
Comprehensive Analysis of VEGFR2 Expression in HPV-Positive and -Negative OPSCC Reveals Differing VEGFR2 Expression Patterns

 , , , , , ,
, , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
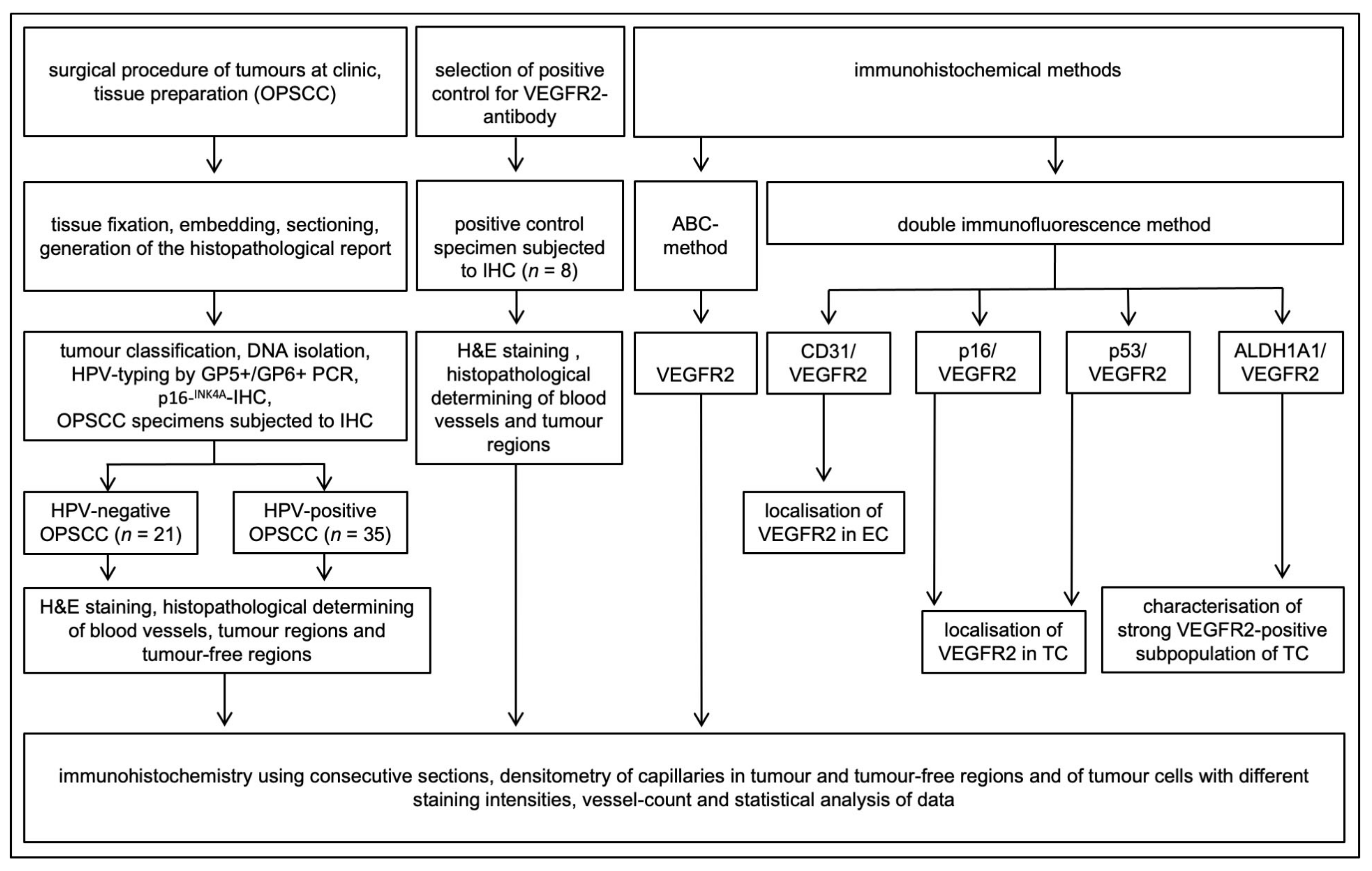
2. Materials and Methods
2.1. Patient Material and Ethics Statement
2.2. Tissue Fixation, Embedding and Sectioning
2.3. DNA Isolation and HPV Typing
2.4. Immunohistochemistry
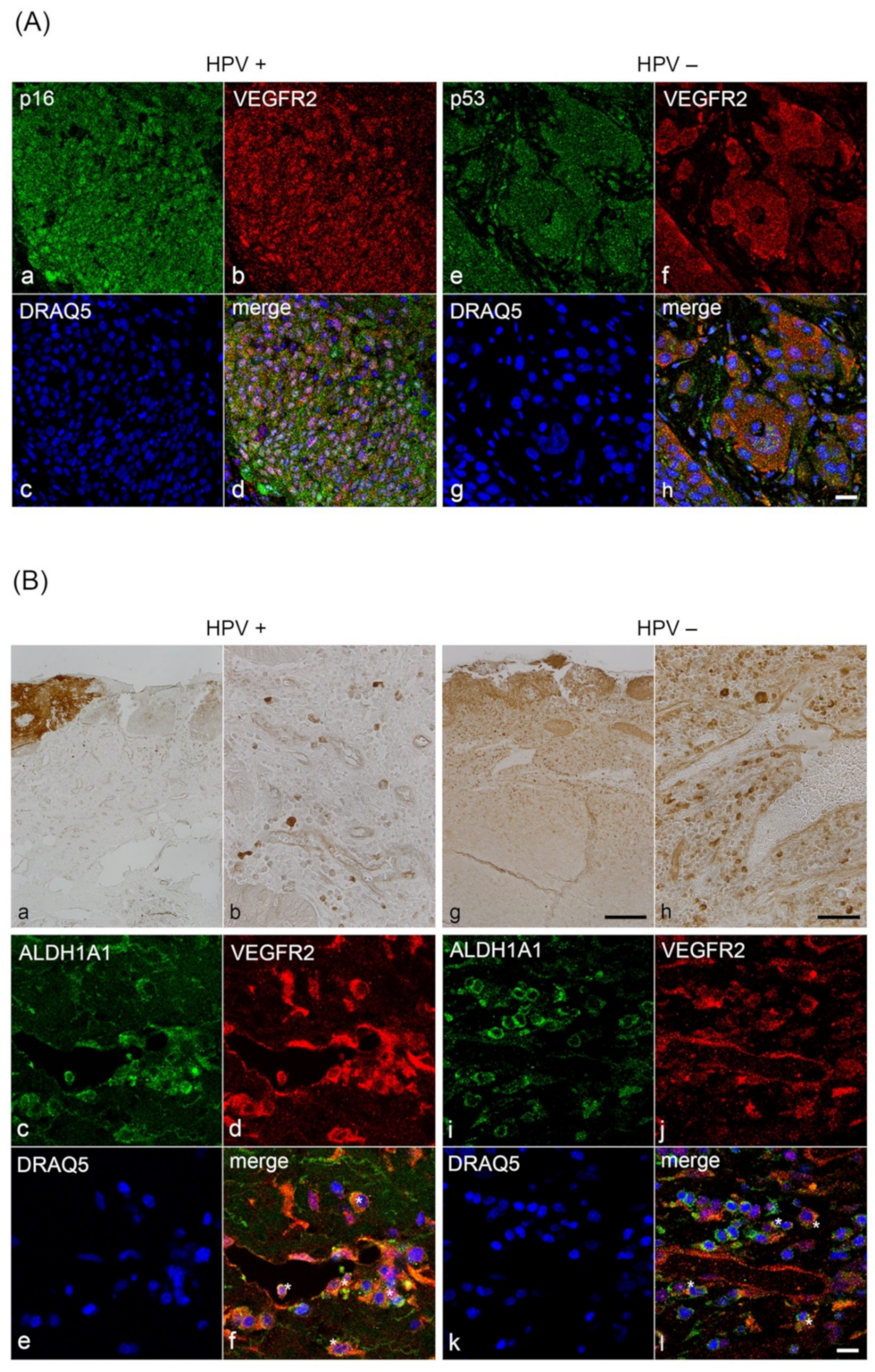
2.5. Double Immunofluorescence Labelling with VEGFR2, CD31, ALDH1A1, p16INK4A and TP53
2.6. Densitometric Quantification of Immunohistochemical Signals
2.7. Cell Culture and Retroviral Transduction
2.8. RNA Isolation, Reverse Transcription and Real-Time Quantitative PCR
2.9. Statistics
3. Results
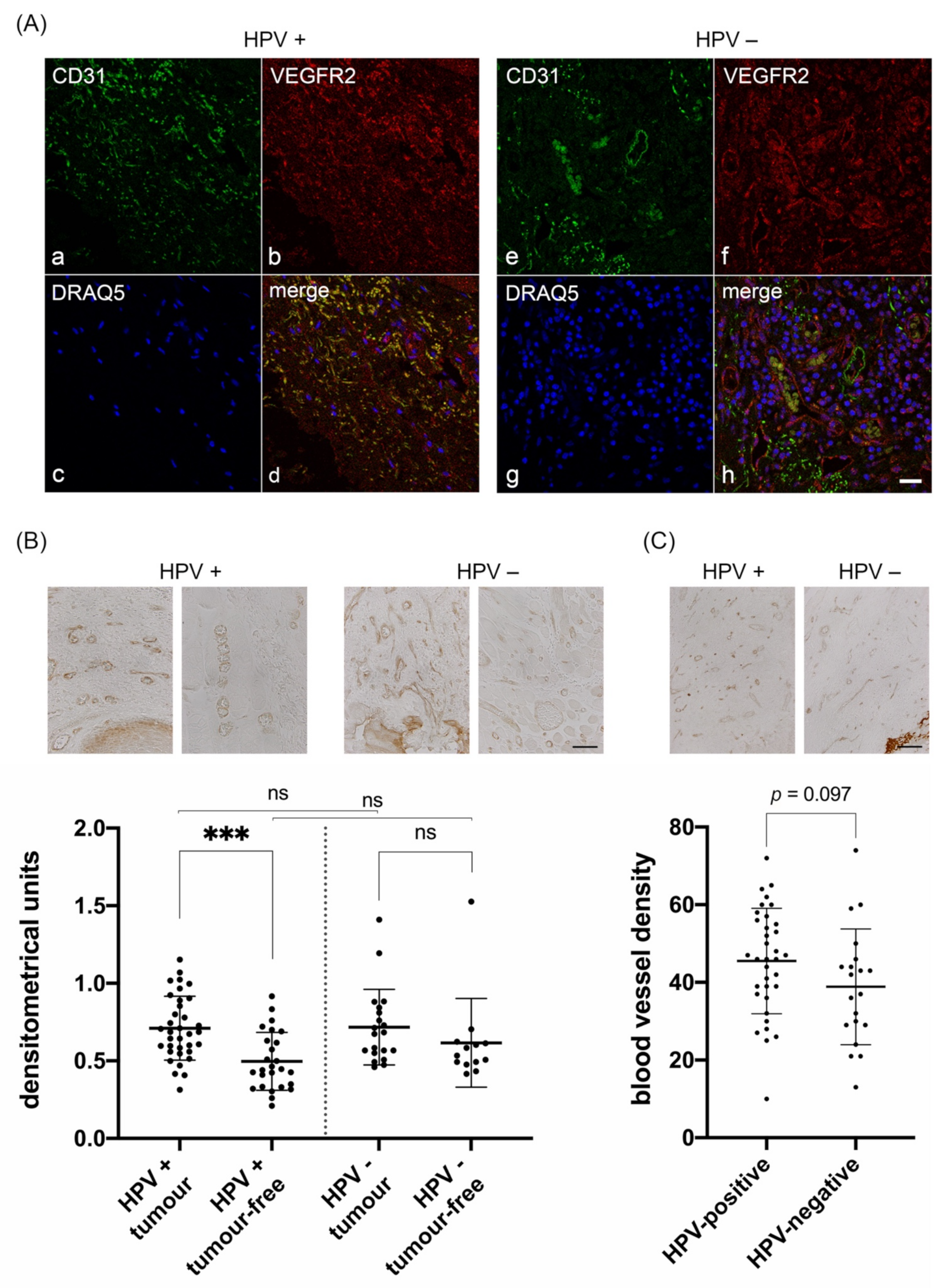
3.1. Characterization of VEGFR2 Expression in Blood Vessels of HPV-Positive and HPV-Negative OPSCC
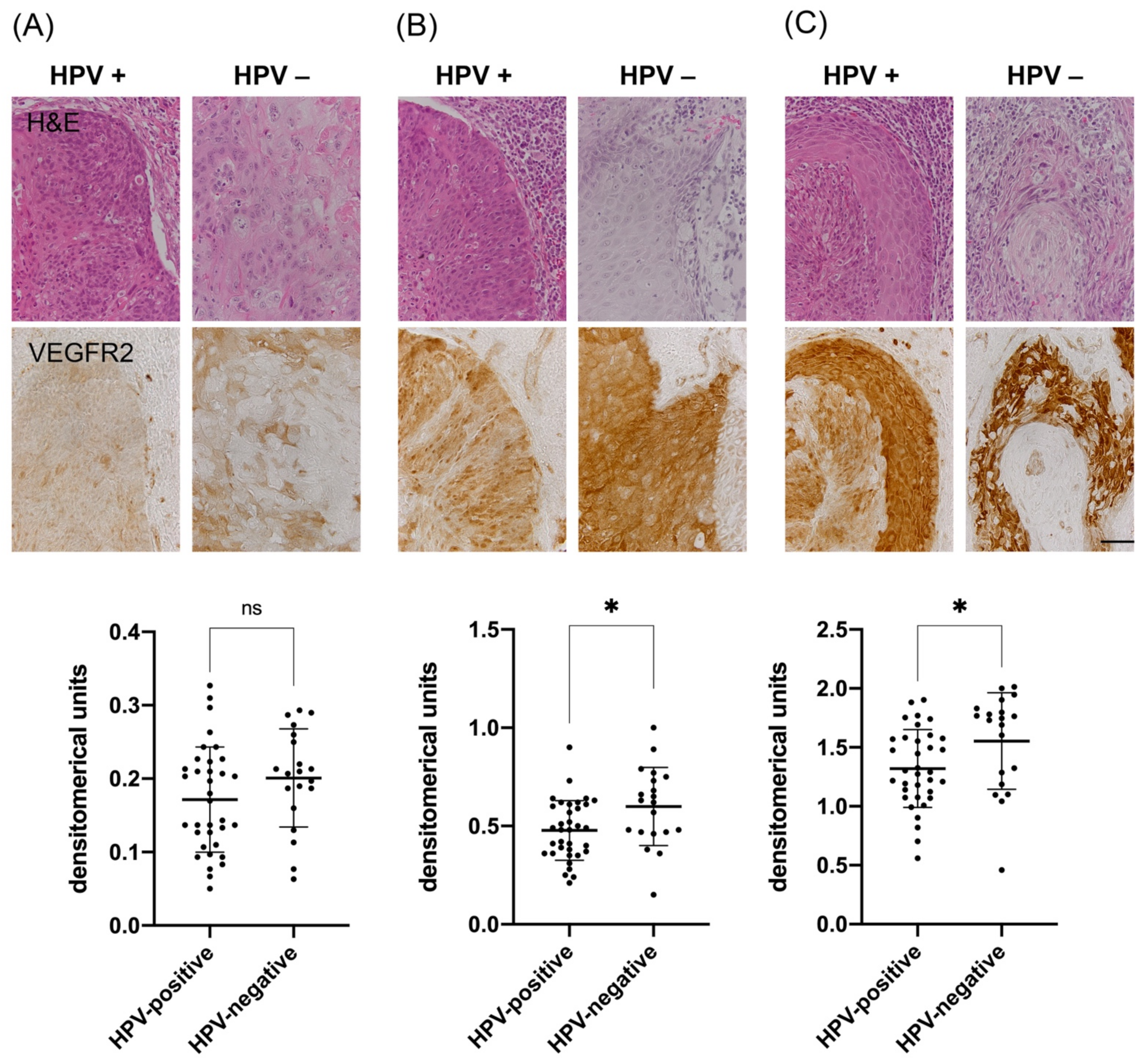
3.2. Analysis of VEGFR2 Staining Intensity in Tumor Cells of HPV-Positive and HPV-Negative OPSCC
3.3. Correlation of VEGFR2, NRF2 and AKR1C3 with Clinicopathological Data
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chaturvedi, A.K.; Engels, E.A.; Pfeiffer, R.M.; Hernandez, B.Y.; Xiao, W.; Kim, E.; Jiang, B.; Goodman, M.T.; Sibug-Saber, M.; Cozen, W.; et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 2011, 29, 4294–4301. [Google Scholar] [CrossRef]
- CDC. Cancers Associated with Human Papillomavirus, United States—2012–2016. USCS Data Brief, no 10. Available online: https://www.cdc.gov/cancer/uscs/about/data-briefs/no10-hpv-assoc-cancers-UnitedStates-2012-2016.htm (accessed on 8 February 2020).
- Leemans, C.R.; Snijders, P.J.F.; Brakenhoff, R.H. The molecular landscape of head and neck cancer. Nat. Rev. Cancer 2018, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Tribius, S. HPV and oropharyngeal squamous cell cancer in the 8th edition of the TNM classification. Laryngorhinootologie 2018, 97, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Molec. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Karaman, S.; Leppänen, V.-M.; Alitalo, K. Vascular endothelial growth factor signaling in development and disease. Development 2018, 145, dev151019. [Google Scholar] [CrossRef] [Green Version]
