
The Hepatic Sinusoid in Chronic Liver Disease: The Optimal Milieu for Cancer

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Cells from the Hepatic Sinusoid
2.1. Liver Sinusoidal Endothelial Cells
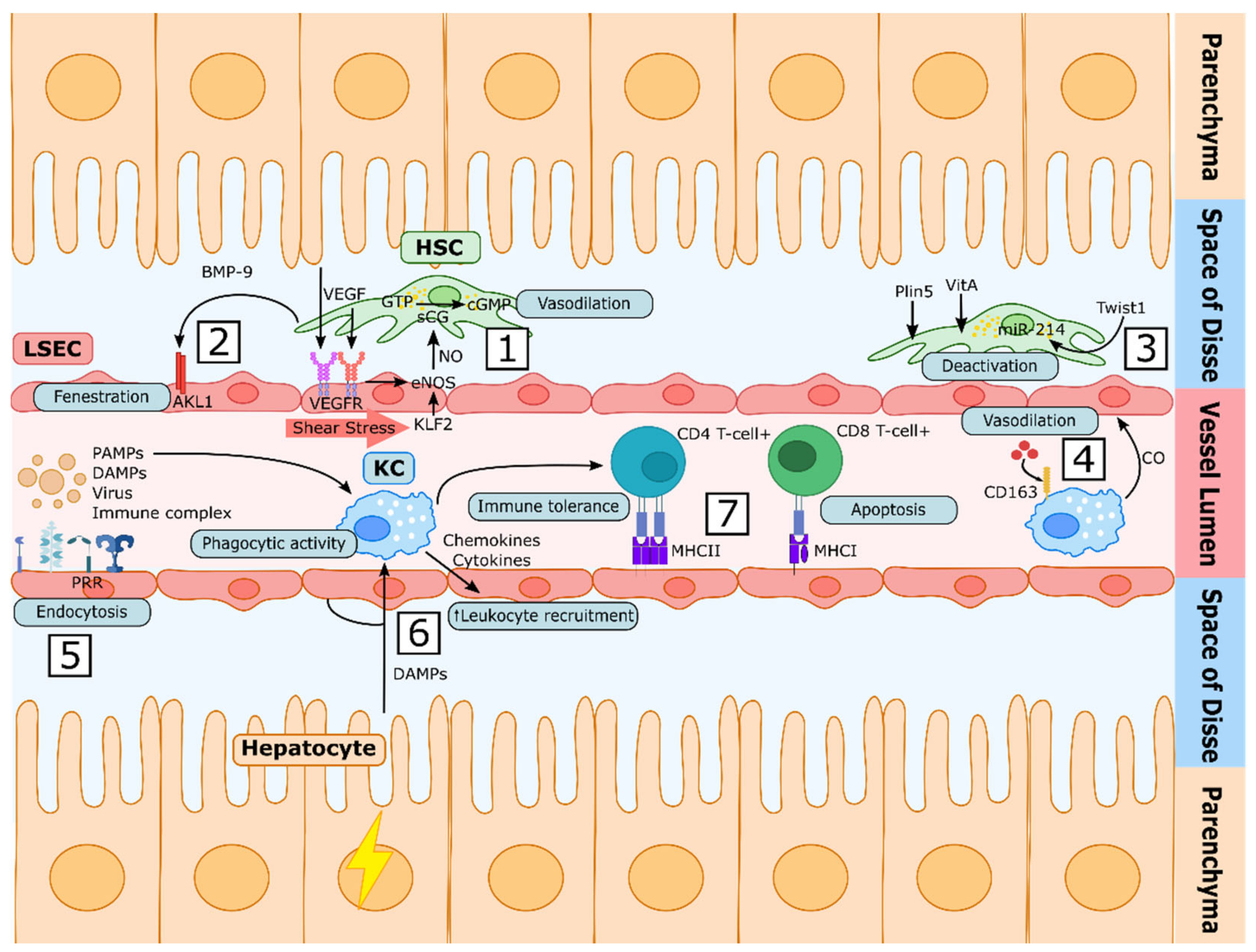
2.1.1. LSEC Functions
Sieving Function
Modulation of Vascular Tone
Endocytic Capacity
Immune Hepatic Tolerance
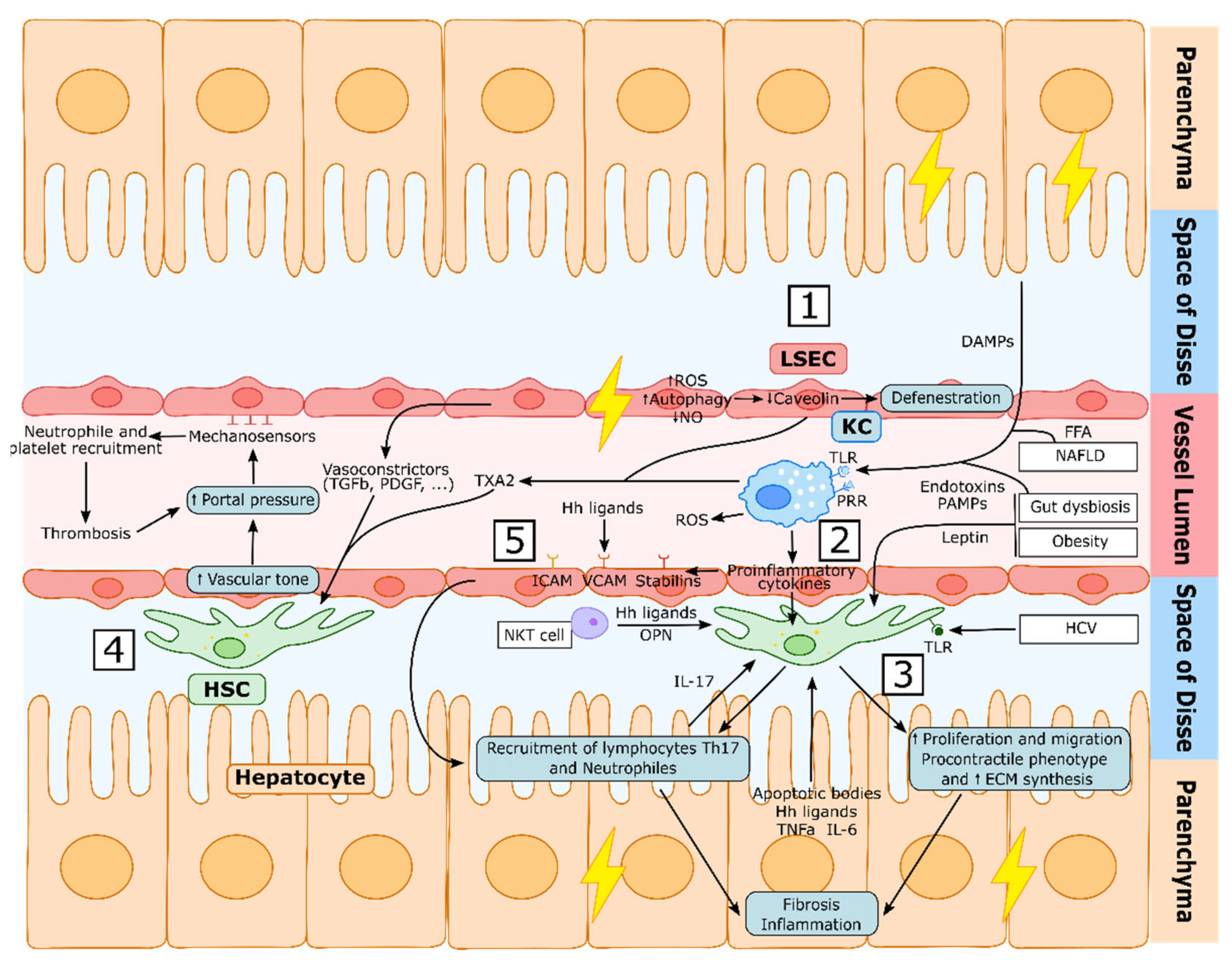
2.1.2. LSEC Capillarization in Liver Injury
2.2. Hepatic Stellate Cells
2.2.1. HSC Functions
Vitamin A Storage and Metabolism
Immunoregulation
HSC Activation in Liver Injury
2.3. Kupffer Cells
2.3.1. Kupffer Cells’ Functions
2.3.2. Kupffer Cells in CLD
3. Cellular Communication in the Liver Sinusoid
Sinusoidal Communication in CLD
4. Therapeutic Approaches for Chronic Liver Disease
4.1. Vasomodulation
4.1.1. Inhibition of Vasoconstriction
4.1.2. Induction of Vasodilation
4.1.3. Targeting other Vascular Alterations
4.2. Cell Death and Inflammation
4.3. Strategies Targeting Fibrogenesis
4.4. Lifestyle and Dietary Interventions
4.4.1. Microbiota
4.4.2. Diet and Nutraceuticals
4.5. Sinusoidal Cell-Targeted Therapies
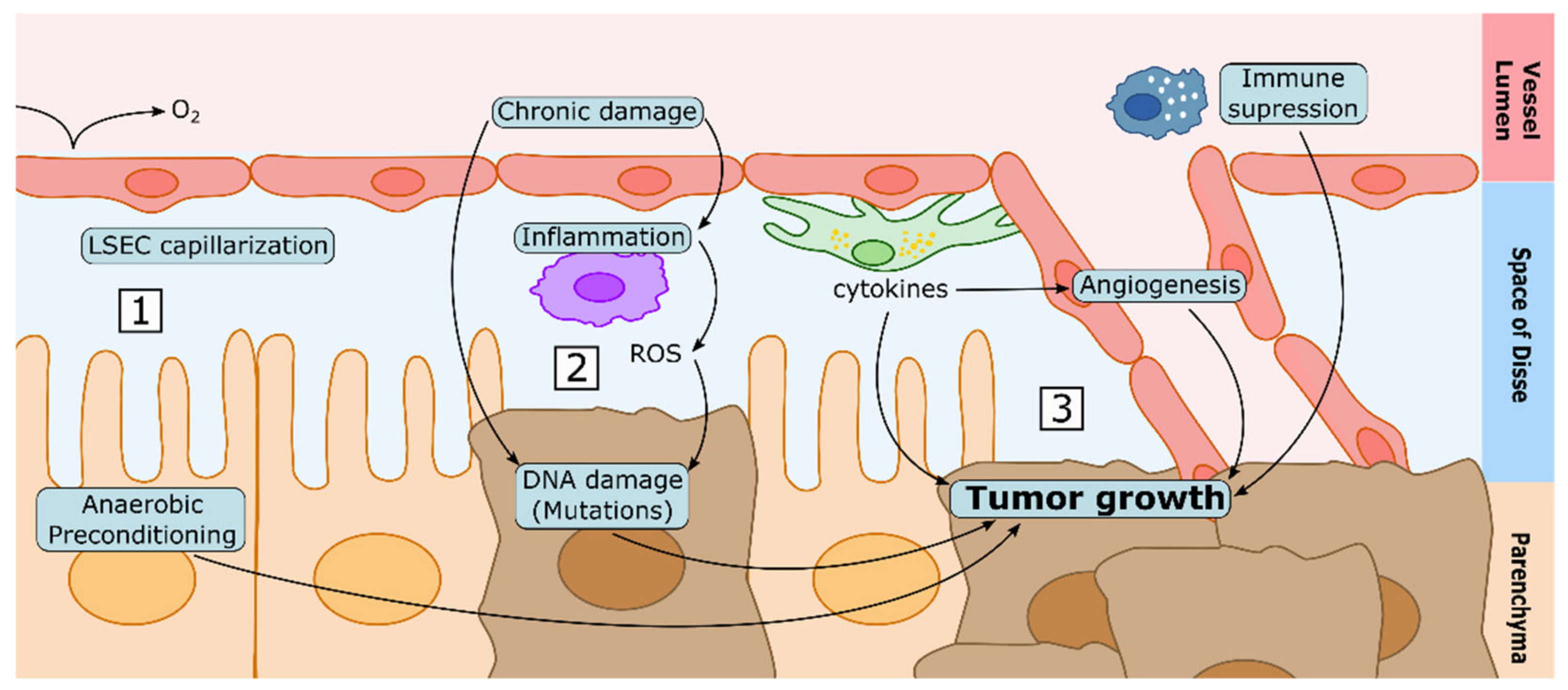
5. CLD and the Sinusoidal Microenvironment in the Development of HCC
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McCuskey, R.S.S. The hepatic microvascular system in health and its response to toxicants. Anat. Rec. 2008, 291, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Wisse, E.; de Zanger, R.B.; Charels, K.; van der Smissen, P.; McCuskey, R.S. The liver sieve: Considerations concerning the structure and function of endothelial fenestrae, the sinusoidal wall and the space of disse. Hepatology 1985, 5, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Parola, M.; Pinzani, M. Liver fibrosis: Pathophysiology, pathogenetic targets and clinical issues. Mol. Aspects Med. 2019, 65, 37–55. [Google Scholar] [CrossRef] [PubMed]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Craig, A.J.; von Felden, J.; Garcia-Lezana, T.; Sarcognato, S.; Villanueva, A. Tumour evolution in hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Groszmann, R.J.; Shah, V.H. Evolution in the understanding of the pathophysiological basis of portal hypertension: How changes in paradigm are leading to successful new treatments. J. Hepatol. 2015, 62, S121–S130. [Google Scholar] [CrossRef] [Green Version]
- Møller, S.; Bendtsen, F. The pathophysiology of arterial vasodilatation and hyperdynamic circulation in cirrhosis. Liver Int. 2018, 38, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Gracia-Sancho, J.; Marrone, G.; Fernández-Iglesias, A. Hepatic microcirculation and mechanisms of portal hypertension. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 221–234. [Google Scholar] [CrossRef]
- Braet, F.; Wisse, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 1, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeve, D.L.; Garcia-Tsao, G. Vascular Liver Disease; Springer: New York, NY, USA, 2011; pp. 3–40. [Google Scholar]
- Wisse, E. An ultrastructural characterization of the endothelial cell in the rat liver sinusoid under normal and various experimental conditions, as a contribution to the distinction between endothelial and Kupffer cells. J. Ultrastruct. Res. 1972, 38, 528–562. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Maeso-Díaz, R.; Fernández-Iglesias, A.; Navarro-Zornoza, M.; Bosch, J. New cellular and molecular targets for the treatment of portal hypertension. Hepatol. Int. 2015, 9, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, K.K.; Simon-Santamaria, J.; McCuskey, R.S.; Smedsrød, B. Liver Sinusoidal Endothelial Cells. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; Volume 5, pp. 1751–1774. [Google Scholar]
- Gracia-Sancho, J.; Caparrós, E.; Fernández-Iglesias, A.; Francés, R. Role of liver sinusoidal endothelial cells in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 411–431. [Google Scholar] [CrossRef]
- Mönkemöller, V.; Øie, C.; Hübner, W.; Huser, T.; McCourt, P. Multimodal super-resolution optical microscopy visualizes the close connection between membrane and the cytoskeleton in liver sinusoidal endothelial cell fenestrations. Sci. Rep. 2015, 5, 16279. [Google Scholar] [CrossRef]
- Wisse, E.; Jacobs, F.; Topal, B.; Frederik, P.; De Geest, B. The size of endothelial fenestrae in human liver sinusoids: Implications for hepatocyte-directed gene transfer. Gene Ther. 2008, 15, 1193–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal-Vanaclocha, F.; Barberá-Guillem, E. Fenestration patterns in endothelial cells of rat liver sinusoids. J. Ultrastruct. Res. Mol. Struct. Res. 1985, 90, 115–123. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Ji, S.; Thurman, R.G. Centrilobular injury following hypoxia in isolated, perfused rat liver. Science 1981, 213, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; Roessner, U.; Warren, A.; Fraser, R.; Le Couteur, D.G. A sieve-raft hypothesis for the regulation of endothelial fenestrations. Comput. Struct. Biotechnol. J. 2013, 8, e201308003. [Google Scholar] [CrossRef] [Green Version]
- DeLeve, L.D. Liver sinusoidal endothelial cells in hepatic fibrosis. Hepatology 2015, 61, 1740–1746. [Google Scholar] [CrossRef] [Green Version]
- Miyao, M.; Kotani, H.; Ishida, T.; Kawai, C.; Manabe, S.; Abiru, H.; Tamaki, K. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab. Investig. 2015, 95, 1130–1144. [Google Scholar] [CrossRef] [Green Version]
