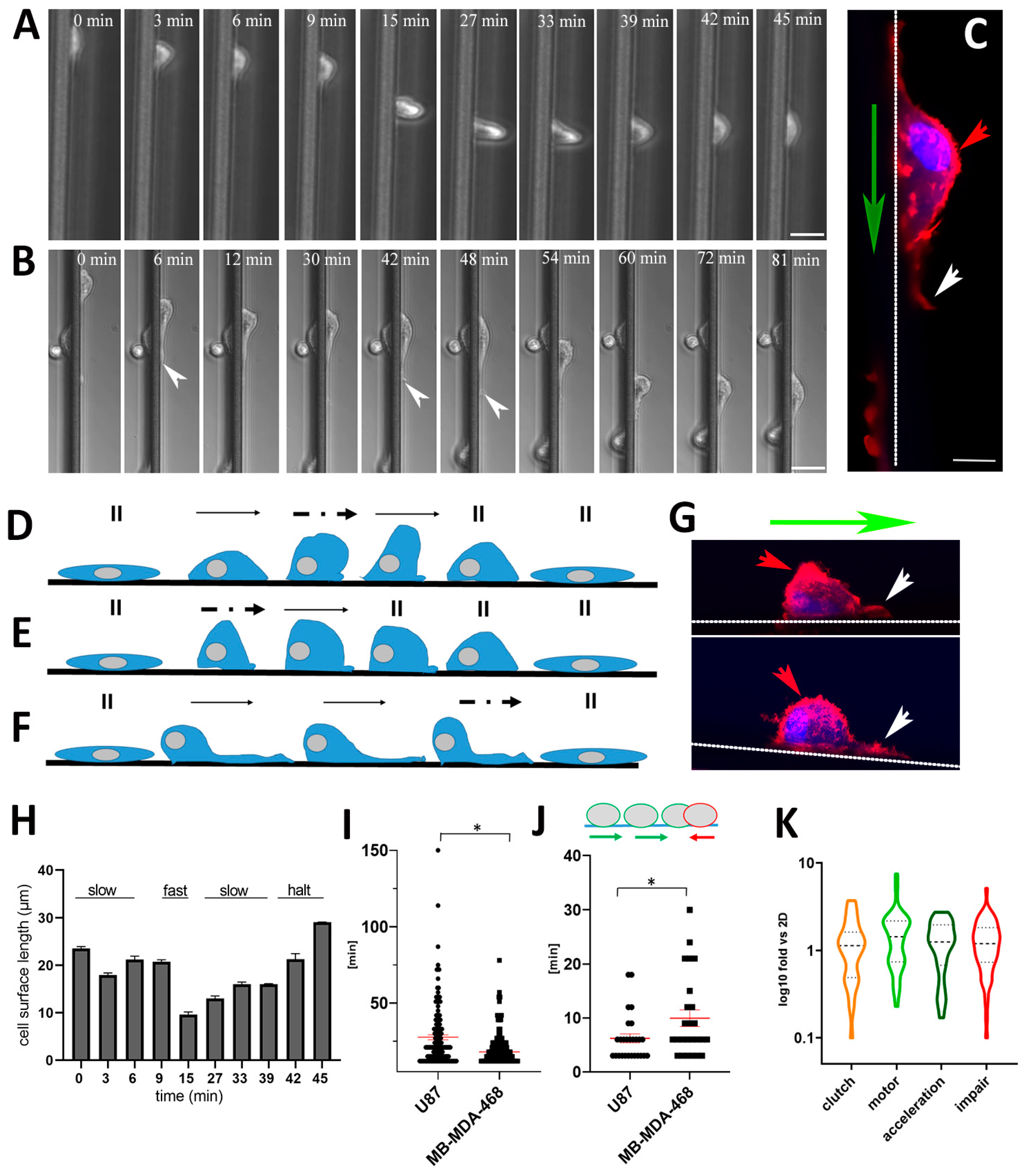
3.3. GBM and Breast Cancer Cell Migratory Behavior with 3D-Aligned Microfibers
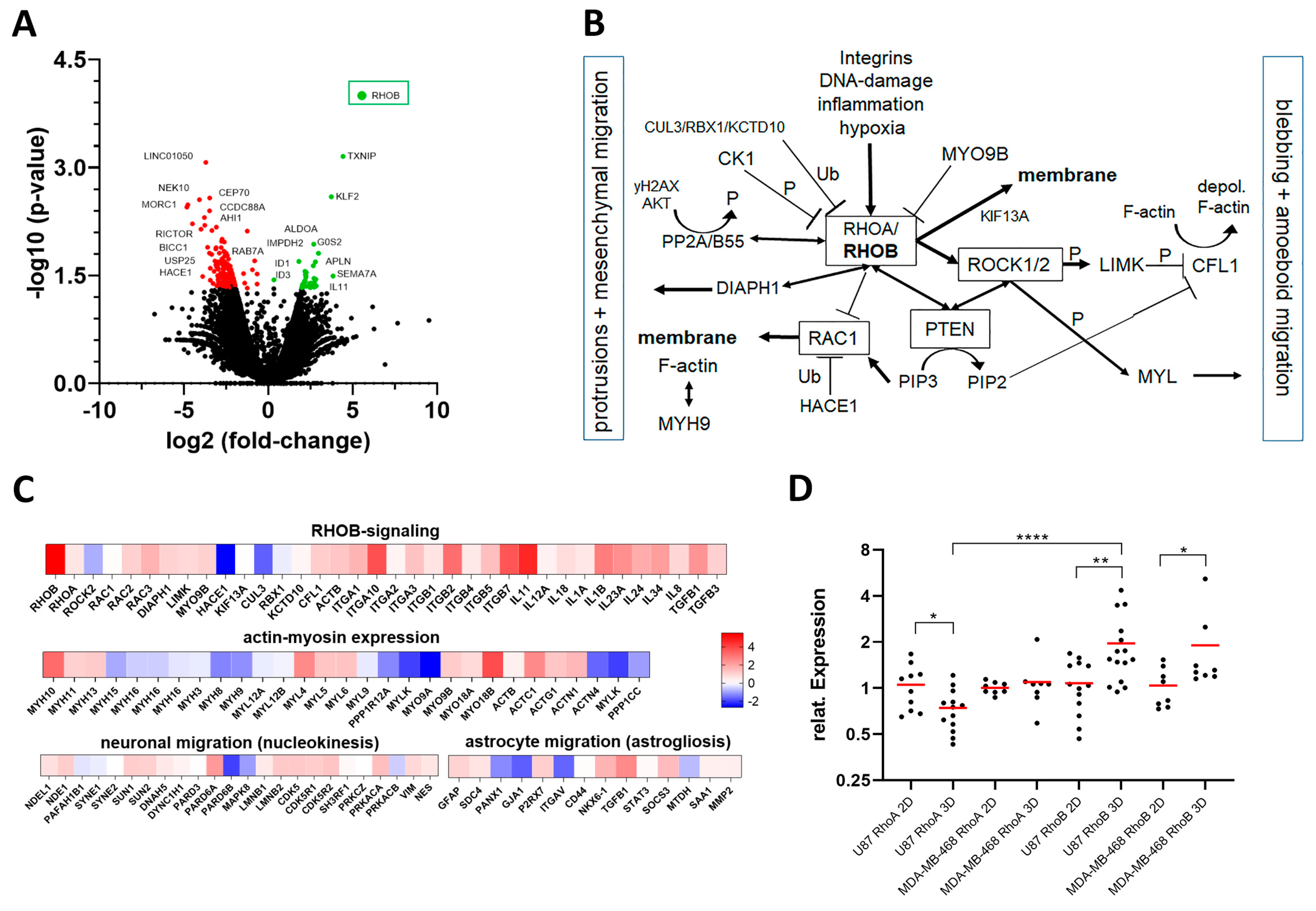
3.4. RHOB as a Novel Regulator of Migration
3.5. RHOB Signaling as a Regulator of Cell Morphology
3.6. Skating Snail-Like Cell Migration and the Motor Clutch Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedl, P.; Sahai, E.; Weiss, S.; Yamada, K.M. New dimensions in cell migration. Nat. Rev. Mol. Cell Biol. 2012, 13, 743–747. [Google Scholar] [CrossRef] [PubMed]
- Papusheva, E.; Heisenberg, C.P. Spatial organization of adhesion: Force-dependent regulation and function in tissue morphogenesis. EMBO J. 2010, 29, 2753–2768. [Google Scholar] [CrossRef] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Pedron, S.; Becka, E.; Harley, B.A. Regulation of glioma cell phenotype in 3D matrices by hyaluronic acid. Biomaterials 2013, 34, 7408–7417. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pointer, K.B.; Clark, P.A.; Schroeder, A.B.; Salamat, M.S.; Eliceiri, K.W.; Kuo, J.S. Association of collagen architecture with glioblastoma patient survival. J. Neurosurg. 2017, 126, 1812–1821. [Google Scholar] [CrossRef] [Green Version]
- Yousif, L.F.; Di Russo, J.; Sorokin, L. Laminin isoforms in endothelial and perivascular basement membranes. Cell Adhes. Migr. 2013, 7, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Mair, D.B.; Ames, H.M.; Li, R. Mechanisms of invasion and motility of high-grade gliomas in the brain. Mol. Biol. Cell 2018, 29, 2509–2515. [Google Scholar] [CrossRef]
- Vega, F.M.; Fruhwirth, G.; Ng, T.; Ridley, A.J. RhoA and RhoC have distinct roles in migration and invasion by acting through different targets. J. Cell Biol. 2011, 193, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Rossi, J.; Giaccherini, L.; Cavallieri, F.; Napoli, M.; Moratti, C.; Froio, E.; Serra, S.; Fraternali, A.; Ghadirpour, R.; Cozzi, S.; et al. Extracranial metastases in secondary glioblastoma multiforme: A case report. BMC Neurol. 2020, 20, 382. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Kanayama, T.; Noguchi, K.; Niwa, A.; Miyai, M.; Kawaguchi, M.; Ishida, K.; Hatano, Y.; Niwa, M.; Tomita, H. Treatment Strategies Based on Histological Targets against Invasive and Resistant Glioblastoma. J. Oncol. 2019, 2019, 2964783. [Google Scholar] [CrossRef] [PubMed]
- Thakkar, J.P.; Dolecek, T.A.; Horbinski, C.; Ostrom, Q.T.; Lightner, D.D.; Barnholtz-Sloan, J.S.; Villano, J.L. Epidemiologic and molecular prognostic review of glioblastoma. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1985–1996. [Google Scholar] [CrossRef] [Green Version]
- Scherer, H.J. Structural development in gliomas. Am. J. Cancer 1938, 34, 333–351. [Google Scholar]