- Kowanetz, M.; Ferrara, N. Vascular endothelial growth factor signaling pathways: Therapeutic perspective. Clin. Cancer Res. 2006, 12, 5018–5022. [Google Scholar] [CrossRef] [Green Version]
- Neuchrist, C.; Erovic, B.M.; Handisurya, A.; Steiner, G.E.; Rockwell, P.; Gedlicka, C.; Burian, M. Vascular endothelial growth factor receptor 2 (VEGFR2) expression in squamous cell carcinomas of the head and neck. Laryngoscope 2001, 111, 1834–1841. [Google Scholar] [CrossRef]
- Christopoulos, A.; Ahn, S.M.; Klein, J.D.; Kim, S. Biology of vascular endothelial growth factor and its receptors in head and neck cancer: Beyond angiogenesis. Head Neck 2011, 33, 1220–1229. [Google Scholar] [CrossRef]
- López-Ocejo, O.; Viloria-Petit, A.; Bequet-Romero, M.; Mukhopadhyay, D.; Rak, J.; Kerbel, R.S. Oncogenes and tumor angiogenesis: The HPV-16 E6 oncoprotein activates the vascular endothelial growth factor (VEGF) gene promoter in a p53 independent manner. Oncogene 2000, 19, 4611–4620. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zhang, Q.; Nishitani, J.; Brown, J.; Shi, S.; Le, A.D. Overexpression of human papillomavirus type 16 oncoproteins enhances hypoxia-inducible factor 1 alpha protein accumulation and vascular endothelial growth factor expression in human cervical carcinoma cells. Clin. Cancer Res. 2007, 13, 2568–2576. [Google Scholar] [CrossRef] [Green Version]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [Green Version]
- Penning, T.M. Aldo-keto reductase regulation by the Nrf2 system: Implications for stress response, chemotherapy drug resistance, and carcinogenesis. Chem. Res. Toxicol. 2017, 30, 162–176. [Google Scholar] [CrossRef]
