
Impact of Chromatin Dynamics and DNA Repair on Genomic Stability and Treatment Resistance in Pediatric High-Grade Gliomas


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
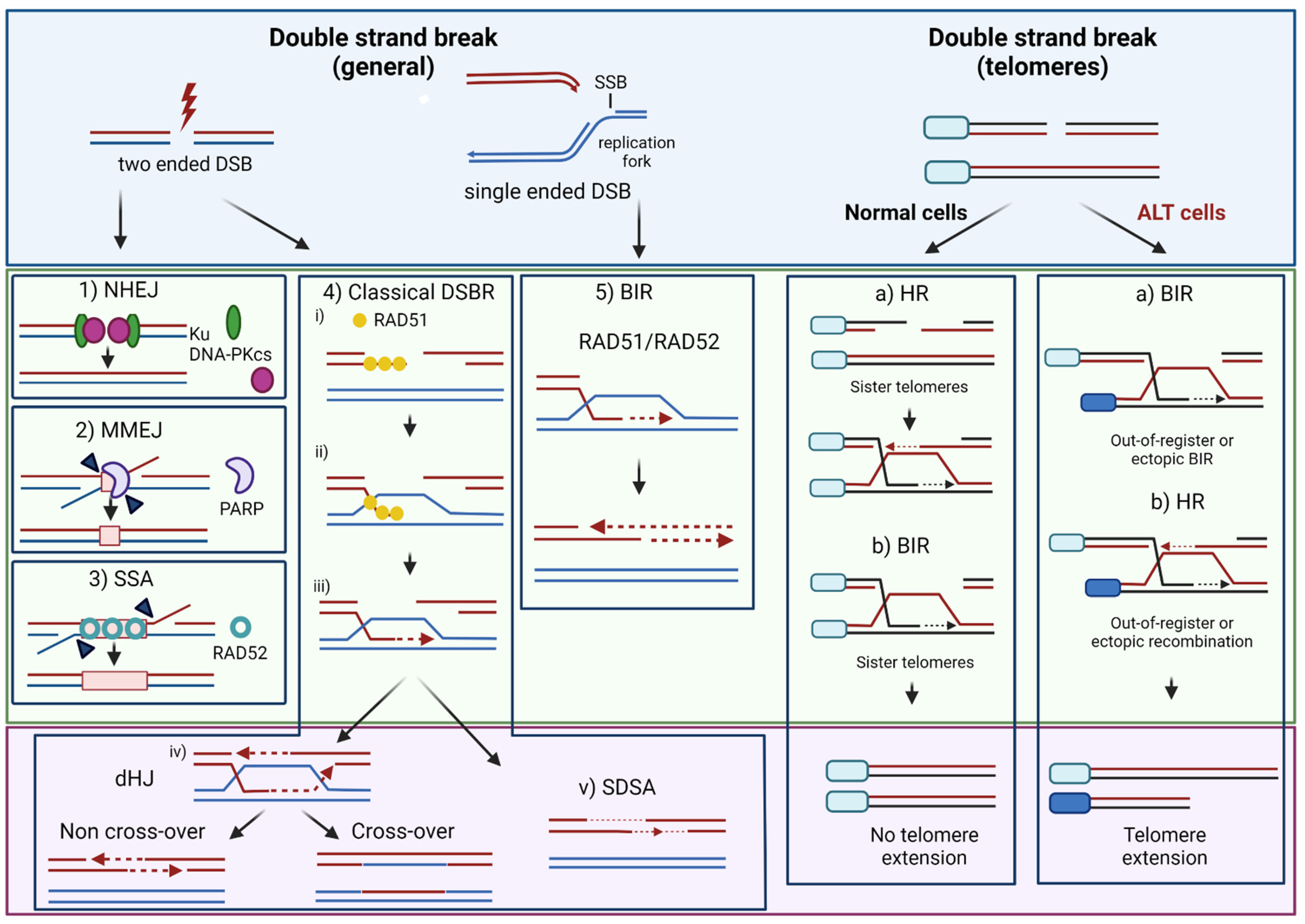
2. DNA Repair Mechanisms Promoting Resistance to IR and Alkylating Agent Therapy
3. Telomere Maintenance Mechanisms
3.1. Telomerase Reactivation
3.2. Alternative Lengthening of Telomeres (ALT)
3.3. DNA Repair at Telomeres
3.3.1. Telomere Uncapping and DSB Repair
3.3.2. Base Excision Repair
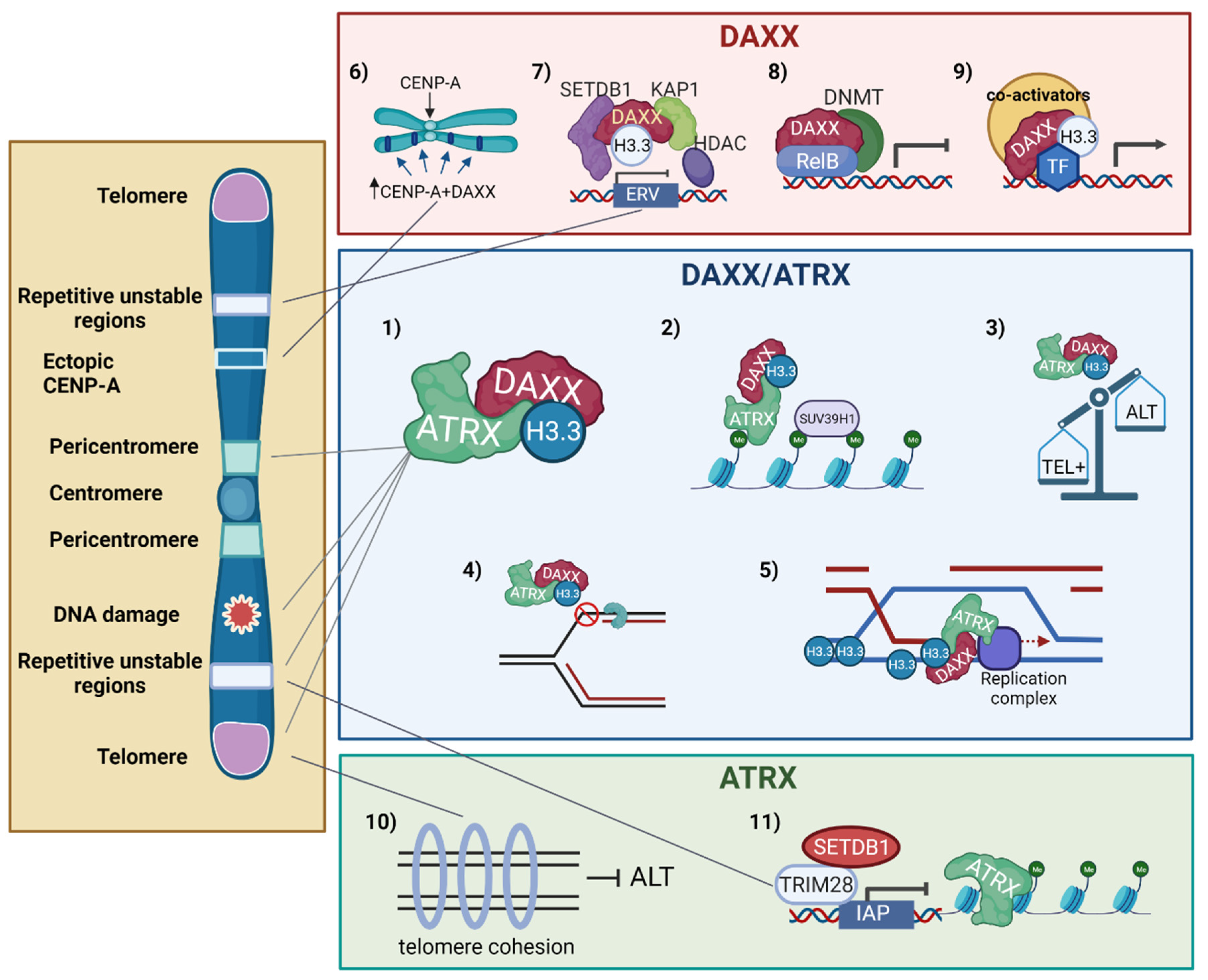
4. The ATRX/DAXX Histone H3.3 Chaperone in Chromatin Dynamics
5. Impact of Mutations in H3.3/ATRX/DAXX on DNA Repair and Telomere Dynamics
5.1. H3.3 and Its Chaperones in DNA Repair
5.2. H3.3 and Its Chaperones in Telomere Dynamics and Telomeric DNA Repair
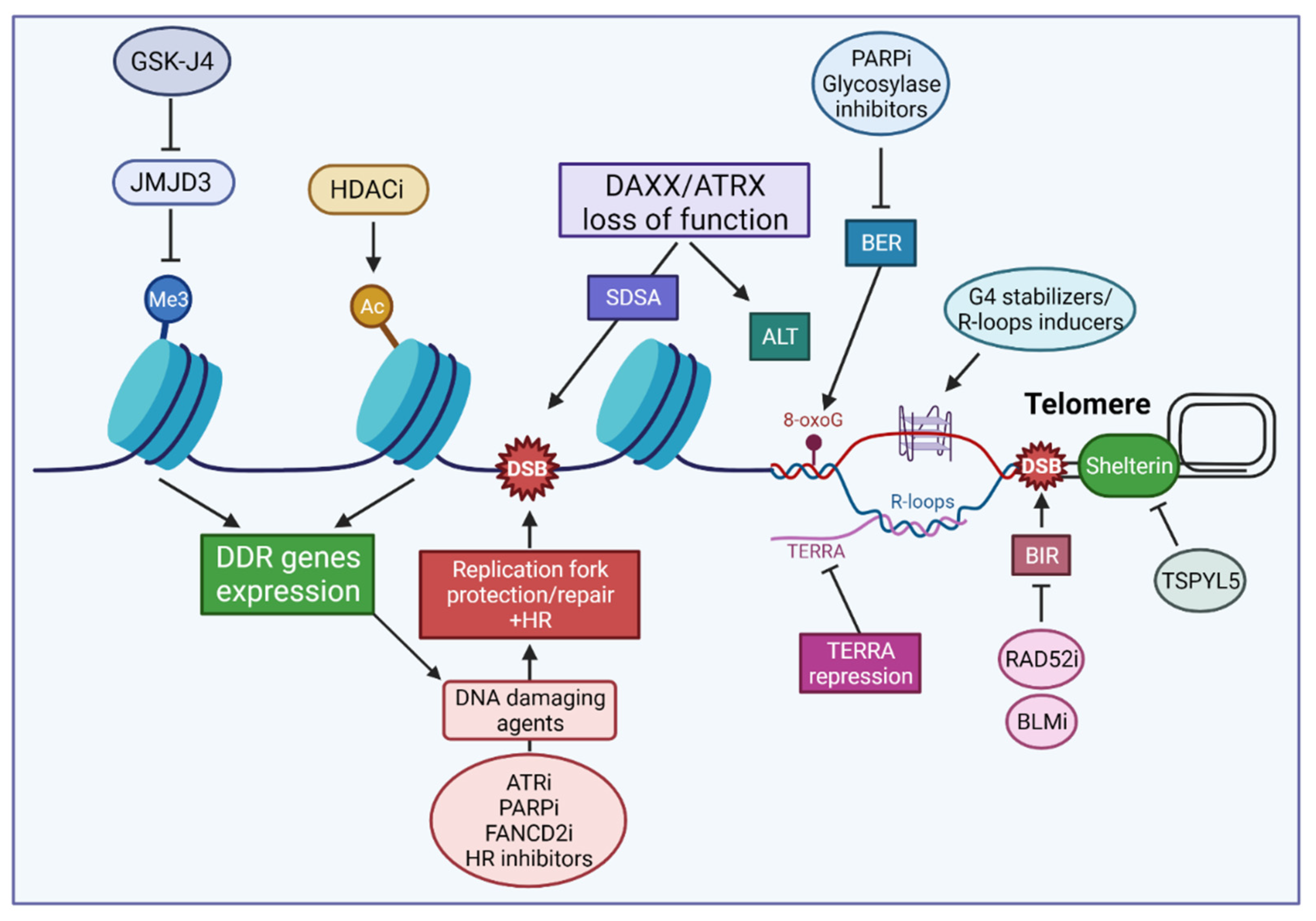
6. Strategies Targeting Chromatin Dynamics and DNA Repair in pHGGs
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Juratli, T.A.; Qin, N.; Cahill, D.P.; Filbin, M.G. Molecular pathogenesis and therapeutic implications in pediatric high-grade gliomas. Pharmacol. Ther. 2018, 182, 70–79. [Google Scholar] [CrossRef]
- Hoffman, L.M.; Veldhuijzen van Zanten, S.E.M.; Colditz, N.; Baugh, J.; Chaney, B.; Hoffmann, M.; Lane, A.; Fuller, C.; Miles, L.; Hawkins, C.; et al. Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report From the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 2018, 36, 1963–1972. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [Green Version]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef]
- Yuen, B.T.; Knoepfler, P.S. Histone H3.3 mutations: A variant path to cancer. Cancer Cell 2013, 24, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 Mutations: An Updated View of Their Role in Chromatin Deregulation and Cancer. Cancers 2019, 11, 660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, A.D.; Banaszynski, L.A.; Noh, K.M.; Lewis, P.W.; Elsaesser, S.J.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Ray-Gallet, D.; Woolfe, A.; Vassias, I.; Pellentz, C.; Lacoste, N.; Puri, A.; Schultz, D.C.; Pchelintsev, N.A.; Adams, P.D.; Jansen, L.E.; et al. Dynamics of histone H3 deposition in vivo reveal a nucleosome gap-filling mechanism for H3.3 to maintain chromatin integrity. Mol. Cell 2011, 44, 928–941. [Google Scholar] [CrossRef] [Green Version]
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagami, H.; Ray-Gallet, D.; Almouzni, G.; Nakatani, Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 2004, 116, 51–61. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [Green Version]
- Dyer, M.A.; Qadeer, Z.A.; Valle-Garcia, D.; Bernstein, E. ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harb. Perspect. Med. 2017, 7, a026567. [Google Scholar] [CrossRef]
- Grill, J.; Massimino, M.; Bouffet, E.; Azizi, A.A.; McCowage, G.; Canete, A.; Saran, F.; Le Deley, M.C.; Varlet, P.; Morgan, P.S.; et al. Phase II, Open-Label, Randomized, Multicenter Trial (HERBY) of Bevacizumab in Pediatric Patients With Newly Diagnosed High-Grade Glioma. J. Clin. Oncol. 2018, 36, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Nunez, F.M.; Gauss, J.C.; Thompson, S.; Brumley, E.; Lowenstein, P.; Castro, M.G. Hemispherical Pediatric High-Grade Glioma: Molecular Basis and Therapeutic Opportunities. Int. J. Mol. Sci. 2020, 21, 9654. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.; Stoller, S.; Grotzer, M.; Stucklin, A.G.; Nazarian, J.; Mueller, S. Pediatric hemispheric high-grade glioma: Targeting the future. Cancer Metastasis Rev. 2020, 39, 245–260. [Google Scholar] [CrossRef]
- El-Khouly, F.E.; Veldhuijzen van Zanten, S.E.M.; Santa-Maria Lopez, V.; Hendrikse, N.H.; Kaspers, G.J.L.; Loizos, G.; Sumerauer, D.; Nysom, K.; Pruunsild, K.; Pentikainen, V.; et al. Diagnostics and treatment of diffuse intrinsic pontine glioma: Where do we stand? J. Neurooncol. 2019, 145, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Aziz-Bose, R.; Monje, M. Diffuse intrinsic pontine glioma: Molecular landscape and emerging therapeutic targets. Curr. Opin. Oncol. 2019, 31, 522–530. [Google Scholar] [CrossRef]
- Metselaar, D.S.; du Chatinier, A.; Stuiver, I.; Kaspers, G.J.L.; Hulleman, E. Radiosensitization in Pediatric High-Grade Glioma: Targets, Resistance and Developments. Front. Oncol. 2021, 11, 662209. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.; Schmiegelow, K.; Hamerlik, P. Radio-Resistance and DNA Repair in Pediatric Diffuse Midline Gliomas. Cancers 2020, 12, 2813. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, R.D.; Mitchell, M.; Lindahl, T. Human DNA repair genes, 2005. Mutat. Res. 2005, 577, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Stadler, J.; Richly, H. Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. Int. J. Mol. Sci. 2017, 18, 1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trenner, A.; Sartori, A.A. Harnessing DNA Double-Strand Break Repair for Cancer Treatment. Front. Oncol. 2019, 9, 1388. [Google Scholar] [CrossRef]
- Seol, J.H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. 2018, 809, 81–87. [Google Scholar] [CrossRef]
- Patterson-Fortin, J.; D’Andrea, A.D. Exploiting the Microhomology-Mediated End-Joining Pathway in Cancer Therapy. Cancer Res. 2020, 80, 4593–4600. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannunzio, N.R.; Watanabe, G.; Lieber, M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10512–10523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasanuma, H.; Yamada, S.; Tsuda, M.; Takeda, S. Restoration of ligatable “clean” double-strand break ends is the rate-limiting step in the rejoining of ionizing-radiation-induced DNA breakage. DNA Repair 2020, 93, 102913. [Google Scholar] [CrossRef] [PubMed]
- Ronato, D.A.; Mersaoui, S.Y.; Busatto, F.F.; Affar, E.B.; Richard, S.; Masson, J.Y. Limiting the DNA Double-Strand Break Resectosome for Genome Protection. Trends Biochem. Sci. 2020, 45, 779–793. [Google Scholar] [CrossRef] [PubMed]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [Green Version]