- Pasarín, M.; La Mura, V.; Gracia-Sancho, J.; García-Calderó, H.; Rodríguez-Vilarrupla, A.; García-Pagán, J.C.; Bosch, J.; Abraldes, J.G. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PLoS ONE 2012, 7, e32785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manicardi, N.; Fernández-Iglesias, A.; Abad-Jordà, L.; Royo, F.; Azkargorta, M.; Ortega-Ribera, M.; Sanfeliu-Redondo, D.; Martínez-Alcocer, A.; Elortza, F.; Hessheimer, A.J.; et al. Transcriptomic profiling of the liver sinusoidal endothelium during cirrhosis reveals stage-specific secretory signature. Cancers 2021, 13, 2688. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Choi, S.S.; Syn, W.-K.; Michelotti, G.A.; Swiderska, M.; Karaca, G.; Chan, I.S.; Chen, Y.; Diehl, A.M. Hedgehog signalling regulates liver sinusoidal endothelial cell capillarisation. Gut 2013, 62, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gu, T.; Li, B.; Li, F.; Ma, Z.; Zhang, Q.; Cai, X.; Lu, L. Delta-like ligand 4/DLL4 regulates the capillarization of liver sinusoidal endothelial cell and liver fibrogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1663–1675. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Russo, L.; García-Calderó, H.; García-Pagán, J.C.; García-Cardeña, G.; Bosch, J. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut 2011, 60, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.; Haddad, F.G.; Garcia-Cardena, G.; Frangos, J.A.; Mennone, A.; Groszmann, R.J.; Sessa, W.C. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J. Clin. Investig. 1997, 100, 2923–2930. [Google Scholar] [CrossRef] [Green Version]
- DeLeve, L.D.; Wang, X.; Guo, Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008, 48, 920–930. [Google Scholar] [CrossRef]
- Elvevold, K.H.; Nedredal, G.I.; Revhaug, A.; Smedsrød, B. Scavenger properties of cultivated pig liver endothelial cells. Comp. Hepatol. 2004, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sørensen, K.K.; McCourt, P.; Berg, T.; Crossley, C.; Le Couteur, D.; Wake, K.; Smedsrød, B. The scavenger endothelial cell: A new player in homeostasis and immunity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 303, R1217–R1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLeve, L.D.; Maretti-Mira, A.C. Liver Sinusoidal Endothelial Cell: An Update. Semin. Liver Dis. 2017, 37, 377–387. [Google Scholar] [CrossRef]
- Breiner, K.M.M.; Schaller, H.; Knolle, P.A.A. Endothelial cell-mediated uptake of a hepatitis B virus: A new concept of liver targeting of hepatotropic microorganisms. Hepatology 2001, 34, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, L.P.; Mohanty, S.; Kim, J.; Clark, K.R.; Robinson, J.M.; Anderson, C.L. Rapid and efficient clearance of blood-borne virus by liver sinusoidal endothelium. PLoS Pathog. 2011, 7, 1002281. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Yao, Z.; Cheplowitz, A.M.; Suer, O.; Phillips, G.S.; Kwiek, J.J.; Rajaram, M.V.S.; Kim, J.; Robinson, J.M.; Ganesan, L.P.; et al. Mouse liver sinusoidal endothelium eliminates HIV-like particles from blood at a rate of 100 million per minute by a second-order kinetic process. Front. Immunol. 2017, 8, 35. [Google Scholar] [CrossRef]
- Oie, C.I.; Wolfson, D.L.L.; Yasunori, T.; Dumitriu, G.; Sorensen, K.K.; McCourt, P.A.A.; Ahluwalia, B.S.S.; Smedsrod, B.; Øie, C.I.; Wolfson, D.L.L.; et al. Liver sinusoidal endothelial cells contribute to the uptake and degradation of entero bacterial viruses. Sci. Rep. 2020, 10, 898. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [Green Version]
- Heymann, F.; Tacke, F. Immunology in the liver-from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Cantor, H.M.; Dumont, A.E. Hepatic suppression of sensitization to antigen absorbed into the portal system. Nature 1967, 215, 744–745. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Limmer, A. Neighborhood politics: The immunoregulatory function of organ-resident liver endothelial cells. Trends Immunol. 2001, 22, 432–437. [Google Scholar] [CrossRef]
- Limmer, A.; Ohl, J.; Kurts, C.; Ljunggren, H.G.; Reiss, Y.; Groettrup, M.; Momburg, F.; Arnold, B.; Knolle, P.A. Efficient presentation of exogenous antigen by liver endothelial cells to CD8+ T cells results in antigen-specific T-cell tolerance. Nat. Med. 2000, 6, 1348–1354. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, D.; Roska, A.K.; Lipsky, P.E. Liver sinusoidal lining cells express class II major histocompatibility antigens but are poor stimulators of fresh allogeneic T lymphocytes. J. Immunol. 1986, 137, 1803–1810. [Google Scholar]
- Knolle, P.A.; Schmitt, E.; Jin, S.; Germann, T.; Duchmann, R.; Hegenbarth, S.; Gerken, G.; Lohse, A.W. Induction of cytokine production in naive CD4+ T cells by antigen-presenting murine liver sinusoidal endothelial cells but failure to induce differentiation toward T(h1) cells. Gastroenterology 1999, 116, 1428–1440. [Google Scholar] [CrossRef]
- Diehl, L.; Schurich, A.; Grochtmann, R.; Hegenbarth, S.; Chen, L.; Knolle, P.A. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology 2007, 47, 296–305. [Google Scholar] [CrossRef]
- Guixé-Muntet, S.; de Mesquita, F.C.; Vila, S.; Hernandez-Gea, V.; Peralta, C.; Garcia-Pagan, J.C.; Bosch, J.; Gracia-Sancho, J. Cross-talk between autophagy and KLF2 determines endothelial cell phenotype and microvascular function in acute liver injury. J. Hepatol. 2017, 66, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Ruart, M.; Chavarria, L.; Campreciós, G.; Suárez-Herrera, N.; Montironi, C.; Guixé-Muntet, S.; Bosch, J.; Friedman, S.L.; Garcia-Pagán, J.C.; Hernández-Gea, V. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J. Hepatol. 2019, 70, 458–469. [Google Scholar] [CrossRef]
- Luo, X.; Wang, D.; Zhu, X.; Wang, G.; You, Y.; Ning, Z.; Li, Y.; Jin, S.; Huang, Y.; Hu, Y.; et al. Autophagic degradation of caveolin-1 promotes liver sinusoidal endothelial cells defenestration article. Cell Death Dis. 2018, 9, 576. [Google Scholar] [CrossRef] [Green Version]
- Luo, X.X.; Wang, D.; Luo, X.X.; Zhu, X.; Wang, G.; Ning, Z.; Li, Y.; Ma, X.; Yang, R.; Jin, S.; et al. Caveolin 1-related autophagy initiated by aldosterone-induced oxidation promotes liver sinusoidal endothelial cells defenestration. Redox Biol. 2017, 13, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Laviña, B.; Rodríguez-Vilarrupla, A.; García-Calderó, H.; Bosch, J.; García-Pagán, J.C. Enhanced vasoconstrictor prostanoid production by sinusoidal endothelial cells increases portal perfusion pressure in cirrhotic rat livers. J. Hepatol. 2007, 47, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Graupera, M.; March, S.; Engel, P.; Rodés, J.; Bosch, J.; García-Pagán, J.C. Sinusoidal endothelial COX-1-derived prostanoids modulate the hepatic vascular tone of cirrhotic rat livers. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G763–G770. [Google Scholar] [CrossRef] [Green Version]