- Huebbers, C.U.; Verhees, F.; Poluschkin, L.; Olthof, N.C.; Kolligs, J.; Siefer, O.G.; Henfling, M.; Ramaekers, F.C.S.; Preuss, S.F.; Beutner, D.; et al. Upregulation of AKR1C1 and AKR1C3 expression in OPSCC with integrated HPV16 and HPV-negative tumors is an indicator of poor prognosis. Int. J. Cancer 2019, 144, 2465–2477. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.D.; Vucic, E.A.; Thu, K.L.; Pikor, L.A.; Lam, S.; Lam, W.L. Disruption of KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex components by multiple genetic mechanisms: Association with poor prognosis in head and neck cancer. Head Neck 2015, 37, 727–734. [Google Scholar] [CrossRef]
- Meyer, M.F.; Huebbers, C.U.; Siefer, O.G.; Vent, J.; Engbert, I.; Eslick, G.D.; Valter, M.; Klussmann, J.P.; Preuss, S.F. Prevalence and risk factors for oral human papillomavirus infection in 129 women screened for cervical HPV infection. Oral Oncol. 2014, 50, 27–31. [Google Scholar] [CrossRef]
- Korkmaz, Y.; Roggendorf, H.C.; Siefer, O.G.; Seehawer, J.; Imhof, T.; Plomann, M.; Bloch, W.; Friebe, A.; Huebbers, C.U. Downregulation of the α1- and β1-subunit of sGC in arterial smooth muscle cells of OPSCC Is HPV-independent. J. Dent. Res. 2018, 97, 1214–1221. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Hufbauer, M.; Biddle, A.; Borgogna, C.; Gariglio, M.; Doorbar, J.; Storey, A.; Pfister, H.; Mackenzie, I.; Akgul, B. Expression of betapapillomavirus oncogenes increases the number of keratinocytes with stem cell-like properties. J. Virol. 2013, 87, 12158–12165. [Google Scholar] [CrossRef] [Green Version]
- Hufbauer, M.; Maltseva, M.; Meinrath, J.; Lechner, A.; Beutner, D.; Huebbers, C.U.; Akgul, B. HPV16 increases the number of migratory cancer stem cells and modulates their miRNA expression profile in oropharyngeal cancer. Int. J. Cancer 2018, 143, 1426–1439. [Google Scholar] [CrossRef] [Green Version]
- Halbert, C.L.; Demers, G.W.; Galloway, D.A. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J. Virol. 1992, 66, 2125–2134. [Google Scholar] [CrossRef] [Green Version]