- Van Dyck, E.; Stasiak, A.Z.; Stasiak, A.; West, S.C. Visualization of recombination intermediates produced by RAD52-mediated single-strand annealing. EMBO Rep. 2001, 2, 905–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motycka, T.A.; Bessho, T.; Post, S.M.; Sung, P.; Tomkinson, A.E. Physical and functional interaction between the XPF/ERCC1 endonuclease and hRad52. J. Biol. Chem. 2004, 279, 13634–13639. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wang, H.; Jehi, S.; Li, J.; Liu, S.; Wang, Z.; Truong, L.; Chiba, T.; Wang, Z.; Wu, X. PIF1 helicase promotes break-induced replication in mammalian cells. EMBO J. 2021, 40, e104509. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Malkova, A. Break-induced replication mechanisms in yeast and mammals. Curr. Opin. Genet. Dev. 2021, 71, 163–170. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Takai, K.; Huh, M.S.; Picketts, D.J.; de Lange, T. ATRX affects the repair of telomeric DSBs by promoting cohesion and a DAXX-dependent activity. PLoS Biol. 2020, 18, e3000594. [Google Scholar] [CrossRef] [Green Version]
- Krokan, H.E.; Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Rev. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Mitra, S. MGMT: A personal perspective. DNA Repair 2007, 6, 1064–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Sturm, D.; Bender, S.; Jones, D.T.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korshunov, A.; Capper, D.; Reuss, D.; Schrimpf, D.; Ryzhova, M.; Hovestadt, V.; Sturm, D.; Meyer, J.; Jones, C.; Zheludkova, O.; et al. Histologically distinct neuroepithelial tumors with histone 3 G34 mutation are molecularly similar and comprise a single nosologic entity. Acta Neuropathol. 2016, 131, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Lin, B.; Cowan, A.; Heinen, C.D. ATR-Chk1 activation mitigates replication stress caused by mismatch repair-dependent processing of DNA damage. Proc. Natl. Acad. Sci. USA 2018, 115, 1523–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiros, S.; Roos, W.P.; Kaina, B. Processing of O6-methylguanine into DNA double-strand breaks requires two rounds of replication whereas apoptosis is also induced in subsequent cell cycles. Cell Cycle 2010, 9, 168–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2016, 38, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Ensminger, M.; Iloff, L.; Ebel, C.; Nikolova, T.; Kaina, B.; Lbrich, M. DNA breaks and chromosomal aberrations arise when replication meets base excision repair. J. Cell Biol. 2014, 206, 29–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell 2019, 73, 398–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Gelot, C.; Magdalou, I.; Lopez, B.S. Replication stress in Mammalian cells and its consequences for mitosis. Genes 2015, 6, 267–298. [Google Scholar] [CrossRef]
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving replication fork integrity and competence via the homologous recombination pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef]
- Arnaudeau, C.; Lundin, C.; Helleday, T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J. Mol. Biol. 2001, 307, 1235–1245. [Google Scholar] [CrossRef]
- Helleday, T.; Lo, J.; van Gent, D.C.; Engelward, B.P. DNA double-strand break repair: From mechanistic understanding to cancer treatment. DNA Repair 2007, 6, 923–935. [Google Scholar] [CrossRef]
- Sotiriou, S.K.; Kamileri, I.; Lugli, N.; Evangelou, K.; Da-Re, C.; Huber, F.; Padayachy, L.; Tardy, S.; Nicati, N.L.; Barriot, S.; et al. Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol. Cell 2016, 64, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Balmus, G.; Pilger, D.; Coates, J.; Demir, M.; Sczaniecka-Clift, M.; Barros, A.C.; Woods, M.; Fu, B.; Yang, F.; Chen, E.; et al. ATM orchestrates the DNA-damage response to counter toxic non-homologous end-joining at broken replication forks. Nat. Commun. 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajesh, P.; Rajesh, C.; Wyatt, M.D.; Pittman, D.L. RAD51D protects against MLH1-dependent cytotoxic responses to O(6)-methylguanine. DNA Repair 2010, 9, 458–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Nikolova, T.; Quiros, S.; Naumann, S.C.; Kiedron, O.; Zdzienicka, M.Z.; Kaina, B. Brca2/Xrcc2 dependent HR, but not NHEJ, is required for protection against O(6)-methylguanine triggered apoptosis, DSBs and chromosomal aberrations by a process leading to SCEs. DNA Repair 2009, 8, 72–86. [Google Scholar] [CrossRef]
- Sharma, A.B.; Erasimus, H.; Pinto, L.; Caron, M.C.; Gopaul, D.; Peterlini, T.; Neumann, K.; Nazarov, P.V.; Fritah, S.; Klink, B.; et al. XAB2 promotes Ku eviction from single-ended DNA double-strand breaks independently of the ATM kinase. Nucleic Acids Res. 2021, 49, 9906–9925. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Abbotts, R.; Wilson, D.M., III. Coordination of DNA single strand break repair. Free Radic Biol. Med. 2017, 107, 228–244. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, A.; Lord, C.J. Synthetic lethal therapies for cancer: What’s next after PARP inhibitors? Nat. Rev. Clin. Oncol. 2018, 15, 564–576. [Google Scholar] [CrossRef]
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; de Lange, T. Mammalian telomeres end in a large duplex loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef] [Green Version]
- Bettin, N.; Oss Pegorar, C.; Cusanelli, E. The Emerging Roles of TERRA in Telomere Maintenance and Genome Stability. Cells 2019, 8, 246. [Google Scholar] [CrossRef] [Green Version]
- Chu, H.P.; Cifuentes-Rojas, C.; Kesner, B.; Aeby, E.; Lee, H.G.; Wei, C.; Oh, H.J.; Boukhali, M.; Haas, W.; Lee, J.T. TERRA RNA Antagonizes ATRX and Protects Telomeres. Cell 2017, 170, 86–101.e16. [Google Scholar] [CrossRef] [Green Version]
- Abedalthagafi, M.; Phillips, J.J.; Kim, G.E.; Mueller, S.; Haas-Kogen, D.A.; Marshall, R.E.; Croul, S.E.; Santi, M.R.; Cheng, J.; Zhou, S.; et al. The alternative lengthening of telomere phenotype is significantly associated with loss of ATRX expression in high-grade pediatric and adult astrocytomas: A multi-institutional study of 214 astrocytomas. Mod. Pathol. 2013, 26, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Schoeftner, S.; Blasco, M.A. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 2008, 10, 228–236. [Google Scholar] [CrossRef]