- Farin, A.; Suzuki, S.O.; Weiker, M.; Goldman, J.E.; Bruce, J.N.; Canoll, P. Transplanted glioma cells migrate and proliferate on host brain vasculature: A dynamic analysis. Glia 2006, 53, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, S.V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force fluctuations within focal adhesions mediate ECM-rigidity sensing to guide directed cell migration. Cell 2012, 151, 1513–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- SenGupta, S.; Parent, C.A.; Bear, J.E. The principles of directed cell migration. Nat. Rev. Mol. Cell Biol. 2021, 22, 529–547. [Google Scholar] [CrossRef]
- Caballero, D.; Comelles, J.; Piel, M.; Voituriez, R.; Riveline, D. Ratchetaxis: Long-Range Directed Cell Migration by Local Cues. Trends Cell Biol. 2015, 25, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, D.H.; Kim, H.N.; Wang, C.J.; Kwak, M.K.; Hur, E.; Suh, K.Y.; An, S.S.; Levchenko, A. Directed migration of cancer cells guided by the graded texture of the underlying matrix. Nat. Mater. 2016, 15, 792–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.; Ma, X.; Du, Y.; Feng, J. Understanding Patterns of Brain Metastasis in Triple-Negative Breast Cancer and Exploring Potential Thhuaerapeutic Targets. OncoTargets Ther. 2021, 14, 589–607. [Google Scholar] [CrossRef]
- Brosnan, E.M.; Anders, C.K. Understanding patterns of brain metastasis in breast cancer and designing rational therapeutic strategies. Ann. Transl. Med. 2018, 6, 163. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R.; Yang, X.; Downes, C.P.; Weijer, C.J. PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr. Biol. 2007, 17, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerisch, G.; Schroth-Diez, B.; Muller-Taubenberger, A.; Ecke, M. PIP3 waves and PTEN dynamics in the emergence of cell polarity. Biophys. J. 2012, 103, 1170–1178. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.L.; Burkhard, C.; Schuler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, K.; Kumon, Y.; Harada, H.; Kohno, S.; Nagato, S.; Teraoka, M.; Fujiwara, S.; Nakagawa, K.; Hamada, K.; Ohnishi, T. PTEN gene transfer suppresses the invasive potential of human malignant gliomas by regulating cell invasion-related molecules. Int. J. Oncol. 2006, 29, 73–81. [Google Scholar] [CrossRef]
- Witzel, I.; Oliveira-Ferrer, L.; Pantel, K.; Muller, V.; Wikman, H. Breast cancer brain metastases: Biology and new clinical perspectives. Breast Cancer Res. 2016, 18, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csolle, M.P.; Ooms, L.M.; Papa, A.; Mitchell, C.A. PTEN and Other PtdIns(3,4,5)P3 Lipid Phosphatases in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 9189. [Google Scholar] [CrossRef]
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24. [Google Scholar] [CrossRef] [Green Version]