- Amitrano, L.; Guardascione, M.A.; Brancaccio, V.; Margaglione, M.; Manguso, F.; Iannaccone, L.; Grandone, E.; Balzano, A. Risk factors and clinical presentation of portal vein thrombosis in patients with liver cirrhosis. J. Hepatol. 2004, 40, 736–741. [Google Scholar] [CrossRef]
- Hilscher, M.B.; Sehrawat, T.; Arab Verdugo, J.P.; Zeng, Z.; Gao, J.; Liu, M.; Kostallari, E.; Gao, Y.; Simonetto, D.A.; Yaqoob, U.; et al. Mechanical Stretch Increases Expression of CXCL1 in Liver Sinusoidal Endothelial Cells to Recruit Neutrophils, Generate Sinusoidal Microthombi, and Promote Portal Hypertension. Gastroenterology 2019, 157, 193–209. [Google Scholar] [CrossRef]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef]
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine Receptor CXCR6-Dependent Hepatic NK T Cell Accumulation Promotes Inflammation and Liver Fibrosis. J. Immunol. 2013, 190, 5226–5236. [Google Scholar] [CrossRef] [Green Version]
- Shetty, S.; Bruns, T.; Weston, C.J.; Stamataki, Z.; Oo, Y.H.; Long, H.M.; Reynolds, G.M.; Pratt, G.; Moss, P.; Jalkanen, S.; et al. Recruitment mechanisms of primary and malignant B cells to the human liver. Hepatology 2012, 56, 1521–1531. [Google Scholar] [CrossRef] [PubMed]
- Lalor, P.F.; Shields, P.; Grant, A.J.; Adams, D.H.; Hields, P.S.; Grant, A.J.; Adams, D.H.; Shields, P.; Grant, A.J.; Adams, D.H.; et al. Recruitment of lymphocytes to the human liver. Immunol. Cell Biol. 2002, 80, 52–64. [Google Scholar] [CrossRef]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Caparrós, E.; Juanola, O.; Gómez-Hurtado, I.; Puig-Kroger, A.; Piñero, P.; Zapater, P.; Linares, R.; Tarín, F.; Martínez-López, S.; Gracia-Sancho, J.; et al. Liver Sinusoidal Endothelial Cells Contribute to Hepatic Antigen-Presenting Cell Function and Th17 Expansion in Cirrhosis. Cells 2020, 9, 1227. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, C.R.; Pinzani, M. Stellate Cells in Health and Disease; Academic Press: Cambridge, MA, USA, 2015; ISBN 9780128005446. [Google Scholar]
- Kostallari, E.; Shah, V.H. Pericytes in the liver. In Pericyte Biology in Different Organs; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1122, pp. 153–167. [Google Scholar]
- Harrison, E.H.; Curley, R.W. Carotenoids and Retinoids: Nomenclature, Chemistry, and Analysis. In The Biochemistry of Retinoid Signaling II; Subcellular Biochemistry; Springer: Dordrecht, The Netherlands, 2016; Volume 81, pp. 1–19. [Google Scholar] [CrossRef]
- Blaner, W.S.; Li, Y.; Brun, P.J.; Yuen, J.J.; Lee, S.A.; Clugston, R.D. Vitamin A absorption, storage and mobilization. Subcell. Biochem. 2016, 81, 95–125. [Google Scholar] [CrossRef]
- Goodman, D.S.; Huang, H.S.; Shiratori, T. Tissue distribution and metabolism of newly absorbed vitamin A in the rat. J. Lipid Res. 1965, 6, 390–396. [Google Scholar] [CrossRef]
- Blaner, W.S.; Hendriks, H.F.J.; Brouwer, A.; de Leeuw, A.M.; Knook, D.L.; Goodman, D.S. Retinoids, retinoid-binding proteins, and retinyl palmitate hydrolase distributions in different types of rat liver cells. J. Lipid Res. 1985, 26, 1241–1251. [Google Scholar] [CrossRef]
- Casini, A.; Pellegrini, G.; Ceni, E.; Salzano, R.; Parola, M.; Robino, G.; Milani, S.; Dianzani, M.U.; Surrenti, C. Human hepatic stellate cells express class I alcohol dehydrogenase and aldehyde dehydrogenase but not cytochrome P4502E1. J. Hepatol. 1998, 28, 40–45. [Google Scholar] [CrossRef]
- Kluwe, J.; Wongsiriroj, N.; Troeger, J.S.; Gwak, G.Y.; Dapito, D.H.; Pradere, J.P.; Jiang, H.; Siddiqi, M.; Piantedosi, R.; O’Byrne, S.M.; et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut 2011, 60, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Yoneda, A.; Sakai-Sawada, K.; Niitsu, Y.; Tamura, Y. Vitamin A and insulin are required for the maintenance of hepatic stellate cell quiescence. Exp. Cell Res. 2016, 341, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Chen, A. Perilipin 5 restores the formation of lipid droplets in activated hepatic stellate cells and inhibits their activation. Lab. Investig. 2016, 96, 791–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirazzi, C.; Valenti, L. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef] [Green Version]
- Winau, F.; Quack, C.; Darmoise, A.; Kaufmann, S.H. Starring stellate cells in liver immunology. Curr. Opin. Immunol. 2008, 20, 68–74. [Google Scholar] [CrossRef]
- Paik, Y.H.; Schwabe, R.F.; Bataller, R.; Russo, M.P.; Jobin, C.; Brenner, D.A. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology 2003, 37, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Brun, P.; Castagliuolo, I.; Pinzani, M.; Palù, G.; Martines, D. Exposure to bacterial cell wall products triggers an inflammatory phenotype in hepatic stellate cells. Am. J. Physiol. Liver Physiol. 2005, 289, G571–G578. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [Green Version]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Marrone, G.; Russo, L.; Rosado, E.; Hide, D.; García-Cardeña, G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial-stellate cell deactivation induced by statins. J. Hepatol. 2013, 58, 98–103. [Google Scholar] [CrossRef]
- Du, Z.; Lin, Z.; Wang, Z.; Liu, D.; Tian, D.; Xia, L. SPOCK1 overexpression induced by platelet-derived growth factor- BB promotes hepatic stellate cell activation and liver fi brosis through the integrin α 5 β 1/PI3K/Akt signaling pathway. Lab. Investig. 2020, 100, 1042–1056. [Google Scholar] [CrossRef]
- Guixé-Muntet, S.; Ortega-Ribera, M.; Wang, C.; Selicean, S.; Andreu, I.; Kechagia, J.Z.; Fondevila, C.; Roca-Cusachs, P.; Dufour, J.-F.; Bosch, J.; et al. Nuclear deformation mediates liver cell mechanosensing in cirrhosis. JHEP Rep. 2020, 2, 100145. [Google Scholar] [CrossRef] [PubMed]
- Thoen, L.F.R.; Guimarães, E.L.M.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Enriori, P.J.; Evans, A.E.; Sinnayah, P.; Cowley, M.A. Leptin Resistance and Obesity. Obesity 2006, 14, 254S–258S. [Google Scholar] [CrossRef]