- Deng, Z.; Norseen, J.; Wiedmer, A.; Riethman, H.; Lieberman, P.M. TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol. Cell 2009, 35, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Montero, J.J.; Lopez de Silanes, I.; Grana, O.; Blasco, M.A. Telomeric RNAs are essential to maintain telomeres. Nat. Commun. 2016, 7, 12534. [Google Scholar] [CrossRef] [Green Version]
- Lopez de Silanes, I.; Grana, O.; De Bonis, M.L.; Dominguez, O.; Pisano, D.G.; Blasco, M.A. Identification of TERRA locus unveils a telomere protection role through association to nearly all chromosomes. Nat. Commun. 2014, 5, 4723. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.L.; Centore, R.C.; O’Sullivan, R.J.; Rai, R.; Tse, A.; Songyang, Z.; Chang, S.; Karlseder, J.; Zou, L. TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 2011, 471, 532–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suva, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.A.; Upton, H.E.; Vogan, J.M.; Collins, K. Telomerase Mechanism of Telomere Synthesis. Annu. Rev. Biochem. 2017, 86, 439–460. [Google Scholar] [CrossRef] [Green Version]
- Cifuentes-Rojas, C.; Shippen, D.E. Telomerase regulation. Mutat. Res. 2012, 730, 20–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W.; Wright, W.E. Senescence and immortalization: Role of telomeres and telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Boldrini, L.; Pistolesi, S.; Gisfredi, S.; Ursino, S.; Ali, G.; Pieracci, N.; Basolo, F.; Parenti, G.; Fontanini, G. Telomerase activity and hTERT mRNA expression in glial tumors. Int. J. Oncol. 2006, 28, 1555–1560. [Google Scholar] [CrossRef]
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M.; et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Diplas, B.H.; He, X.; Brosnan-Cashman, J.A.; Liu, H.; Chen, L.H.; Wang, Z.; Moure, C.J.; Killela, P.J.; Loriaux, D.B.; Lipp, E.S.; et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat. Commun. 2018, 9, 2087. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Pickett, H.A. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends Genet. 2017, 33, 921–932. [Google Scholar] [CrossRef]
- Koelsche, C.; Sahm, F.; Capper, D.; Reuss, D.; Sturm, D.; Jones, D.T.; Kool, M.; Northcott, P.A.; Wiestler, B.; Bohmer, K.; et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013, 126, 907–915. [Google Scholar] [CrossRef] [Green Version]
- Olympios, N.; Gilard, V.; Marguet, F.; Clatot, F.; Di Fiore, F.; Fontanilles, M. TERT Promoter Alterations in Glioblastoma: A Systematic Review. Cancers 2021, 13, 1147. [Google Scholar] [CrossRef] [PubMed]
- Minasi, S.; Baldi, C.; Gianno, F.; Antonelli, M.; Buccoliero, A.M.; Pietsch, T.; Massimino, M.; Buttarelli, F.R. Alternative lengthening of telomeres in molecular subgroups of paediatric high-grade glioma. Childs Nerv. Syst. 2021, 37, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.D.; Leao, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolonio, J.D.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 1801. [Google Scholar] [CrossRef]
- Castelo-Branco, P.; Choufani, S.; Mack, S.; Gallagher, D.; Zhang, C.; Lipman, T.; Zhukova, N.; Walker, E.J.; Martin, D.; Merino, D.; et al. Methylation of the TERT promoter and risk stratification of childhood brain tumours: An integrative genomic and molecular study. Lancet Oncol. 2013, 14, 534–542. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Zhang, J.M.; Zou, L. Alternative lengthening of telomeres: From molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020, 10, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, T.; Gracias, D.; Shepherd, S.; Clynes, D. Alternative Lengthening of Telomeres in Pediatric Cancer: Mechanisms to Therapies. Front. Oncol. 2019, 9, 1518. [Google Scholar] [CrossRef]
- Sommer, A.; Royle, N.J. ALT: A Multi-Faceted Phenomenon. Genes 2020, 11, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, S.M.; O’Sullivan, R.J. Alternative Lengthening of Telomeres: Building Bridges To Connect Chromosome Ends. Trends Cancer 2020, 6, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Verma, P.; Greenberg, R.A. Noncanonical views of homology-directed DNA repair. Genes Dev. 2016, 30, 1138–1154. [Google Scholar] [CrossRef] [Green Version]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Roumelioti, F.M.; Sotiriou, S.K.; Katsini, V.; Chiourea, M.; Halazonetis, T.D.; Gagos, S. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016, 17, 1731–1737. [Google Scholar] [CrossRef]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative Lengthening of Telomeres Mediated by Mitotic DNA Synthesis Engages Break-Induced Replication Processes. Mol. Cell Biol. 2017, 37, e00226-17. [Google Scholar] [CrossRef] [Green Version]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, B.; Arora, R.; Bione, S.; Azzalin, C.M. TERRA transcription destabilizes telomere integrity to initiate break-induced replication in human ALT cells. Nat. Commun. 2021, 12, 3760. [Google Scholar] [CrossRef]
- Longhese, M.P. DNA damage response at functional and dysfunctional telomeres. Genes Dev. 2008, 22, 125–140. [Google Scholar] [CrossRef] [Green Version]
- Fouquerel, E.; Parikh, D.; Opresko, P. DNA damage processing at telomeres: The ends justify the means. DNA Repair 2016, 44, 159–168. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Doksani, Y.; de Lange, T. The role of double-strand break repair pathways at functional and dysfunctional telomeres. Cold Spring Harb. Perspect. Biol. 2014, 6, a016576. [Google Scholar] [CrossRef]
- Lazzerini-Denchi, E.; Sfeir, A. Stop pulling my strings—What telomeres taught us about the DNA damage response. Nat. Rev. Mol. Cell Biol. 2016, 17, 364–378. [Google Scholar] [CrossRef]
- Rybanska-Spaeder, I.; Ghosh, R.; Franco, S. 53BP1 mediates the fusion of mammalian telomeres rendered dysfunctional by DNA-PKcs loss or inhibition. PLoS ONE 2014, 9, e108731. [Google Scholar] [CrossRef]
- Rai, R.; Zheng, H.; He, H.; Luo, Y.; Multani, A.; Carpenter, P.B.; Chang, S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010, 29, 2598–2610. [Google Scholar] [CrossRef] [Green Version]
- Mao, P.; Liu, J.; Zhang, Z.; Zhang, H.; Liu, H.; Gao, S.; Rong, Y.S.; Zhao, Y. Homologous recombination-dependent repair of telomeric DSBs in proliferating human cells. Nat. Commun. 2016, 7, 12154. [Google Scholar] [CrossRef] [Green Version]