- Hufbauer, M.; Cooke, J.; van der Horst, G.T.; Pfister, H.; Storey, A.; Akgul, B. Human papillomavirus mediated inhibition of DNA damage sensing and repair drives skin carcinogenesis. Mol. Cancer 2015, 14, 183. [Google Scholar] [CrossRef] [Green Version]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008, 266, 37–52. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Wagner, S.; Ma, C.; Coordes, A.; Gekeler, J.; Klussmann, J.P.; Hummel, M.; Kaufmann, A.M.; Albers, A.E. Prognostic significance of ALDH1A1-positive cancer stem cells in patients with locally advanced, metastasized head and neck squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2014, 140, 1151–1158. [Google Scholar] [CrossRef]
- Pinheiro, C.; Garcia, E.A.; Morais-Santos, F.; Moreira, M.A.; Almeida, F.M.; Jube, L.F.; Queiroz, G.S.; Paula, E.C.; Andreoli, M.A.; Villa, L.L.; et al. Reprogramming energy metabolism and inducing angiogenesis: Co-expression of monocarboxylate transporters with VEGF family members in cervical adenocarcinomas. BMC Cancer 2015, 15, 835. [Google Scholar] [CrossRef] [Green Version]
- Chandel, V.; Raj, S.; Kumar, P.; Gupta, S.; Dhasmana, A.; Kesari, K.K.; Ruokolainen, J.; Mehra, P.; Das, B.C.; Kamal, M.A.; et al. Metabolic regulation in HPV associated head and neck squamous cell carcinoma. Life Sci. 2020, 258, 118236. [Google Scholar] [CrossRef]
- Lalla, R.V.; Boisoneau, D.S.; Spiro, J.D.; Kreutzer, D.L. Expression of vascular endothelial growth factor receptors on tumor cells in head and neck squamous cell carcinoma. Arch. Otolaryngol. Head Neck Surg. 2003, 129, 882. [Google Scholar] [CrossRef] [Green Version]
- Newman, P.J. The biology of PECAM-1. J. Clin. Investig. 1997, 99, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Bosmuller, H.; Pfefferle, V.; Bittar, Z.; Scheble, V.; Horger, M.; Sipos, B.; Fend, F. Microvessel density and angiogenesis in primary hepatic malignancies: Differential expression of CD31 and VEGFR-2 in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Pathol. Res. Pract. 2018, 214, 1136–1141. [Google Scholar] [CrossRef]