- Saxena, N.K.; Anania, F.A. Adipocytokines and hepatic fibrosis. Trends Endocrinol. Metab. 2015, 26, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.S.; Syn, W.K.; Karaca, G.F.; Omenetti, A.; Moylan, C.A.; Witek, R.P.; Agboola, K.M.; Jung, Y.; Michelotti, G.A.; Diehl, A.M. Leptin promotes the myofibroblastic phenotype in hepatic stellate cells by activating the Hedgehog pathway. J. Biol. Chem. 2010, 285, 36551–36560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Leclercq, I.; Brymora, J.M.; Xu, N.; Ramezani-Moghadam, M.; London, R.M.; Brigstock, D.; George, J. Kupffer Cells Mediate Leptin-Induced Liver Fibrosis. Gastroenterology 2009, 137, 713–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Minicis, S.; Rychlicki, C.; Agostinelli, L.; Saccomanno, S.; Candelaresi, C.; Trozzi, L.; Mingarelli, E.; Facinelli, B.; Magi, G.; Palmieri, C.; et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 2014, 59, 1738–1749. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, X.J.; Li, H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 2019, 70, 1026–1037. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syn, W.K.; Agboola, K.M.; Swiderska, M.; Michelotti, G.A.; Liaskou, E.; Pang, H.; Xie, G.; Philips, G.; Chan, I.S.; Karaca, G.F.; et al. NKT-associated hedgehog and osteopontin drive fibrogenesis in non-alcoholic fatty liver disease. Gut 2012, 61, 1323–1329. [Google Scholar] [CrossRef]
- Tan, Z.; Qian, X.; Jiang, R.; Liu, Q.; Wang, Y.; Chen, C.; Wang, X.; Ryffel, B.; Sun, B. IL-17A Plays a Critical Role in the Pathogenesis of Liver Fibrosis through Hepatic Stellate Cell Activation. J. Immunol. 2013, 191, 1835–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776.e3. [Google Scholar] [CrossRef] [Green Version]
- Beringer, A.; Miossec, P. IL-17 and TNF-α co-operation contributes to the proinflammatory response of hepatic stellate cells. Clin. Exp. Immunol. 2019, 198, 111–120. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.; Liu, J.; She, W.M.; Shi, J.M.; Li, J.; Wang, J.Y.; Jiang, W. Activated hepatic stellate cells directly induce pathogenic Th17 cells in chronic hepatitis B virus infection. Exp. Cell Res. 2017, 359, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, J.; Wang, X.; Sang, M.; Ho, W. Hepatic stellate cells, liver innate immunity, and hepatitis C virus. J. Gastroenterol. Hepatol. 2013, 28, 112–115. [Google Scholar] [CrossRef] [Green Version]
- Shahin, K.; Hosseini, S.Y.; Jamali, H.; Karimi, M.-H.; Azarpira, N.; Zeraatian, M. The enhancing impact of amino termini of hepatitis C virus core protein on activation of hepatic stellate cells. Gastroenterol. Hepatol. Bed Bench 2020, 13, 57. [Google Scholar] [PubMed]
- Wu, C.F.; Lin, Y.L.; Huang, Y.T. Hepatitis C virus core protein stimulates fibrogenesis in hepatic stellate cells involving the obese receptor. J. Cell. Biochem. 2013, 114, 541–550. [Google Scholar] [CrossRef]
- Coenen, M.; Nischalke, H.D.; Krämer, B.; Langhans, B.; Glässner, A.; Schulte, D.; Körner, C.; Sauerbruch, T.; Nattermann, J.; Spengler, U. Hepatitis C virus core protein induces fibrogenic actions of hepatic stellate cells via toll-like receptor 2. Lab. Investig. 2011, 91, 1375–1382. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [Green Version]
- Yavuz, B.G.; Pestana, R.C.; Abugabal, Y.I.; Krishnan, S.; Chen, J.; Hassan, M.M.; Wolff, R.A.; Rashid, A.; Amin, H.M.; Kaseb, A.O. Origin and role of hepatic myofibroblasts in hepatocellular carcinoma. Oncotarget 2020, 11, 1186–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; van Rooijen, N.; Ochando, J.C.; Randolph, G.J.; et al. Liver Inflammation Abrogates Immunological Tolerance Induced by Kupffer Cells. Hepatology 2015, 62, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Munniksma, J.; Noteborn, M.; Kooistra, T.; Stienstra, S.; Bouma, J.M.W.; Gruber, M.; Brouwer, A.; Praaning-Van Dalent, D.; Knookt, D.L. Fluid endocytosis by rat liver and spleen Experiments with 125I-labelled poly(vinylpyrrolidone) in vivo. Biochem. J. 1980, 192, 613–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, S.H.; Cousens, L.P.; van Rooijen, N.; Döpp, E.A.; Carlos, T.M.; Wing, E.J. Complementary Adhesion Molecules Promote Neutrophil- Kupffer Cell Interaction and the Elimination of Bacteria Taken Up by the Liver. J. Immunol. 2002, 168, 308–315. [Google Scholar] [CrossRef] [PubMed]
- You, Q.; Cheng, L.; Kedl, R.M.; Ju, C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology 2008, 48, 978–990. [Google Scholar] [CrossRef]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Zannetti, C.; Roblot, G.; Charrier, E.; Ainouze, M.; Tout, I.; Briat, F.; Isorce, N.; Faure-Dupuy, S.; Michelet, M.; Marotel, M.; et al. Characterization of the Inflammasome in Human Kupffer Cells in Response to Synthetic Agonists and Pathogens. J. Immunol. 2016, 197, 356–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362. [Google Scholar] [CrossRef]
- Steib, C.J.; Gerbes, A.L. Signaling pathways in liver diseases kupffer cells. In Signaling Pathways in Liver Diseases; Springer: Berlin/Heidelberg, Germany, 2010; pp. 69–78. ISBN 9783642001499. [Google Scholar]
- Li, P.; He, K.; Li, J.; Liu, Z.; Gong, J. The role of Kupffer cells in hepatic diseases. Mol. Immunol. 2017, 85, 222–229. [Google Scholar] [CrossRef]
- Shan, Z.; Ju, C. Hepatic Macrophages in Liver Injury. Front. Immunol. 2020, 11, 322. [Google Scholar] [CrossRef]
- Nagpal, R.; Newman, T.M.; Wang, S.; Jain, S.; Lovato, J.F.; Yadav, H. Obesity-Linked Gut Microbiome Dysbiosis Associated with Derangements in Gut Permeability and Intestinal Cellular Homeostasis Independent of Diet. J. Diabetes Res. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Luck, H.; Tsai, S.; Winer, S. The intestinal immune system in obesity and insulin resistance. Cell Metab. 2016, 23, 413–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wigg, A.J.; Roberts-Thomson, I.C.; Grose, R.H.; Cummins, A.G.; Dymock, R.B.; McCarthy, P.J. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor α in the pathogenesis of non-alcoholic steatohepatitis. Gut 2001, 48, 206–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorribas, M.; Jakob, M.O.; Yilmaz, B.; Li, H.; Stutz, D.; Noser, Y.; de Gottardi, A.; Moghadamrad, S.; Hassan, M.; Albillos, A.; et al. FXR modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. J. Hepatol. 2019, 71, 1126–1140. [Google Scholar] [CrossRef]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- Xu, F.L.; You, H.B.; Li, X.H.; Chen, X.F.; Liu, Z.J.; Gong, J.P. Glycine attenuates endotoxin-induced liver injury by downregulating TLR4 signaling in Kupffer cells. Am. J. Surg. 2008, 196, 139–148. [Google Scholar] [CrossRef]