- Cadet, J.; Wagner, J.R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, S.; Hiraku, Y.; Oikawa, S. Mechanism of guanine-specific DNA damage by oxidative stress and its role in carcinogenesis and aging. Mutat. Res. 2001, 488, 65–76. [Google Scholar] [CrossRef]
- Oikawa, S.; Kawanishi, S. Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. 1999, 453, 365–368. [Google Scholar] [CrossRef] [Green Version]
- Fouquerel, E.; Lormand, J.; Bose, A.; Lee, H.T.; Kim, G.S.; Li, J.; Sobol, R.W.; Freudenthal, B.D.; Myong, S.; Opresko, P.L. Oxidative guanine base damage regulates human telomerase activity. Nat. Struct. Mol. Biol. 2016, 23, 1092–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, A.M.; Zhou, J.; Wallace, S.S.; Burrows, C.J. A Role for the Fifth G-Track in G-Quadruplex Forming Oncogene Promoter Sequences during Oxidative Stress: Do These “Spare Tires” Have an Evolved Function? ACS Cent. Sci. 2015, 1, 226–233. [Google Scholar] [CrossRef]
- Jia, P.; Her, C.; Chai, W. DNA excision repair at telomeres. DNA Repair 2015, 36, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Dianov, G.; Lindahl, T. Reconstitution of the DNA base excision-repair pathway. Curr. Biol. 1994, 4, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Whitaker, A.M.; Schaich, M.A.; Smith, M.R.; Flynn, T.S.; Freudenthal, B.D. Base excision repair of oxidative DNA damage: From mechanism to disease. Front. Biosci. 2017, 22, 1493–1522. [Google Scholar] [CrossRef] [Green Version]
- Barnes, R.P.; Fouquerel, E.; Opresko, P.L. The impact of oxidative DNA damage and stress on telomere homeostasis. Mech. Ageing Dev. 2019, 177, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Richter, T.; von Zglinicki, T. A continuous correlation between oxidative stress and telomere shortening in fibroblasts. Exp. Gerontol. 2007, 42, 1039–1042. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Rhee, D.B.; Lu, J.; Bohr, C.T.; Zhou, F.; Vallabhaneni, H.; de Souza-Pinto, N.C.; Liu, Y. Characterization of oxidative guanine damage and repair in mammalian telomeres. PLoS Genet. 2010, 6, e1000951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Tan, R.; Xu, J.; LaFace, J.; Gao, Y.; Xiao, Y.; Attar, M.; Neumann, C.; Li, G.M.; Su, B.; et al. Targeted DNA damage at individual telomeres disrupts their integrity and triggers cell death. Nucleic Acids Res. 2015, 43, 6334–6347. [Google Scholar] [CrossRef] [PubMed]
- Fouquerel, E.; Barnes, R.P.; Uttam, S.; Watkins, S.C.; Bruchez, M.P.; Opresko, P.L. Targeted and Persistent 8-Oxoguanine Base Damage at Telomeres Promotes Telomere Loss and Crisis. Mol. Cell 2019, 75, 117–130.e6. [Google Scholar] [CrossRef]
- Zhou, J.; Chan, J.; Lambele, M.; Yusufzai, T.; Stumpff, J.; Opresko, P.L.; Thali, M.; Wallace, S.S. NEIL3 Repairs Telomere Damage during S Phase to Secure Chromosome Segregation at Mitosis. Cell Rep. 2017, 20, 2044–2056. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Fleming, A.M.; Averill, A.M.; Burrows, C.J.; Wallace, S.S. The NEIL glycosylases remove oxidized guanine lesions from telomeric and promoter quadruplex DNA structures. Nucleic Acids Res. 2015, 43, 7171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Liu, M.; Fleming, A.M.; Burrows, C.J.; Wallace, S.S. Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J. Biol. Chem. 2013, 288, 27263–27272. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.M.; Zhu, J.; Howpay Manage, S.A.; Burrows, C.J. Human NEIL3 Gene Expression Regulated by Epigenetic-Like Oxidative DNA Modification. J. Am. Chem. Soc. 2019, 141, 11036–11049. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Raja, S.; Van Houten, B. The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic Acids Res. 2020, 48, 11227–11243. [Google Scholar] [CrossRef]
- Mahmud, I.; Liao, D. DAXX in cancer: Phenomena, processes, mechanisms and regulation. Nucleic Acids Res. 2019, 47, 7734–7752. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, M.; Amato, R.; Sgura, A.; Antoccia, A.; Berardinelli, F. The Multiple Facets of ATRX Protein. Cancers 2021, 13, 2211. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Garcia-Fabiani, M.B.; Carney, S.; Altshuler, D.; Nunez, F.J.; Mendez, F.M.; Nunez, F.; Lowenstein, P.R.; Castro, M.G. Mutant ATRX: Uncovering a new therapeutic target for glioma. Expert Opin. Ther. Targets 2018, 22, 599–613. [Google Scholar] [CrossRef]
- Dhayalan, A.; Tamas, R.; Bock, I.; Tattermusch, A.; Dimitrova, E.; Kudithipudi, S.; Ragozin, S.; Jeltsch, A. The ATRX-ADD domain binds to H3 tail peptides and reads the combined methylation state of K4 and K9. Hum. Mol. Genet. 2011, 20, 2195–2203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwase, S.; Xiang, B.; Ghosh, S.; Ren, T.; Lewis, P.W.; Cochrane, J.C.; Allis, C.D.; Picketts, D.J.; Patel, D.J.; Li, H.; et al. ATRX ADD domain links an atypical histone methylation recognition mechanism to human mental-retardation syndrome. Nat. Struct. Mol. Biol. 2011, 18, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.B.; Dimitrov, S.; Hamiche, A.; Van Dyck, E. Centromeric and ectopic assembly of CENP-A chromatin in health and cancer: Old marks and new tracks. Nucleic Acids Res. 2019, 47, 1051–1069. [Google Scholar] [CrossRef]
- Barra, V.; Fachinetti, D. The dark side of centromeres: Types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 2018, 9, 4340. [Google Scholar] [CrossRef] [Green Version]
- Strom, A.R.; Biggs, R.J.; Banigan, E.J.; Wang, X.; Chiu, K.; Herman, C.; Collado, J.; Yue, F.; Ritland Politz, J.C.; Tait, L.J.; et al. HP1alpha is a chromatin crosslinker that controls nuclear and mitotic chromosome mechanics. Elife 2021, 10. [Google Scholar] [CrossRef]
- Eckert, C.A.; Gravdahl, D.J.; Megee, P.C. The enhancement of pericentromeric cohesin association by conserved kinetochore components promotes high-fidelity chromosome segregation and is sensitive to microtubule-based tension. Genes Dev. 2007, 21, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Smurova, K.; De Wulf, P. Centromere and Pericentromere Transcription: Roles and Regulation … in Sickness and in Health. Front. Genet. 2018, 9, 674. [Google Scholar] [CrossRef] [Green Version]