- Hanns, E.; Job, S.; Coliat, P.; Wasylyk, C.; Ramolu, L.; Pencreach, E.; Suarez-Carmona, M.; Herfs, M.; Ledrappier, S.; Macabre, C.; et al. Human papillomavirus-related tumours of the oropharynx display a lower tumour hypoxia signature. Oral Oncol. 2015, 51, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Bohman, S.; Dixelius, J.; Berge, T.; Dimberg, A.; Magnusson, P.; Wang, L.; Wikner, C.; Qi, J.H.; Wernstedt, C.; et al. VEGF receptor-2 Y951 signaling and a role for the adapter molecule TSAd in tumor angiogenesis. EMBO J. 2005, 24, 2342–2353. [Google Scholar] [CrossRef] [PubMed]
- Le Buanec, H.; D’Anna, R.; Lachgar, A.; Zagury, J.F.; Bernard, J.; Ittele, D.; d’Alessio, P.; Hallez, S.; Giannouli, C.; Burny, A.; et al. HPV-16 E7 but not E6 oncogenic protein triggers both cellular immunosuppression and angiogenic processes. Biomed. Pharmacother. 1999, 53, 424–431. [Google Scholar] [CrossRef]
- Chen, W.; Li, F.; Mead, L.; White, H.; Walker, J.; Ingram, D.A.; Roman, A. Human papillomavirus causes an angiogenic switch in keratinocytes which is sufficient to alter endothelial cell behavior. Virology 2007, 367, 168–174. [Google Scholar] [CrossRef] [Green Version]
- Sturgis, E.M.; Ang, K.K. The epidemic of HPV-associated oropharyngeal cancer is here: Is it time to change our treatment paradigms? J. Natl. Compr. Cancer Netw. 2011, 9, 665–673. [Google Scholar] [CrossRef] [Green Version]
- Vassilakopoulou, M.; Psyrri, A.; Argiris, A. Targeting angiogenesis in head and neck cancer. Oral Oncol. 2015, 51, 409–415. [Google Scholar] [CrossRef]
- Hsu, H.W.; Wall, N.R.; Hsueh, C.T.; Kim, S.; Ferris, R.L.; Chen, C.S.; Mirshahidi, S. Combination antiangiogenic therapy and radiation in head and neck cancers. Oral Oncol. 2014, 50, 19–26. [Google Scholar] [CrossRef]
- Li, J.; Huang, S.; Armstrong, E.A.; Fowler, J.F.; Harari, P.M. Angiogenesis and radiation response modulation after vascular endothelial growth factor receptor-2 (VEGFR2) blockade. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 1477–1485. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Farhang Ghahremani, M.; Goossens, S.; Haigh, J.J. The p53 family and VEGF regulation: "It’s complicated". Cell Cycle 2013, 12, 1331–1332. [Google Scholar] [CrossRef] [Green Version]