- Hohensee, I.; Chuang, H.N.; Grottke, A.; Werner, S.; Schulte, A.; Horn, S.; Lamszus, K.; Bartkowiak, K.; Witzel, I.; Westphal, M.; et al. PTEN mediates the cross talk between breast and glial cells in brain metastases leading to rapid disease progression. Oncotarget 2017, 8, 6155–6168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikman, H.; Lamszus, K.; Detels, N.; Uslar, L.; Wrage, M.; Benner, C.; Hohensee, I.; Ylstra, B.; Eylmann, K.; Zapatka, M.; et al. Relevance of PTEN loss in brain metastasis formation in breast cancer patients. Breast Cancer Res. 2012, 14, R49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamo, B.; Deal, A.M.; Burrows, E.; Geradts, J.; Hamilton, E.; Blackwell, K.L.; Livasy, C.; Fritchie, K.; Prat, A.; Harrell, J.C.; et al. Phosphatidylinositol 3-kinase pathway activation in breast cancer brain metastases. Breast Cancer Res. 2011, 13, R125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, C.D.; Ridley, A.J. Rho GTPase signaling complexes in cell migration and invasion. J. Cell Biol. 2018, 217, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Dong, X.; Wang, Z.; Liu, W.; Deng, N.; Ding, Y.; Tang, L.; Hla, T.; Zeng, R.; Li, L.; et al. Regulation of PTEN by Rho small GTPases. Nat. Cell Biol. 2005, 7, 399–404. [Google Scholar] [CrossRef]
- Gong, X.; Didan, Y.; Lock, J.G.; Stromblad, S. KIF13A-regulated RhoB plasma membrane localization governs membrane blebbing and blebby amoeboid cell migration. EMBO J. 2018, 37, e98994. [Google Scholar] [CrossRef] [PubMed]
- Loessner, D.; Rockstroh, A.; Shokoohmand, A.; Holzapfel, B.M.; Wagner, F.; Baldwin, J.; Boxberg, M.; Schmalfeldt, B.; Lengyel, E.; Clements, J.A.; et al. A 3D tumor microenvironment regulates cell proliferation, peritoneal growth and expression patterns. Biomaterials 2019, 190-191, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Hutmacher, D.W.; Loessner, D.; Rizzi, S.; Kaplan, D.L.; Mooney, D.J.; Clements, J.A. Can tissue engineering concepts advance tumor biology research? Trends Biotechnol. 2010, 28, 125–133. [Google Scholar] [CrossRef]
- Kim, J.; Bakirci, E.; O’Neill, K.L.; Hrynevich, A.; Dalton, P.D. Fiber Bridging during Melt Electrowriting of Poly(epsilon-Caprolactone) and the Influence of Fiber Diameter and Wall Height. Macromol. Mater. Eng. 2021, 306. [Google Scholar] [CrossRef]
- Pham, Q.P.; Sharma, U.; Mikos, A.G. Electrospinning of polymeric nanofibers for tissue engineering applications: A review. Tissue Eng. 2006, 12, 1197–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, T.M.; Hutmacher, D.W.; Dalton, P.D. The Next Frontier in Melt Electrospinning: Taming the Jet. Adv. Funct. Mater. 2019, 29, 1904664. [Google Scholar] [CrossRef] [Green Version]
- Visser, J.; Melchels, F.P.; Jeon, J.E.; van Bussel, E.M.; Kimpton, L.S.; Byrne, H.M.; Dhert, W.J.; Dalton, P.D.; Hutmacher, D.W.; Malda, J. Reinforcement of hydrogels using three-dimensionally printed microfibres. Nat. Commun. 2015, 6, 6933. [Google Scholar] [CrossRef] [PubMed]
- Janzen, D.; Bakirci, E.; Wieland, A.; Martin, C.; Dalton, P.D.; Villmann, C. Cortical Neurons form a Functional Neuronal Network in a 3D Printed Reinforced Matrix. Adv. Healthc. Mater. 2020, 9, e1901630. [Google Scholar] [CrossRef] [PubMed]