- Zeller, P.; Gasser, S.M. The Importance of Satellite Sequence Repression for Genome Stability. Cold Spring Harb. Symp. Quant. Biol. 2017, 82, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, C.M.; Lupski, J.R. Mechanisms underlying structural variant formation in genomic disorders. Nat. Rev. Genet. 2016, 17, 224–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Liu, B.; Jin, W.; Wu, X.; Zhou, M.; Liu, V.Z.; Goel, A.; Shen, Z.; Zheng, L.; Shen, B. hDNA2 nuclease/helicase promotes centromeric DNA replication and genome stability. EMBO J. 2018, 37, e96729. [Google Scholar] [CrossRef]
- Racca, C.; Britton, S.; Hedouin, S.; Francastel, C.; Calsou, P.; Larminat, F. BRCA1 prevents R-loop-associated centromeric instability. Cell Death Dis. 2021, 12, 896. [Google Scholar] [CrossRef]
- Tsouroula, K.; Furst, A.; Rogier, M.; Heyer, V.; Maglott-Roth, A.; Ferrand, A.; Reina-San-Martin, B.; Soutoglou, E. Temporal and Spatial Uncoupling of DNA Double Strand Break Repair Pathways within Mammalian Heterochromatin. Mol. Cell 2016, 63, 293–305. [Google Scholar] [CrossRef] [Green Version]
- Eichler, E.E. Masquerading repeats: Paralogous pitfalls of the human genome. Genome Res. 1998, 8, 758–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Q.; Kim, H.; Huang, R.; Lu, W.; Tang, M.; Shi, F.; Yang, D.; Zhang, X.; Huang, J.; Liu, D.; et al. The Daxx/Atrx Complex Protects Tandem Repetitive Elements during DNA Hypomethylation by Promoting H3K9 Trimethylation. Cell Stem Cell 2015, 17, 273–286. [Google Scholar] [CrossRef] [Green Version]
- Jang, C.W.; Shibata, Y.; Starmer, J.; Yee, D.; Magnuson, T. Histone H3.3 maintains genome integrity during mammalian development. Genes Dev. 2015, 29, 1377–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunleavy, E.M.; Almouzni, G.; Karpen, G.H. H3.3 is deposited at centromeres in S phase as a placeholder for newly assembled CENP-A in G(1) phase. Nucleus 2011, 2, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.A.; Kernohan, K.D.; Jiang, Y.; Berube, N.G. ATRX promotes gene expression by facilitating transcriptional elongation through guanine-rich coding regions. Hum. Mol. Genet. 2015, 24, 1824–1835. [Google Scholar] [CrossRef] [Green Version]
- Lacoste, N.; Woolfe, A.; Tachiwana, H.; Garea, A.V.; Barth, T.; Cantaloube, S.; Kurumizaka, H.; Imhof, A.; Almouzni, G. Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol. Cell 2014, 53, 631–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puto, L.A.; Reed, J.C. Daxx represses RelB target promoters via DNA methyltransferase recruitment and DNA hypermethylation. Genes Dev. 2008, 22, 998–1010. [Google Scholar] [CrossRef] [Green Version]
- Hollenbach, A.D.; McPherson, C.J.; Mientjes, E.J.; Iyengar, R.; Grosveld, G. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J. Cell Sci. 2002, 115, 3319–3330. [Google Scholar] [CrossRef] [PubMed]
- Boellmann, F.; Guettouche, T.; Guo, Y.; Fenna, M.; Mnayer, L.; Voellmy, R. DAXX interacts with heat shock factor 1 during stress activation and enhances its transcriptional activity. Proc. Natl. Acad. Sci. USA 2004, 101, 4100–4105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelper, D.; Huang, H.; Jain, A.Y.; Patel, D.J.; Lewis, P.W. Structural and mechanistic insights into ATRX-dependent and -independent functions of the histone chaperone DAXX. Nat. Commun. 2017, 8, 1193. [Google Scholar] [CrossRef] [Green Version]
- Wasylishen, A.R.; Sun, C.; Moyer, S.M.; Qi, Y.; Chau, G.P.; Aryal, N.K.; McAllister, F.; Kim, M.P.; Barton, M.C.; Estrella, J.S.; et al. Daxx maintains endogenous retroviral silencing and restricts cellular plasticity in vivo. Sci. Adv. 2020, 6, eaba8415. [Google Scholar] [CrossRef]
- Sadic, D.; Schmidt, K.; Groh, S.; Kondofersky, I.; Ellwart, J.; Fuchs, C.; Theis, F.J.; Schotta, G. Atrx promotes heterochromatin formation at retrotransposons. EMBO Rep. 2015, 16, 836–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, S.; Polo, S.E.; Almouzni, G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell 2013, 155, 94–106. [Google Scholar] [CrossRef] [Green Version]
- Frey, A.; Listovsky, T.; Guilbaud, G.; Sarkies, P.; Sale, J.E. Histone H3.3 is required to maintain replication fork progression after UV damage. Curr. Biol. 2014, 24, 2195–2201. [Google Scholar] [CrossRef] [Green Version]
- Huh, M.S.; Ivanochko, D.; Hashem, L.E.; Curtin, M.; Delorme, M.; Goodall, E.; Yan, K.; Picketts, D.J. Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis. 2016, 7, e2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell 2016, 61, 547–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Tyler, J.K. Nucleosome disassembly during human non-homologous end joining followed by concerted HIRA- and CAF-1-dependent reassembly. Elife 2016, 5, e15129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koschmann, C.; Lowenstein, P.R.; Castro, M.G. ATRX mutations and glioblastoma: Impaired DNA damage repair, alternative lengthening of telomeres, and genetic instability. Mol. Cell Oncol. 2016, 3, e1167158. [Google Scholar] [CrossRef] [Green Version]
- Juhasz, S.; Elbakry, A.; Mathes, A.; Lobrich, M. ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol. Cell 2018, 71, 11–24.e7. [Google Scholar] [CrossRef] [Green Version]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Lobrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [Green Version]
- Elbakry, A.; Juhasz, S.; Mathes, A.; Lobrich, M. DNA repair synthesis and histone deposition partner during homologous recombination. Mol. Cell Oncol. 2018, 5, e1511210. [Google Scholar] [CrossRef]
- Oh, B.K.; Choi, Y.; Bae, J.; Lee, W.M.; Hoh, J.K.; Choi, J.S. Increased amounts and stability of telomeric repeat-containing RNA (TERRA) following DNA damage induced by etoposide. PLoS ONE 2019, 14, e0225302. [Google Scholar] [CrossRef]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vries, N.A.; Hulsman, D.; Akhtar, W.; de Jong, J.; Miles, D.C.; Blom, M.; van Tellingen, O.; Jonkers, J.; van Lohuizen, M. Prolonged Ezh2 Depletion in Glioblastoma Causes a Robust Switch in Cell Fate Resulting in Tumor Progression. Cell Rep. 2015, 10, 383–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crea, F.; Hurt, E.M.; Farrar, W.L. Clinical significance of Polycomb gene expression in brain tumors. Mol. Cancer 2010, 9, 265. [Google Scholar] [CrossRef] [Green Version]