- Blazquez, C.; Cook, N.; Micklem, K.; Harris, A.L.; Gatter, K.C.; Pezzella, F. Phosphorylated KDR can be located in the nucleus of neoplastic cells. Cell Res. 2006, 16, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Domingues, I.; Rino, J.; Demmers, J.A.; de Lanerolle, P.; Santos, S.C. VEGFR2 translocates to the nucleus to regulate its own transcription. PLoS ONE 2011, 6, e25668. [Google Scholar] [CrossRef] [Green Version]
- Meyer, R.D.; Srinivasan, S.; Singh, A.J.; Mahoney, J.E.; Gharahassanlou, K.R.; Rahimi, N. PEST motif serine and tyrosine phosphorylation controls vascular endothelial growth factor receptor 2 stability and downregulation. Mol. Cell Biol. 2011, 31, 2010–2025. [Google Scholar] [CrossRef] [Green Version]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef] [Green Version]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen synthase kinase-3β inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol Chem 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [Green Version]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Pan, C.; Sun, J.; Gilbert, C.; Drews-Elger, K.; Azzam, D.J.; Picon-Ruiz, M.; Kim, M.; Ullmer, W.; El-Ashry, D.; et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 2015, 34, 3107–3119. [Google Scholar] [CrossRef]
- Zhang, M.; Kumar, B.; Piao, L.; Xie, X.; Schmitt, A.; Arradaza, N.; Cippola, M.; Old, M.; Agrawal, A.; Ozer, E.; et al. Elevated intrinsic cancer stem cell population in human papillomavirus-associated head and neck squamous cell carcinoma. Cancer 2014, 120, 992–1001. [Google Scholar] [CrossRef]
- Reid, P.; Wilson, P.; Li, Y.; Marcu, L.G.; Staudacher, A.H.; Brown, M.P.; Bezak, E. In vitro investigation of head and neck cancer stem cell proportions and their changes following X-ray irradiation as a function of HPV status. PLoS ONE 2017, 12, e0186186. [Google Scholar] [CrossRef]
- Jinesh, G.G.; Manyam, G.C.; Mmeje, C.O.; Baggerly, K.A.; Kamat, A.M. Surface PD-L1, E-cadherin, CD24, and VEGFR2 as markers of epithelial cancer stem cells associated with rapid tumorigenesis. Sci. Rep. 2017, 7, 9602. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
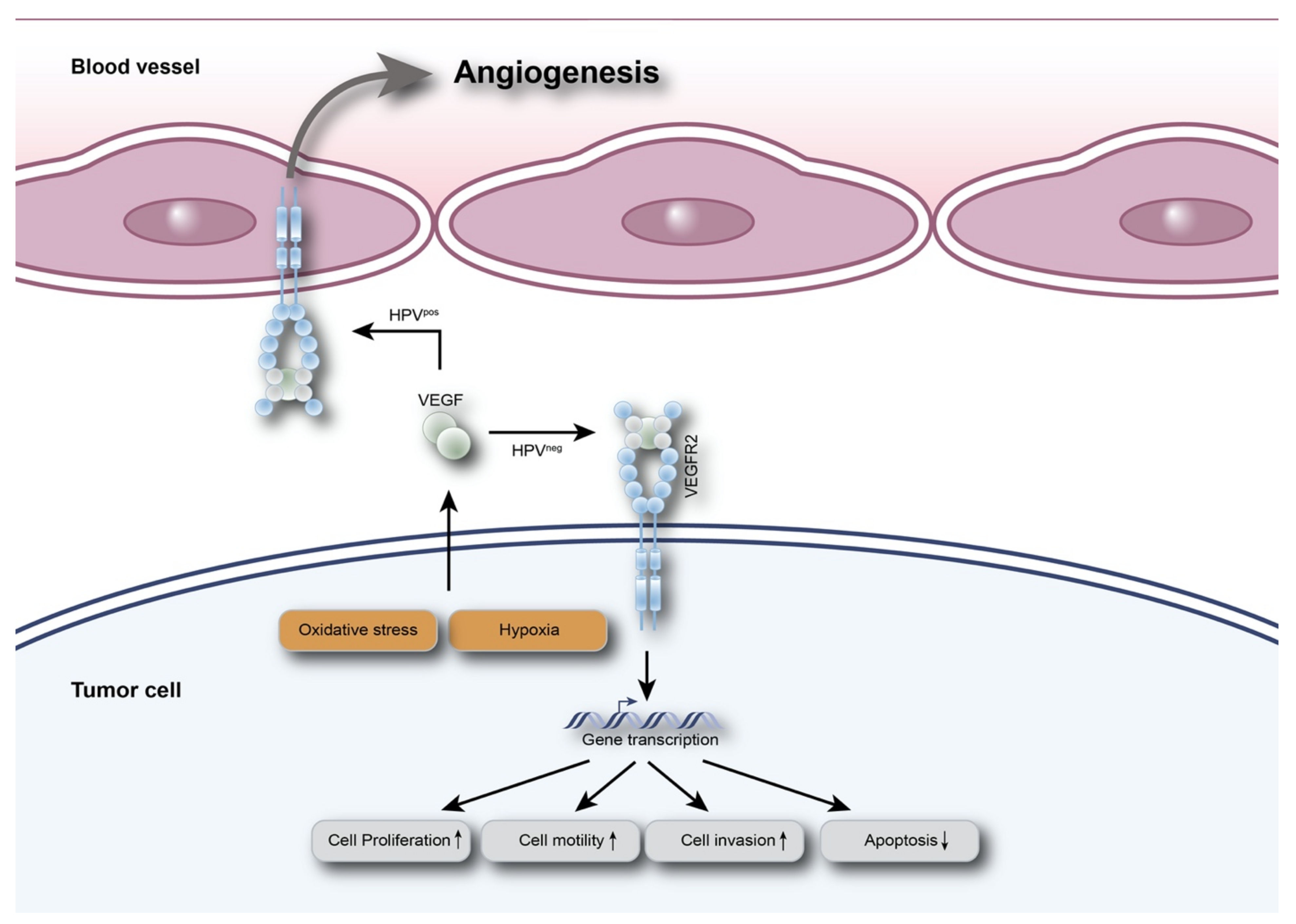{kind=link}