- Weigand, A.; Boos, A.M.; Tasbihi, K.; Beier, J.P.; Dalton, P.D.; Schrauder, M.; Horch, R.E.; Beckmann, M.W.; Strissel, P.L.; Strick, R. Selective isolation and characterization of primary cells from normal breast and tumors reveal plasticity of adipose derived stem cells. Breast Cancer Res. 2016, 18, 32. [Google Scholar] [CrossRef] [Green Version]
- Furnari, F.B.; Lin, H.; Huang, H.S.; Cavenee, W.K. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc. Natl. Acad. Sci. USA 1997, 94, 12479–12484. [Google Scholar] [CrossRef] [Green Version]
- Hollestelle, A.; Elstrodt, F.; Nagel, J.H.; Kallemeijn, W.W.; Schutte, M. Phosphatidylinositol-3-OH kinase or RAS pathway mutations in human breast cancer cell lines. Mol. Cancer Res. 2007, 5, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosshaus, C.; Bakirci, E.; Berthel, M.; Hrynevich, A.; Kade, J.C.; Hochleitner, G.; Groll, J.; Dalton, P.D. Melt Electrospinning of Nanofibers from Medical-Grade Poly(epsilon-Caprolactone) with a Modified Nozzle. Small 2020, 16, e2003471. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakirci, E.; Schaefer, N.; Dahri, O.; Hrynevich, A.; Strissel, P.; Strick, R.; Dalton, P.D.; Villmann, C. Melt Electrowritten In Vitro Radial Device to Study Cell Growth and Migration. Adv. Biosyst. 2020, 4, e2000077. [Google Scholar] [CrossRef]
- Jain, A.; Betancur, M.; Patel, G.D.; Valmikinathan, C.M.; Mukhatyar, V.J.; Vakharia, A.; Pai, S.B.; Brahma, B.; MacDonald, T.J.; Bellamkonda, R.V. Guiding intracortical brain tumour cells to an extracortical cytotoxic hydrogel using aligned polymeric nanofibres. Nat. Mater. 2014, 13, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.M.; Wang, H.B.; Dembo, M.; Wang, Y.L. Cell movement is guided by the rigidity of the substrate. Biophys. J. 2000, 79, 144–152. [Google Scholar] [CrossRef] [Green Version]
- DuChez, B.J.; Doyle, A.D.; Dimitriadis, E.K.; Yamada, K.M. Durotaxis by Human Cancer Cells. Biophys. J. 2019, 116, 670–683. [Google Scholar] [CrossRef] [Green Version]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; de Toledo, M.; Anguille, C.; Roux, P. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J. Cell Biol. 2007, 178, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.M.; Sixt, M. Mechanisms of 3D cell migration. Nat. Rev. Mol. Cell Biol. 2019, 20, 738–752. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, T.; Fujita, Y.; Kishima, H.; Yamashita, T. Methylation dependent down-regulation of G0S2 leads to suppression of invasion and improved prognosis of IDH1-mutant glioma. PLoS ONE 2018, 13, e0206552. [Google Scholar] [CrossRef] [PubMed]
- Podgorska, M.; Pietraszek-Gremplewicz, K.; Nowak, D. Apelin Effects Migration and Invasion Abilities of Colon Cancer Cells. Cells 2018, 7, 113. [Google Scholar] [CrossRef] [Green Version]