- Entz-Werle, N.; Poidevin, L.; Nazarov, P.V.; Poch, O.; Lhermitte, B.; Chenard, M.P.; Burckel, H.; Guerin, E.; Fuchs, Q.; Castel, D.; et al. A DNA Repair and Cell Cycle Gene Expression Signature in Pediatric High-Grade Gliomas: Prognostic and Therapeutic Value. Cancers 2021, 13, 2252. [Google Scholar] [CrossRef]
- Delaney, K.; Strobino, M.; Wenda, J.M.; Pankowski, A.; Steiner, F.A. H3.3K27M-induced chromatin changes drive ectopic replication through misregulation of the JNK pathway in C. elegans. Nat. Commun. 2019, 10, 2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitanaka, C.; Sato, A.; Okada, M. JNK Signaling in the Control of the Tumor-Initiating Capacity Associated with Cancer Stem Cells. Genes Cancer 2013, 4, 388–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, M.; Sato, A.; Shibuya, K.; Watanabe, E.; Seino, S.; Suzuki, S.; Seino, M.; Narita, Y.; Shibui, S.; Kayama, T.; et al. JNK contributes to temozolomide resistance of stem-like glioblastoma cells via regulation of MGMT expression. Int. J. Oncol. 2014, 44, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, K.; Sato, A.; Okada, M.; Shibuya, K.; Seino, S.; Suzuki, K.; Watanabe, E.; Narita, Y.; Shibui, S.; Kayama, T.; et al. Targeting JNK for therapeutic depletion of stem-like glioblastoma cells. Sci. Rep. 2012, 2, 516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Huang, Y.; Mao, G.; Yang, S.; Rennert, G.; Gu, L.; Li, H.; Li, G.M. Cancer-driving H3G34V/R/D mutations block H3K36 methylation and H3K36me3-MutSalpha interaction. Proc. Natl. Acad. Sci. USA 2018, 115, 9598–9603. [Google Scholar] [CrossRef] [Green Version]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, C.C.; Deegan, R.S.; Subramanian, L.; Gal, C.; Sarkar, S.; Blaikley, E.J.; Walker, C.; Hulme, L.; Bernhard, E.; Codlin, S.; et al. A histone H3K36 chromatin switch coordinates DNA double-strand break repair pathway choice. Nat. Commun. 2014, 5, 4091. [Google Scholar] [CrossRef] [Green Version]
- Jha, D.K.; Strahl, B.D. An RNA polymerase II-coupled function for histone H3K36 methylation in checkpoint activation and DSB repair. Nat. Commun. 2014, 5, 3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, R.K.; Jablonowski, C.M.; Fernandez, A.G.; Lowe, B.R.; Henry, R.A.; Finkelstein, D.; Barnum, K.J.; Pidoux, A.L.; Kuo, Y.M.; Huang, J.; et al. Histone H3G34R mutation causes replication stress, homologous recombination defects and genomic instability in S. pombe. Elife 2017, 6, e27406. [Google Scholar] [CrossRef]
- Lowe, B.R.; Yadav, R.K.; Henry, R.A.; Schreiner, P.; Matsuda, A.; Fernandez, A.G.; Finkelstein, D.; Campbell, M.; Kallappagoudar, S.; Jablonowski, C.M.; et al. Surprising phenotypic diversity of cancer-associated mutations of Gly 34 in the histone H3 tail. Elife 2021, 10, e65369. [Google Scholar] [CrossRef]
- Niraj, J.; Farkkila, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef]
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef]
- Yost, K.E.; Clatterbuck Soper, S.F.; Walker, R.L.; Pineda, M.A.; Zhu, Y.J.; Ester, C.D.; Showman, S.; Roschke, A.V.; Waterfall, J.J.; Meltzer, P.S. Rapid and reversible suppression of ALT by DAXX in osteosarcoma cells. Sci. Rep. 2019, 9, 4544. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, R.J.; Arnoult, N.; Lackner, D.H.; Oganesian, L.; Haggblom, C.; Corpet, A.; Almouzni, G.; Karlseder, J. Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat. Struct. Mol. Biol. 2014, 21, 167–174. [Google Scholar] [CrossRef]
- Fan, Q.; Zhang, F.; Barrett, B.; Ren, K.; Andreassen, P.R. A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 2009, 37, 1740–1754. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Xiang, T.; Pandita, T.K.; Gonzalez-Suarez, I.; Gonzalo, S.; Harris, C.C.; Yang, Q. Telomere recombination requires the MUS81 endonuclease. Nat. Cell Biol. 2009, 11, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, J.W.; Ghosal, G.; Wang, W.; Shen, X.; Wang, J.; Li, L.; Chen, J. Alpha thalassemia/mental retardation syndrome X-linked gene product ATRX is required for proper replication restart and cellular resistance to replication stress. J. Biol. Chem. 2013, 288, 6342–6350. [Google Scholar] [CrossRef] [Green Version]
- Lerner, L.K.; Sale, J.E. Replication of G Quadruplex DNA. Genes 2019, 10, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, M.J.; Lower, K.M.; Voon, H.P.; Hughes, J.R.; Garrick, D.; Viprakasit, V.; Mitson, M.; De Gobbi, M.; Marra, M.; Morris, A.; et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 2010, 143, 367–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Y.C.; Sundaresan, A.; O’Hara, R.; Gant, V.U.; Li, M.; Martire, S.; Warshaw, J.N.; Basu, A.; Banaszynski, L.A. ATRX promotes heterochromatin formation to protect cells from G-quadruplex DNA-mediated stress. Nat. Commun. 2021, 12, 3887. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Chang, E.Y.C.; Lim, J.; Kwan, H.H.; Monchaud, D.; Yip, S.; Stirling, P.C.; Wong, J.M.Y. G-quadruplexes mark alternative lengthening of telomeres. NAR Cancer 2021, 3, zcab031. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.; Voon, H.P.J.; Xella, B.; Scott, C.; Clynes, D.; Babbs, C.; Ayyub, H.; Kerry, J.; Sharpe, J.A.; Sloane-Stanley, J.A.; et al. The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 2017, 18, 914–928. [Google Scholar] [CrossRef]
- Arora, R.; Lee, Y.; Wischnewski, H.; Brun, C.M.; Schwarz, T.; Azzalin, C.M. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat. Commun. 2014, 5, 5220. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Deng, Z.; Zhang, L.; Wu, C.; Jin, Y.; Hwang, I.; Vladimirova, O.; Xu, L.; Yang, L.; Lu, B.; et al. ATRX loss induces telomere dysfunction and necessitates induction of alternative lengthening of telomeres during human cell immortalization. EMBO J. 2019, 38, e96659. [Google Scholar] [CrossRef]