| Clinico Pathological Feature | Total | HPV—Status | VEGFR2—Staining | NRF2—Staining | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HPV—positive | HPV—negative | VEGFR+ | VEGFR2− | NRF2+ | NRF2− | ||||||||||||
| n | % | n | % | n | % | X2 | n | % | n | % | X2 | n | % | n | % | X2 | |
| Mean age (years) | 60.2 | – | 59.9 | – | 60.5 | – | 0.838 | 59.2 | – | 61.0 | – | 0.517 | 62.9 | – | 58.8 | – | 0.177 |
| Gender | |||||||||||||||||
| Male | 43 | 76.8 | 26 | 46.8 | 17 | 30.0 | 23 | 41.1 | 19 | 33.9 | 17 | 30.3 | 26 | 46.4 | |||
| Female | 13 | 23.2 | 9 | 16.1 | 4 | 7.1 | 0.747 | 2 | 3.6 | 11 | 19.6 | 0.023 | 2 | 3.6 | 11 | 19.6 | 0.098 |
| T classification | |||||||||||||||||
| pT1 and pT2 | 30 | 53.6 | 20 | 35.7 | 10 | 17.9 | 13 | 23.6 | 16 | 29.1 | 8 | 14.3 | 22 | 39.3 | |||
| pT3 and pT4 | 26 | 46.4 | 15 | 26.8 | 11 | 19.6 | 0.584 | 12 | 21.8 | 14 | 25.5 | 1.00 | 11 | 19.6 | 15 | 26.8 | 0.265 |
| N classification | |||||||||||||||||
| pN0 | 14 | 25.0 | 9 | 16.1 | 5 | 8.9 | 4 | 7.3 | 9 | 16.4 | 4 | 7.1 | 10 | 17.9 | |||
| pN1-3 | 42 | 75.0 | 26 | 46.4 | 16 | 28.6 | 1.00 | 21 | 38.2 | 21 | 38.2 | 0.341 | 15 | 26.8 | 27 | 48.2 | 0.751 |
| M classification | |||||||||||||||||
| pM0 | 54 | 98.2 | 35 | 63.6 | 19 | 34.5 | 24 | 44.4 | 29 | 53.7 | 18 | 32.7 | 36 | 65.5 | |||
| pM1 | 1 | 1.8 | 0 | 0.0 | 1 | 1.8 | 0.364 | 0 | 0.0 | 1 | 1.9 | 1.00 | 1 | 1.8 | 0 | 0.0 | 0.345 |
| Death | |||||||||||||||||
| Yes | 16 | 28.6 | 9 | 16.1 | 7 | 12.5 | 6 | 10.9 | 10 | 18.2 | 12 | 21.4 | 4 | 7.1 | |||
| No | 40 | 71.4 | 26 | 46.4 | 14 | 25.0 | 0.557 | 24 | 43.6 | 15 | 27.3 | 0.140 | 7 | 12.5 | 33 | 58.9 | <0.0001 |
| HPV-status | |||||||||||||||||
| Negative | 21 | 37.5 | 13 | 23.6 | 7 | 12.7 | 7 | 12.5 | 14 | 25.0 | |||||||
| Positive | 35 | 62.5 | 12 | 21.8 | 23 | 41.8 | 0.048 | 12 | 21.4 | 23 | 41.1 | 1.00 | |||||
| NRF2-staining | |||||||||||||||||
| Negative | 36 | 65.5 | 23 | 41.1 | 14 | 25.0 | 14 | 25.5 | 22 | 40.0 | |||||||
| Positive | 19 | 34.5 | 12 | 21.4 | 7 | 12.5 | 1.00 | 11 | 20.0 | 8 | 14.5 | 0.256 | |||||
| AKR1C3-staining | |||||||||||||||||