- Seki, E.; Tsutsui, H.; Nakano, H.; Tsuji, N.M.; Hoshino, K.; Adachi, O.; Adachi, K.; Futatsugi, S.; Kuida, K.; Takeuchi, O.; et al. Lipopolysaccharide-Induced IL-18 Secretion from Murine Kupffer Cells Independently of Myeloid Differentiation Factor 88 That Is Critically Involved in Induction of Production of IL-12 and IL-1β. J. Immunol. 2001, 166, 2651–2657. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Deng, X.; Liu, Y.; Tan, Q.; Huang, G.; Che, Q.; Guo, J.; Su, Z. Kupffer cells in non-alcoholic fatty liver disease: Friend or foe? Int. J. Biol. Sci. 2020, 16, 2367–2378. [Google Scholar] [CrossRef]
- Mihm, S. Danger-associated molecular patterns (DAMPs): Molecular triggers for sterile inflammation in the liver. Int. J. Mol. Sci. 2018, 19, 3104. [Google Scholar] [CrossRef] [Green Version]
- Huebener, P.; Pradere, J.P.; Hernandez, C.; Gwak, G.Y.; Caviglia, J.M.; Mu, X.; Loike, J.D.; Jenkins, R.E.; Antoine, D.J.; Schwabe, R.F. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Investig. 2015, 125, 539–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Hou, W.; Zhang, Q.; Kang, R.; Fan, X.G.; Tang, D. Emerging role of high-mobility group box 1 (HMGB1) in liver diseases. Mol. Med. 2013, 19, 357–366. [Google Scholar] [CrossRef]
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003, 38, 1188–1198. [Google Scholar] [CrossRef] [PubMed]
- Pradere, J.-P.P.; Kluwe, J.; De Minicis, S.; Jiao, J.-J.J.; Gwak, G.-Y.Y.; Dapito, D.H.; Jang, M.-K.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- Kiagiadaki, F.; Kampa, M.; Voumvouraki, A.; Castanas, E.; Kouroumalis, E.; Notas, G. Activin-A causes Hepatic stellate cell activation via the induction of TNFα and TGFβ in Kupffer cells. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Pose, E.; Coll, M.; Martínez-Sánchez, C.; Zeng, Z.; Surewaard, B.G.J.; Català, C.; Velasco, M.; Lozano, J.; Ariño, S.; Fuster, D.; et al. PD-L1 is overexpressed in liver macrophages in chronic liver diseases and its blockade improves the antibacterial activity against infections. Hepatology 2020, 74, 296–311. [Google Scholar] [CrossRef]
- Marrone, G.; Shah, V.H.; Gracia-Sancho, J. Sinusoidal communication in liver fibrosis and regeneration. J. Hepatol. 2016, 65, 608–617. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, S.A.; Skjeldal, F.; Fønhus, M.S.; Haugen, L.H.; Eskild, W.; Berg, T.; Bakke, O. Receptor-Mediated Endocytosis of VEGF-A in Rat Liver Sinusoidal Endothelial Cells. Biomed. Res. Int. 2019, 2019, 5496197. [Google Scholar] [CrossRef]
- DeLeve, L.D.; Wang, X.; Hu, L.; McCuskey, M.K.; McCuskey, R.S. Rat liver sinusoidal endothelial cell phenotype is maintained by paracrine and autocrine regulation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G757–G763. [Google Scholar] [CrossRef] [Green Version]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; Deleve, L.D. Role of Differentiation of Liver Sinusoidal Endothelial Cells in Progression and Regression of Hepatic Fibrosis in Rats. Gastroenterology 2012, 142, 918–927. [Google Scholar] [CrossRef] [Green Version]
- Graupera, M.; García-Pagán, J.C.; Abraldes, J.G.; Peralta, C.; Bragulat, M.; Corominola, H.; Bosch, J.; Rodés, J. Cyclooxygenase-derived products modulate the increased intrahepatic resistance of cirrhotic rat livers. Hepatology 2003, 37, 172–181. [Google Scholar] [CrossRef]
- Graupera, M.; GarcíaPagán, J.C.; Titos, E.; Claria, J.; Massaguer, A.; Bosch, J.; Rodés, J. 5-Lipoxygenase inhibition reduces intrahepatic vascular resistance of cirrhotic rat livers: A possible role of cysteinyl-leukotrienes. Gastroenterology 2002, 122, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Weisiger, R.A. Endothelin induced contractility of stellate cells from normal and cirrhotic rat liver: Implications for regulation of portal pressure and resistance. Hepatology 1996, 24, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Desroches-Castan, A.; Tillet, E.; Ricard, N.; Ouarné, M.; Mallet, C.; Belmudes, L.; Couté, Y.; Boillot, O.; Scoazec, J.Y.; Bailly, S.; et al. Bone Morphogenetic Protein 9 Is a Paracrine Factor Controlling Liver Sinusoidal Endothelial Cell Fenestration and Protecting Against Hepatic Fibrosis. Hepatology 2019, 70, 1392–1408. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Kuang, H.; Ansari, S.; Liu, T.; Gong, J.; Wang, S.; Zhao, X.Y.; Ji, Y.; Li, C.; Guo, L.; et al. Landscape of Intercellular Crosstalk in Healthy and NASH Liver Revealed by Single-Cell Secretome Gene Analysis. Mol. Cell 2019, 75, 644–660. [Google Scholar] [CrossRef] [PubMed]
- Royo, F.; Schlangen, K.; Palomo, L.; Gonzalez, E.; Conde-Vancells, J.; Berisa, A.; Aransay, A.M.; Falcon-Perez, J.M. Transcriptome of extracellular vesicles released by hepatocytes. PLoS ONE 2013, 8, e68693. [Google Scholar] [CrossRef]
- Jiang, F.; Chen, Q.; Wang, W.; Ling, Y.; Yan, Y.; Xia, P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J. Hepatol. 2019, 72, 156–166. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R.; Kemper, S.; Charrier, A.; Brigstock, D.R. Suppression of fibrogenic signaling in hepatic stellate cells by twist1-dependent microRNA-214 expression: Role of exosomes in horizontal transfer of twist. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G491–G499. [Google Scholar] [CrossRef] [Green Version]
- Marrone, G.; Maeso-Díaz, R.; García-Cardena, G.; Abraldes, J.G.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: Behind the molecular mechanisms of statins. Gut 2015, 64, 1434–1443. [Google Scholar] [CrossRef]