- Tang, M.; Li, Y.; Zhang, Y.; Chen, Y.; Huang, W.; Wang, D.; Zaug, A.J.; Liu, D.; Zhao, Y.; Cech, T.R.; et al. Disease mutant analysis identifies a new function of DAXX in telomerase regulation and telomere maintenance. J. Cell Sci. 2015, 128, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.P.; Figueroa, M.; Mohiuddin, S.; Zaky, W.; Chandra, J. Cutting Edge Therapeutic Insights Derived from Molecular Biology of Pediatric High-Grade Glioma and Diffuse Intrinsic Pontine Glioma (DIPG). Bioengineering 2018, 5, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.J.; Singleton, W.G.; Lowis, S.P.; Malik, K.; Kurian, K.M. Therapeutic Targeting of Histone Modifications in Adult and Pediatric High-Grade Glioma. Front. Oncol. 2017, 7, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathania, M.; De Jay, N.; Maestro, N.; Harutyunyan, A.S.; Nitarska, J.; Pahlavan, P.; Henderson, S.; Mikael, L.G.; Richard-Londt, A.; Zhang, Y.; et al. H3.3(K27M) Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 2017, 32, 684–700.e9. [Google Scholar] [CrossRef] [Green Version]
- Lange, A.M.; Lo, H.W. Inhibiting TRK Proteins in Clinical Cancer Therapy. Cancers 2018, 10, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leszczynska, K.B.; Jayaprakash, C.; Kaminska, B.; Mieczkowski, J. Emerging Advances in Combinatorial Treatments of Epigenetically Altered Pediatric High-Grade H3K27M Gliomas. Front. Genet. 2021, 12, 742561. [Google Scholar] [CrossRef]
- Hashizume, R.; Andor, N.; Ihara, Y.; Lerner, R.; Gan, H.; Chen, X.; Fang, D.; Huang, X.; Tom, M.W.; Ngo, V.; et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nat. Med. 2014, 20, 1394–1396. [Google Scholar] [CrossRef]
- Katagi, H.; Louis, N.; Unruh, D.; Sasaki, T.; He, X.; Zhang, A.; Ma, Q.; Piunti, A.; Shimazu, Y.; Lamano, J.B.; et al. Radiosensitization by Histone H3 Demethylase Inhibition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2019, 25, 5572–5583. [Google Scholar] [CrossRef]
- Meel, M.H.; de Gooijer, M.C.; Metselaar, D.S.; Sewing, A.C.P.; Zwaan, K.; Waranecki, P.; Breur, M.; Buil, L.C.M.; Lagerweij, T.; Wedekind, L.E.; et al. Combined Therapy of AXL and HDAC Inhibition Reverses Mesenchymal Transition in Diffuse Intrinsic Pontine Glioma. Clin. Cancer Res. 2020, 26, 3319–3332. [Google Scholar] [CrossRef] [Green Version]
- Adimoolam, S.; Sirisawad, M.; Chen, J.; Thiemann, P.; Ford, J.M.; Buggy, J.J. HDAC inhibitor PCI-24781 decreases RAD51 expression and inhibits homologous recombination. Proc. Natl. Acad. Sci. USA 2007, 104, 19482–19487. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [Green Version]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [Green Version]
- Mu, M.D.; Qian, Z.M.; Yang, S.X.; Rong, K.L.; Yung, W.H.; Ke, Y. Therapeutic effect of a histone demethylase inhibitor in Parkinson’s disease. Cell Death Dis. 2020, 11, 927. [Google Scholar] [CrossRef] [PubMed]
- Mansor, N.I.; Nordin, N.; Mohamed, F.; Ling, K.H.; Rosli, R.; Hassan, Z. Crossing the Blood-Brain Barrier: A Review on Drug Delivery Strategies for Treatment of the Central Nervous System Diseases. Curr. Drug Deliv. 2019, 16, 698–711. [Google Scholar] [CrossRef]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Stine, C.A.; Munson, J.M. Convection-Enhanced Delivery: Connection to and Impact of Interstitial Fluid Flow. Front. Oncol. 2019, 9, 966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knijnenburg, T.A.; Wang, L.; Zimmermann, M.T.; Chambwe, N.; Gao, G.F.; Cherniack, A.D.; Fan, H.; Shen, H.; Way, G.P.; Greene, C.S.; et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell Rep. 2018, 23, 239–254.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, R.D.; Gajjar, M.K.; Tuckova, L.; Jensen, K.E.; Maya-Mendoza, A.; Holst, C.B.; Mollgaard, K.; Rasmussen, J.S.; Brennum, J.; Bartek, J., Jr.; et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat. Commun. 2016, 7, 13398. [Google Scholar] [CrossRef]
- Toledo, L.I.; Murga, M.; Zur, R.; Soria, R.; Rodriguez, A.; Martinez, S.; Oyarzabal, J.; Pastor, J.; Bischoff, J.R.; Fernandez-Capetillo, O. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 2011, 18, 721–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietlein, F.; Thelen, L.; Reinhardt, H.C. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014, 30, 326–339. [Google Scholar] [CrossRef]
- Chan, D.A.; Giaccia, A.J. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat. Rev. Drug Discov. 2011, 10, 351–364. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.S.; O’Carrigan, B.; Jackson, S.P.; Yap, T.A. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discov. 2017, 7, 20–37. [Google Scholar] [CrossRef] [Green Version]
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2020, 11, 629266. [Google Scholar] [CrossRef] [PubMed]
- Hengel, S.R.; Spies, M.A.; Spies, M. Small-Molecule Inhibitors Targeting DNA Repair and DNA Repair Deficiency in Research and Cancer Therapy. Cell Chem. Biol. 2017, 24, 1101–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourley, C.; Balmana, J.; Ledermann, J.A.; Serra, V.; Dent, R.; Loibl, S.; Pujade-Lauraine, E.; Boulton, S.J. Moving From Poly (ADP-Ribose) Polymerase Inhibition to Targeting DNA Repair and DNA Damage Response in Cancer Therapy. J. Clin. Oncol. 2019, 37, 2257–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toma, M.; Sullivan-Reed, K.; Sliwinski, T.; Skorski, T. RAD52 as a Potential Target for Synthetic Lethality-Based Anticancer Therapies. Cancers 2019, 11, 1561. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.M.; Aguirre, A.J.; Shapiro, G.I.; D’Andrea, A.D. Biomarker-Guided Development of DNA Repair Inhibitors. Mol. Cell 2020, 78, 1070–1085. [Google Scholar] [CrossRef]