| Yes | 38 | 67.9 | 25 | 44.6 | 13 | 23.2 | 15 | 27.3 | 22 | 40.0 | 14 | 25.5 | 22 | 40.0 | |||
| No | 18 | 32.1 | 10 | 17.9 | 8 | 14.3 | 0.558 | 10 | 18.2 | 8 | 14.5 | 0.389 | 11 | 20.0 | 8 | 14.5 | 0.256 |
| Blood vessel density | |||||||||||||||||
| Low | 30 | 54.5 | 15 | 27.3 | 15 | 27.3 | 12 | 21.8 | 18 | 32.7 | 7 | 12.7 | 23 | 41.8 | |||
| High | 25 | 45.5 | 20 | 36.4 | 5 | 9.1 | 0.027 | 13 | 23.6 | 12 | 21.8 | 0.424 | 12 | 21.8 | 13 | 23.6 | 0.087 |
| Smoking | |||||||||||||||||
| Never smoked | 13 | 23.2 | 12 | 21.4 | 1 | 1.8 | 3 | 5.5 | 10 | 18.2 | 3 | 5.4 | 10 | 17.9 | |||
| Smoker | 43 | 76.8 | 23 | 41.1 | 20 | 35.7 | 0.020 | 22 | 40.0 | 20 | 36.4 | 0.110 | 16 | 28.6 | 27 | 48.2 | 0.507 |
| Alcohol | |||||||||||||||||
| No or < 1 glass/day | 43 | 78.2 | 30 | 53.6 | 14 | 25.0 | 17 | 30.9 | 26 | 47.3 | 17 | 30.4 | 27 | 48.2 | |||
| Active, > 1 glass/day | 12 | 21.8 | 5 | 8.9 | 7 | 12.5 | 0.108 | 8 | 14.5 | 4 | 7.3 | 0.114 | 2 | 3.6 | 10 | 17.9 | 0.189 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uzun, S.; Korkmaz, Y.; Wuerdemann, N.; Arolt, C.; Puladi, B.; Siefer, O.G.; Dönmez, H.G.; Hufbauer, M.; Akgül, B.; Klussmann, J.P.; et al. Comprehensive Analysis of VEGFR2 Expression in HPV-Positive and -Negative OPSCC Reveals Differing VEGFR2 Expression Patterns. Cancers 2021, 13, 5221. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205221
Uzun S, Korkmaz Y, Wuerdemann N, Arolt C, Puladi B, Siefer OG, Dönmez HG, Hufbauer M, Akgül B, Klussmann JP, et al. Comprehensive Analysis of VEGFR2 Expression in HPV-Positive and -Negative OPSCC Reveals Differing VEGFR2 Expression Patterns. Cancers. 2021; 13(20):5221. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205221
Chicago/Turabian StyleUzun, Senem, Yüksel Korkmaz, Nora Wuerdemann, Christoph Arolt, Behrus Puladi, Oliver G. Siefer, Hanife G. Dönmez, Martin Hufbauer, Baki Akgül, Jens P. Klussmann, and et al. 2021. "Comprehensive Analysis of VEGFR2 Expression in HPV-Positive and -Negative OPSCC Reveals Differing VEGFR2 Expression Patterns" Cancers 13, no. 20: 5221. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13205221