- Tochio, T.; Tanaka, H.; Nakata, S.; Hosoya, H. Fructose-1,6-bisphosphate aldolase A is involved in HaCaT cell migration by inducing lamellipodia formation. J. Dermatol. Sci. 2010, 58, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Messina, A.; Ferraris, N.; Wray, S.; Cagnoni, G.; Donohue, D.E.; Casoni, F.; Kramer, P.R.; Derijck, A.A.; Adolfs, Y.; Fasolo, A.; et al. Dysregulation of Semaphorin7A/beta1-integrin signaling leads to defective GnRH-1 cell migration, abnormal gonadal development and altered fertility. Hum. Mol. Genet. 2011, 20, 4759–4774. [Google Scholar] [CrossRef] [Green Version]
- Duan, S.; Huang, W.; Liu, X.; Liu, X.; Chen, N.; Xu, Q.; Hu, Y.; Song, W.; Zhou, J. IMPDH2 promotes colorectal cancer progression through activation of the PI3K/AKT/mTOR and PI3K/AKT/FOXO1 signaling pathways. J. Exp. Clin. Cancer Res. 2018, 37, 304. [Google Scholar] [CrossRef] [Green Version]
- Bangasser, B.L.; Odde, D.J. Master equation-based analysis of a motor-clutch model for cell traction force. Cell. Mol. Bioeng. 2013, 6, 449–459. [Google Scholar] [CrossRef] [Green Version]
- Prahl, L.S.; Bangasser, P.F.; Stopfer, L.E.; Hemmat, M.; White, F.M.; Rosenfeld, S.S.; Odde, D.J. Microtubule-Based Control of Motor-Clutch System Mechanics in Glioma Cell Migration. Cell. Rep. 2018, 25, 2591–2604.e2598. [Google Scholar] [CrossRef] [Green Version]
- Simpson, K.J.; Selfors, L.M.; Bui, J.; Reynolds, A.; Leake, D.; Khvorova, A.; Brugge, J.S. Identification of genes that regulate epithelial cell migration using an siRNA screening approach. Nat. Cell Biol. 2008, 10, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Mitchison, T.; Kirschner, M. Cytoskeletal dynamics and nerve growth. Neuron 1988, 1, 761–772. [Google Scholar] [CrossRef]
- Giannone, G.; Dubin-Thaler, B.J.; Rossier, O.; Cai, Y.; Chaga, O.; Jiang, G.; Beaver, W.; Dobereiner, H.G.; Freund, Y.; Borisy, G.; et al. Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 2007, 128, 561–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riveline, D.; Zamir, E.; Balaban, N.Q.; Schwarz, U.S.; Ishizaki, T.; Narumiya, S.; Kam, Z.; Geiger, B.; Bershadsky, A.D. Focal contacts as mechanosensors: Externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J. Cell Biol. 2001, 153, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Hwang, T.L.; Close, T.P.; Grego, J.M.; Brannon, W.L.; Gonzales, F. Predilection of brain metastasis in gray and white matter junction and vascular border zones. Cancer 1996, 77, 1551–1555. [Google Scholar] [CrossRef]
- Lu, Y.B.; Franze, K.; Seifert, G.; Steinhauser, C.; Kirchhoff, F.; Wolburg, H.; Guck, J.; Janmey, P.; Wei, E.Q.; Kas, J.; et al. Viscoelastic properties of individual glial cells and neurons in the CNS. Proc. Natl. Acad. Sci. USA 2006, 103, 17759–17764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahaffy, R.E.; Shih, C.K.; MacKintosh, F.C.; Kas, J. Scanning probe-based frequency-dependent microrheology of polymer gels and biological cells. Phys. Rev. Lett. 2000, 85, 880–883. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.R.; Pinto-Costa, R.; Sousa, S.C.; Sousa, M.M. The Regulation of Axon Diameter: From Axonal Circumferential Contractility to Activity-Dependent Axon Swelling. Front. Mol. Neurosci. 2018, 11, 319. [Google Scholar] [CrossRef]