- Carruthers, R.D.; Ahmed, S.U.; Ramachandran, S.; Strathdee, K.; Kurian, K.M.; Hedley, A.; Gomez-Roman, N.; Kalna, G.; Neilson, M.; Gilmour, L.; et al. Replication Stress Drives Constitutive Activation of the DNA Damage Response and Radioresistance in Glioblastoma Stem-like Cells. Cancer Res. 2018, 78, 5060–5071. [Google Scholar] [CrossRef] [Green Version]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Metselaar, D.S.; Meel, M.H.; Benedict, B.; Waranecki, P.; Koster, J.; Kaspers, G.J.L.; Hulleman, E. Celastrol-induced degradation of FANCD2 sensitizes pediatric high-grade gliomas to the DNA-crosslinking agent carboplatin. eBioMedicine 2019, 50, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Schlacher, K.; Wu, H.; Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 2012, 22, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Van Vuurden, D.G.; Hulleman, E.; Meijer, O.L.; Wedekind, L.E.; Kool, M.; Witt, H.; Vandertop, P.W.; Wurdinger, T.; Noske, D.P.; Kaspers, G.J.; et al. PARP inhibition sensitizes childhood high grade glioma, medulloblastoma and ependymoma to radiation. Oncotarget 2011, 2, 984–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, B.; Kline, C.; Mueller, S. Radiation in Combination With Targeted Agents and Immunotherapies for Pediatric Central Nervous System Tumors—Progress, Opportunities, and Challenges. Front. Oncol. 2021, 11, 674596. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.; Smith, R.; Cahill, D.P.; Stephens, P.; Stevens, C.; Teague, J.; Greenman, C.; Edkins, S.; Bignell, G.; Davies, H.; et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 2006, 66, 3987–3991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, D.P.; Levine, K.K.; Betensky, R.A.; Codd, P.J.; Romany, C.A.; Reavie, L.B.; Batchelor, T.T.; Futreal, P.A.; Stratton, M.R.; Curry, W.T.; et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 2007, 13, 2038–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, S.; Miao, J.; Cahill, D.P.; Iafrate, A.J.; Aldape, K.; Nutt, C.L.; Louis, D.N. MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin. Cancer Res. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, F.; Nagashima, H.; Ning, J.; Koerner, M.V.A.; Wakimoto, H.; Cahill, D.P. Restoration of Temozolomide Sensitivity by PARP Inhibitors in Mismatch Repair Deficient Glioblastoma is Independent of Base Excision Repair. Clin. Cancer Res. 2020, 26, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Elbakry, A.; Juhasz, S.; Chan, K.C.; Lobrich, M. ATRX and RECQ5 define distinct homologous recombination subpathways. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Elbakry, A.; Lobrich, M. Homologous Recombination Subpathways: A Tangle to Resolve. Front. Genet. 2021, 12, 723847. [Google Scholar] [CrossRef]
- Fan, H.C.; Chen, C.M.; Chi, C.S.; Tsai, J.D.; Chiang, K.L.; Chang, Y.K.; Lin, S.Z.; Harn, H.J. Targeting Telomerase and ATRX/DAXX Inducing Tumor Senescence and Apoptosis in the Malignant Glioma. Int. J. Mol. Sci. 2019, 20, 200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asamitsu, S.; Obata, S.; Yu, Z.; Bando, T.; Sugiyama, H. Recent Progress of Targeted G-Quadruplex-Preferred Ligands Toward Cancer Therapy. Molecules 2019, 24, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, G.J.; Parsons, J.L. Base excision repair and its implications to cancer therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar] [CrossRef]
- Mechetin, G.V.; Endutkin, A.V.; Diatlova, E.A.; Zharkov, D.O. Inhibitors of DNA Glycosylases as Prospective Drugs. Int. J. Mol. Sci. 2020, 21, 3118. [Google Scholar] [CrossRef] [PubMed]
- Episkopou, H.; Diman, A.; Claude, E.; Viceconte, N.; Decottignies, A. TSPYL5 Depletion Induces Specific Death of ALT Cells through USP7-Dependent Proteasomal Degradation of POT1. Mol. Cell 2019, 75, 469–482.e6. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Lok, B.H.; Carley, A.C.; Tchang, B.; Powell, S.N. RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene 2013, 32, 3552–3558. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol. Cell Biol. 2013, 33, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, S.; Oaks, J.; Ren, J.; Li, L.; Wu, X. The concerted roles of FANCM and Rad52 in the protection of common fragile sites. Nat. Commun. 2018, 9, 2791. [Google Scholar] [CrossRef]
- Yang, Q.; Li, Y.; Sun, R.; Li, J. Identification of a RAD52 Inhibitor Inducing Synthetic Lethality in BRCA2-Deficient Cancer Cells. Front. Pharmacol. 2021, 12, 637825. [Google Scholar] [CrossRef]
- Gobin, M.; Nazarov, P.V.; Warta, R.; Timmer, M.; Reifenberger, G.; Felsberg, J.; Vallar, L.; Chalmers, A.J.; Herold-Mende, C.C.; Goldbrunner, R.; et al. A DNA Repair and Cell-Cycle Gene Expression Signature in Primary and Recurrent Glioblastoma: Prognostic Value and Clinical Implications. Cancer Res. 2019, 79, 1226–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Werbrouck, C.; Evangelista, C.C.S.; Lobon-Iglesias, M.J.; Barret, E.; Le Teuff, G.; Merlevede, J.; Brusini, R.; Kergrohen, T.; Mondini, M.; Bolle, S.; et al. TP53 Pathway Alterations Drive Radioresistance in Diffuse Intrinsic Pontine Gliomas (DIPG). Clin. Cancer Res. 2019, 25, 6788–6800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Meyer, D.H.; Schumacher, B. H3K4me2 regulates the recovery of protein biosynthesis and homeostasis following DNA damage. Nat. Struct. Mol. Biol. 2020, 27, 1165–1177. [Google Scholar] [CrossRef] [PubMed]
- Chornenkyy, Y.; Agnihotri, S.; Yu, M.; Buczkowicz, P.; Rakopoulos, P.; Golbourn, B.; Garzia, L.; Siddaway, R.; Leung, S.; Rutka, J.T.; et al. Poly-ADP-Ribose Polymerase as a Therapeutic Target in Pediatric Diffuse Intrinsic Pontine Glioma and Pediatric High-Grade Astrocytoma. Mol. Cancer Ther. 2015, 14, 2560–2568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Xu, K.; Zhu, X.; Dunphy, P.S.; Gudenas, B.; Lin, W.; Twarog, N.; Hover, L.D.; Kwon, C.H.; Kasper, L.H.; et al. Patient-derived models recapitulate heterogeneity of molecular signatures and drug response in pediatric high-grade glioma. Nat. Commun. 2021, 12, 4089. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, L.; Baidarjad, H.; Entz-Werlé, N.; Van Dyck, E. Impact of Chromatin Dynamics and DNA Repair on Genomic Stability and Treatment Resistance in Pediatric High-Grade Gliomas. Cancers 2021, 13, 5678. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225678
Pinto L, Baidarjad H, Entz-Werlé N, Van Dyck E. Impact of Chromatin Dynamics and DNA Repair on Genomic Stability and Treatment Resistance in Pediatric High-Grade Gliomas. Cancers. 2021; 13(22):5678. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225678
Chicago/Turabian StylePinto, Lia, Hanane Baidarjad, Natacha Entz-Werlé, and Eric Van Dyck. 2021. "Impact of Chromatin Dynamics and DNA Repair on Genomic Stability and Treatment Resistance in Pediatric High-Grade Gliomas" Cancers 13, no. 22: 5678. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225678