- Fernández-Iglesias, A.; Gracia-Sancho, J. How to face chronic liver disease: The sinusoidal perspective. Front. Med. 2017, 4, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bode, J.G.; Albrecht, U.; Häussinger, D.; Heinrich, P.C.; Schaper, F. Hepatic acute phase proteins—Regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur. J. Cell Biol. 2012, 91, 496–505. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nunez, G. Sterile inflammation: Sensing and reacting to damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeCouter, J.; Moritz, D.R.; Li, B.; Phillips, G.L.; Liang, X.H.; Gerber, H.P.; Hillan, K.J.; Ferraral, N. Angiogenesis-independent endothelial protection of liver: Role of VEGFR-1. Science 2003, 299, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.S.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nature 2010, 468, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Guixé-Muntet, S.; Zhu, C.P.; Xie, W.F.; Gracia-Sancho, J. Novel therapeutics for portal hypertension and fibrosis in chronic liver disease. Pharmacol. Ther. 2020, 215, 107626. [Google Scholar] [CrossRef]
- Selicean, S.; Wang, C.; Guixé-Muntet, S.; Stefanescu, H.; Kawada, N.; Gracia-Sancho, J. Regression of portal hypertension: Underlying mechanisms and therapeutic strategies. Hepatol. Int. 2021, 15, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Baiges, A.; Hernández-Gea, V.; Bosch, J. Pharmacologic prevention of variceal bleeding and rebleeding. Hepatol. Int. 2018, 12, 68–80. [Google Scholar] [CrossRef]
- Trautwein, C.; Friedman, S.L.; Schuppan, D.; Pinzani, M. Hepatic fibrosis: Concept to treatment. J. Hepatol. 2015, 62, S15–S24. [Google Scholar] [CrossRef] [Green Version]
- Vilaseca, M.; Guixé-Muntet, S.; Fernández-Iglesias, A.; Gracia-Sancho, J. Advances in therapeutic options for portal hypertension. Ther. Adv. Gastroenterol. 2018, 11, 175628481881129. [Google Scholar] [CrossRef]
- Testino, G.; Leone, S.; Fagoonee, S.; Pellicano, R. Alcoholic liver fibrosis: Detection and treatment. Minerva Med. 2018, 109, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Schwabl, P.; Laleman, W. Novel treatment options for portal hypertension. Gastroenterol. Rep. 2017, 5, 90–103. [Google Scholar] [CrossRef]
- Singh, S.; Osna, N.A.; Kharbanda, K.K. Treatment options for alcoholic and non-alcoholic fatty liver disease: A review. World J. Gastroenterol. 2017, 23, 6549–6570. [Google Scholar] [CrossRef] [PubMed]
- Graupera, M.; García-Pagán, J.C.; Parés, M.; Abraldes, J.G.; Roselló, J.; Bosch, J.; Rodés, J. Cyclooxygenase-1 inhibition corrects endothelial dysfunction in cirrhotic rat livers. J. Hepatol. 2003, 39, 515–521. [Google Scholar] [CrossRef]
- Rosado, E.; Rodríguez-Vilarrupla, A.; Gracia-Sancho, J.; Tripathi, D.; García-Calderó, H.; Bosch, J.; García-Pagán, J.C. Terutroban, a TP-receptor antagonist, reduces portal pressure in cirrhotic rats. Hepatology 2013, 58, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Cai, M.; Deng, S.; Huang, W.; Huang, J.; Huang, X.; Huang, M.; Wang, Y.; Shuai, X.; Zhu, K. Amelioration of cirrhotic portal hypertension by targeted cyclooxygenase-1 siRNA delivery to liver sinusoidal endothelium with polyethylenimine grafted hyaluronic acid. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 2329–2339. [Google Scholar] [CrossRef]
- Nanji, A.A.; Liong, E.C.; Xiao, J.; Tipoe, G.L. Thromboxane Inhibitors Attenuate Inflammatory and Fibrotic Changes in Rat Liver Despite Continued Ethanol Administrations. Alcohol Clin. Exp. Res. 2013, 37, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Laleman, W.; Van Landeghem, L.; Van der Elst, I.; Zeegers, M.; Fevery, J.; Nevens, F. Nitroflurbiprofen, a nitric oxide-releasing cyclooxygenase inhibitor, improves cirrhotic portal hypertension in rats. Gastroenterology 2007, 132, 709–719. [Google Scholar] [CrossRef] [Green Version]
- Feng, H.Q.; Weymouth, N.D.; Rockey, D.C. Endothelin antagonism in portal hypertensive mice: Implications for endothelin receptor-specific signaling in liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G27–G33. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Takashimizu, S.; Nishizaki, Y.; Kojima, S.; Kagawa, T.; Matsuzaki, S. An endothelin A receptor antagonist induces dilatation of sinusoidal endothelial fenestrae: Implications for endothelin-1 in hepatic microcirculation. J. Gastroenterol. 2007, 42, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, D.; Therapondos, G.; Ferguson, J.W.; Newby, D.E.; Webb, D.J.; Hayes, P.C. Endothelin-1 contributes to maintenance of systemic but not portal haemodynamics in patients with early cirrhosis: A randomised controlled trial. Gut 2006, 55, 1290–1295. [Google Scholar] [CrossRef]
- Zipprich, A.; Schenkel, E.; Gittinger, F.; Winkler, M.; Michl, P.; Ripoll, C. Selective endothelin-a blockade decreases portal pressure in patients with cirrhosis: A pilot study combining a local intraarterial and systemic administration. J. Hepatol. 2016, 64, S247. [Google Scholar] [CrossRef]
- Bravo, M.; Raurell, I.; Barberá, A.; Hide, D.; Gil, M.; Estrella, F.; Salcedo, M.T.; Augustin, S.; Genescá, J.; Martell, M. Synergic effect of atorvastatin and ambrisentan on sinusoidal and hemodynamic alterations in a rat model of NASH. DMM Dis. Model. Mech. 2021, 14, dmm048884. [Google Scholar] [CrossRef] [PubMed]
- Schwabl, P.; Brusilovskaya, K.; Supper, P.; Bauer, D.; Königshofer, P.; Riedl, F.; Hayden, H.; Fuchs, C.D.; Stift, J.; Oberhuber, G.; et al. The soluble guanylate cyclase stimulator riociguat reduces fibrogenesis and portal pressure in cirrhotic rats. Sci. Rep. 2018, 8, 9372. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.C.; Bernier, S.G.; Jacobson, S.; Liu, G.; Zhang, P.Y.; Sarno, R.; Catanzano, V.; Currie, M.G.; Masferrer, J.L. SGC stimulator praliciguat suppresses stellate cell fibrotic transformation and inhibits fibrosis and inflammation in models of NASH. Proc. Natl. Acad. Sci. USA 2019, 166, 11057–11062. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.C.; Yang, Y.Y.; Huang, Y.T.; Lee, F.Y.; Hou, M.C.; Lin, H.C.; Lee, S.D. Administration of a low dose of sildenafil for 1 week decreases intrahepatic resistance in rats with biliary cirrhosis: The role of NO bioavailability. Clin. Sci. 2010, 119, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.M.; Shin, J.H.; Kim, J.M.; Lee, C.H.; Kang, K.K.; Ahn, B.O.; Yoo, M. Effect of udenafil on portal venous pressure and hepatic fibrosis in rats: A novel therapeutic option for portal hypertension. Arzneim.-Forsch. Drug Res. 2009, 59, 641–646. [Google Scholar] [CrossRef]