- Zhang, Y.; Abiraman, K.; Li, H.; Pierce, D.M.; Tzingounis, A.V.; Lykotrafitis, G. Modeling of the axon membrane skeleton structure and implications for its mechanical properties. PLoS Comput. Biol. 2017, 13, e1005407. [Google Scholar] [CrossRef] [Green Version]
- Romano, A.; Scheel, M.; Hirsch, S.; Braun, J.; Sack, I. In vivo waveguide elastography of white matter tracts in the human brain. Magn. Reson. Med. 2012, 68, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Arnett, D.K.; Tyroler, H.A.; Riley, W.A.; Chambless, L.E.; Szklo, M.; Heiss, G. Arterial stiffness and the development of hypertension. The ARIC study. Hypertension 1999, 34, 201–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, B.; Lang, S.; Dominietto, M.; Rudin, M.; Schulz, G.; Deyhle, H.; Germann, M.; Pfeiffer, F.; David, C.; Weitkamp, T. High-resolution tomographic imaging of microvessels. Proc. SPIE 2008, 7078, 70780B. [Google Scholar] [CrossRef] [Green Version]
- Winkler, F.; Kienast, Y.; Fuhrmann, M.; Von Baumgarten, L.; Burgold, S.; Mitteregger, G.; Kretzschmar, H.; Herms, J. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia 2009, 57, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Pogoda, K.; Chin, L.; Georges, P.C.; Byfield, F.J.; Bucki, R.; Kim, R.; Weaver, M.; Wells, R.G.; Marcinkiewicz, C.; Janmey, P.A. Compression stiffening of brain and its effect on mechanosensing by glioma cells. New J. Phys. 2014, 16, 075002. [Google Scholar] [CrossRef] [PubMed]
- Mohiuddin, E.; Wakimoto, H. Extracellular matrix in glioblastoma: Opportunities for emerging therapeutic approaches. Am. J. Cancer Res. 2021, 11, 3742–3754. [Google Scholar]
- Pepin, K.M.; McGee, K.P.; Arani, A.; Lake, D.S.; Glaser, K.J.; Manduca, A.; Parney, I.F.; Ehman, R.L.; Huston, J., 3rd. MR Elastography Analysis of Glioma Stiffness and IDH1-Mutation Status. AJNR Am. J. Neuroradiol. 2018, 39, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Lebelt, A.; Dzieciol, J.; Guzinska-Ustymowicz, K.; Lemancewicz, D.; Zimnoch, L.; Czykier, E. Angiogenesis in gliomas. Folia Histochem. Cytobiol. 2008, 46, 69–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the maligant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lautscham, L.A.; Kammerer, C.; Lange, J.R.; Kolb, T.; Mark, C.; Schilling, A.; Strissel, P.L.; Strick, R.; Gluth, C.; Rowat, A.C.; et al. Migration in Confined 3D Environments Is Determined by a Combination of Adhesiveness, Nuclear Volume, Contractility, and Cell Stiffness. Biophys. J. 2015, 109, 900–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, A.W.; Grant, A.D.; Parker, S.S.; Hill, S.; Whalen, M.B.; Chakrabarti, J.; Harman, M.W.; Roman, M.R.; Forte, B.L.; Gowan, C.C.; et al. Breast tumor stiffness instructs bone metastasis via maintenance of mechanical conditioning. Cell Rep. 2021, 35, 109293. [Google Scholar] [CrossRef] [PubMed]
- Palama, I.E.; D’Amone, S.; Ratano, P.; Donatelli, A.; Liscio, A.; Antonacci, G.; Testini, M.; Di Angelantonio, S.; Ragozzino, D.; Cortese, B. Mechanical Durotactic Environment Enhances Specific Glioblastoma Cell Responses. Cancers 2019, 11, 643. [Google Scholar] [CrossRef] [Green Version]
- Bloomekatz, J.; Grego-Bessa, J.; Migeotte, I.; Anderson, K.V. Pten regulates collective cell migration during specification of the anterior-posterior axis of the mouse embryo. Dev. Biol. 2012, 364, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marhuenda, E.; Fabre, C.; Zhang, C.; Martin-Fernandez, M.; Iskratsch, T.; Saleh, A.; Bauchet, L.; Cambedouzou, J.; Hugnot, J.P.; Duffau, H.; et al. Glioma stem cells invasive phenotype at optimal stiffness is driven by MGAT5 dependent mechanosensing. J. Exp. Clin. Cancer Res. 2021, 40, 139. [Google Scholar] [CrossRef] [PubMed]
- Funamoto, S.; Meili, R.; Lee, S.; Parry, L.; Firtel, R.A. Spatial and temporal regulation of 3-phosphoinositides by PI 3-kinase and PTEN mediates chemotaxis. Cell 2002, 109, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.; Parsons, R.; Yamada, K.M. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Li, G.; Li, R.; Liu, Q.; He, Q.; Zhang, J. Rho-kinase inhibitor, fasudil, suppresses glioblastoma cell line progression in vitro and in vivo. Cancer Biol. Ther. 2010, 9, 875–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balzer, E.M.; Tong, Z.; Paul, C.D.; Hung, W.C.; Stroka, K.M.; Boggs, A.E.; Martin, S.S.; Konstantopoulos, K. Physical confinement alters tumor cell adhesion and migration phenotypes. FASEB J. 2012, 26, 4045–4056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.L.; Kilic, O.; Schiapparelli, P.; Guerrero-Cazares, H.; Kim, D.H.; Sedora-Roman, N.I.; Gupta, S.; O’Donnell, T.; Chaichana, K.L.; Rodriguez, F.J.; et al. Migration Phenotype of Brain-Cancer Cells Predicts Patient Outcomes. Cell Rep. 2016, 15, 2616–2624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef] [PubMed]
- Charras, G.T.; Hu, C.K.; Coughlin, M.; Mitchison, T.J. Reassembly of contractile actin cortex in cell blebs. J. Cell Biol. 2006, 175, 477–490. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Borja, M.; Janssen, L.; Verwoerd, D.; Hordijk, P.; Neefjes, J. RhoB regulates endosome transport by promoting actin assembly on endosomal membranes through Dia1. J. Cell Sci. 2005, 118, 2661–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deakin, N.O.; Turner, C.E. Distinct roles for paxillin and Hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Mol. Biol. Cell 2011, 22, 327–341. [Google Scholar] [CrossRef]