- Uschner, F.E.; Glückert, K.; Paternostro, R.; Gnad, T.; Schierwagen, R.; Mandorfer, M.; Magdaleno, F.; Ortiz, C.; Schwarzkopf, K.; Kamath, P.S.; et al. Combination of phosphodiesterase-5-inhibitors and beta blockers improves experimental portal hypertension and erectile dysfunction. Liver Int. 2020, 40, 2228–2241. [Google Scholar] [CrossRef]
- Deibert, P.; Schumacher, Y.-O.; Ruecker, G.; Opitz, O.G.; Blum, H.E.; Rössle, M.; Kreisel, W. Effect of vardenafil, an inhibitor of phosphodiesterase-5, on portal haemodynamics in normal and cirrhotic liver—Results of a pilot study. Aliment. Pharmacol. Ther. 2006, 23, 121–128. [Google Scholar] [CrossRef]
- Kreisel, W.; Deibert, P.; Kupcinskas, L.; Sumskiene, J.; Appenrodt, B.; Roth, S.; Neagu, M.; Rössle, M.; Zipprich, A.; Caca, K.; et al. The phosphodiesterase-5-inhibitor udenafil lowers portal pressure in compensated preascitic liver cirrhosis. A dose-finding phase-II-study. Dig. Liver Dis. 2015, 47, 144–150. [Google Scholar] [CrossRef] [PubMed]
- García-Calderó, H.; Rodríguez-Vilarrupla, A.; Gracia-Sancho, J.; Diví, M.; Laviña, B.; Bosch, J.; García-Pagán, J.-C. Tempol administration, a superoxide dismutase mimetic, reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J. Hepatol. 2011, 54, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, M.; Rodriguez-Vilarrupla, A.; Gracia-Sancho, J.; Rosado, E.; Mancini, A.; Bosch, J.; Garcia-Pagán, J.C. Recombinant human manganese superoxide dismutase reduces liver fibrosis and portal pressure in CCl4-cirrhotic rats. J. Hepatol. 2013, 58, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Zafra, C.; Abraldes, J.G.; Turnes, J.; Berzigotti, A.; Fernández, M.; García-Pagán, J.C.; Rodés, J.; Bosch, J. Simvastatin Enhances Hepatic Nitric Oxide Production and Decreases the Hepatic Vascular Tone in Patients with Cirrhosis. Gastroenterology 2004, 126, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Abraldes, J.G.; Albillos, A.; Bañares, R.; Turnes, J.; González, R.; García–Pagán, J.C.; Bosch, J. Simvastatin Lowers Portal Pressure in Patients With Cirrhosis and Portal Hypertension: A Randomized Controlled Trial. Gastroenterology 2009, 136, 1651–1658. [Google Scholar] [CrossRef]
- Trebicka, J.; Hennenberg, M.; Laleman, W.; Shelest, N.; Biecker, E.; Schepke, M.; Nevens, F.; Sauerbruch, T.; Heller, J. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology 2007, 46, 242–253. [Google Scholar] [CrossRef]
- Trebicka, J.; Hennenberg, M.; Odenthal, M.; Shir, K.; Klein, S.; Granzow, M.; Vogt, A.; Dienes, H.-P.; Lammert, F.; Reichen, J.; et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J. Hepatol. 2010, 53, 702–712. [Google Scholar] [CrossRef]
- Abraldes, J.G.; Rodríguez-Vilarrupla, A.; Graupera, M.; Zafra, C.; García-Calderó, H.; García-Pagán, J.C.; Bosch, J. Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J. Hepatol. 2007, 46, 1040–1046. [Google Scholar] [CrossRef]
- Huang, H.C.; Wang, S.S.; Lee, J.Y.; Chen, Y.C.; Lee, F.Y.; Lin, H.C.; Chang, C.C.; Lee, S.D. Simvastatin effects on portal-systemic collaterals of portal hypertensive rats. J. Gastroenterol. Hepatol. 2010, 25, 1401–1409. [Google Scholar] [CrossRef]
- Hsu, S.-J.; Wang, S.-S.; Hsin, I.-F.; Huang, H.-C.; Lee, F.-Y.; Lee, J.-Y.; Lin, H.-C.; Chuang, C.-L.; Lee, S.-D. Effects of simvastatin on the portal-systemic collateral vascular response to endothelin-1 and shunting degree in portal hypertensive rats. Scand. J. Gastroenterol. 2013, 48, 831–838. [Google Scholar] [CrossRef]
- Bravo, M.; Raurell, I.; Hide, D.; Fernández-Iglesias, A.; Gil, M.; Barberá, A.; Salcedo, M.T.; Augustin, S.; Genescà, J.; Martell, M. Restoration of liver sinusoidal cell phenotypes by statins improves portal hypertension and histology in rats with NASH. Sci. Rep. 2019, 9, 20183. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, C.; Zhou, J.; Zhen, Z.; Wang, Y.; Shen, C. Simvastatin Ameliorates Liver Fibrosis via Mediating Nitric Oxide Synthase in Rats with Non-Alcoholic Steatohepatitis-Related Liver Fibrosis. PLoS ONE 2013, 8, e76538. [Google Scholar] [CrossRef] [Green Version]
- La Mura, V.; Pasarín, M.; Meireles, C.Z.; Miquel, R.; Rodríguez-Vilarrupla, A.; Hide, D.; Gracia-Sancho, J.; García-Pagán, J.C.; Bosch, J.; Abraldes, J.G. Effects of simvastatin administration on rodents with lipopolysaccharide-induced liver microvascular dysfunction. Hepatology 2013, 57, 1172–1181. [Google Scholar] [CrossRef]
- Tripathi, D.M.; Vilaseca, M.; Lafoz, E.; Garcia-Calderó, H.; Viegas Haute, G.; Fernández-Iglesias, A.; Rodrigues de Oliveira, J.; García-Pagán, J.C.; Bosch, J.; Gracia-Sancho, J. Simvastatin Prevents Progression of Acute on Chronic Liver Failure in Rats With Cirrhosis and Portal Hypertension. Gastroenterology 2018, 155, 1564–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meireles, C.Z.; Pasarin, M.; Lozano, J.J.; García-Calderó, H.; Gracia-Sancho, J.; García-Pagán, J.C.; Bosch, J.; Abraldes, J.G. Simvastatin Attenuates Liver Injury in Rodents with Biliary Cirrhosis Submitted to Hemorrhage/Resuscitation. Shock 2017, 47, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Maeso-Díaz, R.; Ortega-Ribera, M.; Lafoz, E.; JoséLozano, J.; Baiges, A.; Francés, R.; Albillos, A.; Peralta, C.; García-Pagán, J.C.; Bosch, J.; et al. Aging influences hepatic microvascular biology and liver fibrosis in advanced chronic liver disease. Aging Dis. 2019, 10, 684–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbeke, L.; Farre, R.; Trebicka, J.; Komuta, M.; Roskams, T.; Klein, S.; Elst, I.V.; Windmolders, P.; Vanuytsel, T.; Nevens, F.; et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 2014, 59, 2286–2298. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, L.; Mannaerts, I.; Schierwagen, R.; Govaere, O.; Klein, S.; Vander Elst, I.; Windmolders, P.; Farre, R.; Wenes, M.; Mazzone, M.; et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci. Rep. 2016, 6, 33453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerini, F.; Vilaseca, M.; Lafoz, E.; García-Irigoyen, O.; García-Calderó, H.; Tripathi, D.M.; Avila, M.; Reverter, J.C.; Bosch, J.; Gracia-Sancho, J.; et al. Enoxaparin reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J. Hepatol. 2016, 64, 834–842. [Google Scholar] [CrossRef]