- Petrie, R.J.; Harlin, H.M.; Korsak, L.I.; Yamada, K.M. Activating the nuclear piston mechanism of 3D migration in tumor cells. J. Cell Biol. 2017, 216, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.; Swift, J.; Dingal, P.C.; Shah, P.; Shin, J.W.; Discher, D.E. Crawling from soft to stiff matrix polarizes the cytoskeleton and phosphoregulates myosin-II heavy chain. J. Cell Biol. 2012, 199, 669–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gundersen, G.G.; Worman, H.J. Nuclear positioning. Cell 2013, 152, 1376–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maninova, M.; Klimova, Z.; Parsons, J.T.; Weber, M.J.; Iwanicki, M.P.; Vomastek, T. The reorientation of cell nucleus promotes the establishment of front-rear polarity in migrating fibroblasts. J. Mol. Biol. 2013, 425, 2039–2055. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, P.; Rupprecht, J.F.; Wieser, S.; Ruprecht, V.; Benichou, O.; Carpi, N.; Coppey, M.; De Beco, S.; Gov, N.; Heisenberg, C.P.; et al. Actin flows mediate a universal coupling between cell speed and cell persistence. Cell 2015, 161, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, A.D.; Wang, F.W.; Matsumoto, K.; Yamada, K.M. One-dimensional topography underlies three-dimensional fibrillar cell migration. J. Cell Biol. 2009, 184, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Sheets, K.; Elankumaran, S.; Nain, A.S. The mechanistic influence of aligned nanofibers on cell shape, migration and blebbing dynamics of glioma cells. Integr. Biol. 2013, 5, 1036–1044. [Google Scholar] [CrossRef] [Green Version]
- Estabridis, H.M.; Jana, A.; Nain, A.; Odde, D.J. Cell Migration in 1D and 2D Nanofiber Microenvironments. Ann. Biomed. Eng. 2018, 46, 392–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, N.S.; Ridley, A.J. Targeting Rho GTPase Signaling Networks in Cancer. Front. Cell Dev. Biol. 2020, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Svensmark, J.H.; Brakebusch, C. Rho GTPases in cancer: Friend or foe? Oncogene 2019, 38, 7447–7456. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wieland, A.; Strissel, P.L.; Schorle, H.; Bakirci, E.; Janzen, D.; Beckmann, M.W.; Eckstein, M.; Dalton, P.D.; Strick, R. Brain and Breast Cancer Cells with PTEN Loss of Function Reveal Enhanced Durotaxis and RHOB Dependent Amoeboid Migration Utilizing 3D Scaffolds and Aligned Microfiber Tracts. Cancers 2021, 13, 5144. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205144
Wieland A, Strissel PL, Schorle H, Bakirci E, Janzen D, Beckmann MW, Eckstein M, Dalton PD, Strick R. Brain and Breast Cancer Cells with PTEN Loss of Function Reveal Enhanced Durotaxis and RHOB Dependent Amoeboid Migration Utilizing 3D Scaffolds and Aligned Microfiber Tracts. Cancers. 2021; 13(20):5144. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205144
Chicago/Turabian StyleWieland, Annalena, Pamela L. Strissel, Hannah Schorle, Ezgi Bakirci, Dieter Janzen, Matthias W. Beckmann, Markus Eckstein, Paul D. Dalton, and Reiner Strick. 2021. "Brain and Breast Cancer Cells with PTEN Loss of Function Reveal Enhanced Durotaxis and RHOB Dependent Amoeboid Migration Utilizing 3D Scaffolds and Aligned Microfiber Tracts" Cancers 13, no. 20: 5144. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205144