- Vilaseca, M.; García-Calderó, H.; Lafoz, E.; García-Irigoyen, O.; Avila, M.A.; Reverter, J.C.; Bosch, J.; Hernández-Gea, V.; Gracia-Sancho, J.; García-Pagán, J.C. The anticoagulant rivaroxaban lowers portal hypertension in cirrhotic rats mainly by deactivating hepatic stellate cells. Hepatology 2017, 65, 2031–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mejias, M.; Garcia-Pras, E.; Tiani, C.; Miquel, R.; Bosch, J.; Fernandez, M. Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocollateral circulations in portal hypertensive and cirrhotic rats. Hepatology 2009, 49, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Pesce, A.; Ciurleo, R.; Bramanti, A.; Armeli Iapichino, E.C.; Petralia, M.C.; Magro, G.G.; Fagone, P.; Bramanti, P.; Nicoletti, F.; Mangano, K. Effects of Combined Admistration of Imatinib and Sorafenib in a Murine Model of Liver Fibrosis. Molecules 2020, 25, 4310. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Manicardi, N.; Ortega-Ribera, M.; Maeso-Díaz, R.; Guixé-Muntet, S.; Fernández-Iglesias, A.; Hide, D.; García-Calderó, H.; Boyer-Díaz, Z.; Contreras, P.C.; et al. Emricasan Ameliorates Portal Hypertension and Liver Fibrosis in Cirrhotic Rats Through a Hepatocyte-Mediated Paracrine Mechanism. Hepatol. Commun. 2019, 3, 987–1000. [Google Scholar] [CrossRef]
- Garcia-Tsao, G.; Bosch, J.; Kayali, Z.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.; Satapathy, S.K.; Ghabril, M.; Shiffman, M.L.; Younes, Z.H.; et al. Randomized placebo-controlled trial of emricasan for non-alcoholic steatohepatitis-related cirrhosis with severe portal hypertension. J. Hepatol. 2020, 72, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Wong, V.W.S.; Okanoue, T.; Bzowej, N.; Vuppalanchi, R.; Younes, Z.; Kohli, A.; Sarin, S.; Caldwell, S.H.; Alkhouri, N.; et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J. Hepatol. 2020, 73, 26–39. [Google Scholar] [CrossRef]
- Rodríguez-Vilarrupla, A.; Laviña, B.; García-Calderó, H.; Russo, L.; Rosado, E.; Roglans, N.; Bosch, J.; García-Pagán, J.C. PPARα activation improves endothelial dysfunction and reduces fibrosis and portal pressure in cirrhotic rats. J. Hepatol. 2012, 56, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Lefere, S.; Puengel, T.; Hundertmark, J.; Penners, C.; Frank, A.K.; Guillot, A.; de Muynck, K.; Heymann, F.; Adarbes, V.; Defrêne, E.; et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J. Hepatol. 2020, 73, 757–770. [Google Scholar] [CrossRef]
- Boyer-Diaz, Z.; Aristu-Zabalza, P.; Andres-Rozas, M.; Robert, C.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Broqua, P.; Junien, J.; Wettstein, G.; Bosch, J.; et al. Pan-PPAR agonist lanifibranor improves portal hypertension and hepatic fibrosis in experimental advanced chronic liver disease. J. Hepatol. 2020, 74, 1188–1199. [Google Scholar] [CrossRef]
- De Mesquita, F.C.; Guixé-Muntet, S.; Fernández-Iglesias, A.; Maeso-DIáz, R.; Vila, S.; Hide, D.; Ortega-Ribera, M.; Rosa, J.L.; Garciá-Pagán, J.C.; Bosch, J.; et al. Liraglutide improves liver microvascular dysfunction in cirrhosis: Evidence from translational studies. Sci. Rep. 2017, 7, 3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): A multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [Green Version]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, D.M.; Erice, E.; Lafoz, E.; García-Calderó, H.; Sarin, S.K.; Bosch, J.; Gracia-Sancho, J.; García-Pagán, J.C. Metformin reduces hepatic resistance and portal pressure in cirrhotic rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G301–G309. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M.G.; Gracia-Sancho, J.; Marrone, G.; Rodríguez-Vilarrupla, A.; Deulofeu, R.; Abraldes, J.G.; Bosch, J.; García-Pagán, J.C. Leptin receptor blockade reduces intrahepatic vascular resistance and portal pressure in an experimental model of rat liver cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G496–G502. [Google Scholar] [CrossRef] [Green Version]
- Boyer-Diaz, Z.; Domingo, J.C.; de Gregorio, E.; Manicardi, N.; Aristu-Zabalza, P.; Cordobilla, B.; Abad-Jordà, L.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Marí, M.; et al. A nutraceutical rich in docosahexaenoic acid improves portal hypertension in a preclinical model of advanced chronic liver disease. Nutrients 2019, 11, 2358. [Google Scholar] [CrossRef] [Green Version]
- Rincón, D.; Vaquero, J.; Hernando, A.; Galindo, E.; Ripoll, C.; Puerto, M.; Salcedo, M.; Francés, R.; Matilla, A.; Catalina, M.V.; et al. Oral probiotic VSL#3 attenuates the circulatory disturbances of patients with cirrhosis and ascites. Liver Int. 2014, 34, 1504–1512. [Google Scholar] [CrossRef]
- Dhiman, R.K.; Rana, B.; Agrawal, S.; Garg, A.; Chopra, M.; Thumburu, K.K.; Khattri, A.; Malhotra, S.; Duseja, A.; Chawla, Y.K. Probiotic VSL#3 Reduces Liver Disease Severity and Hospitalization in Patients With Cirrhosis: A Randomized, Controlled Trial. Gastroenterology 2014, 147, 1327–1337.e3. [Google Scholar] [CrossRef]
- Gupta, N.; Kumar, A.; Sharma, P.; Garg, V.; Sharma, B.C.; Sarin, S.K. Effects of the adjunctive probiotic VSL#3 on portal haemodynamics in patients with cirrhosis and large varices: A randomized trial. Liver Int. 2013, 33, 1148–1157. [Google Scholar] [CrossRef]
- Gómez-Hurtado, I.; Zapater, P.; Portune, K.; Juanola, O.; Fernández-Iglesias, A.; González-Navajas, J.M.; Gracia-Sancho, J.; Sanz, Y.; Francés, R. Improved hemodynamic and liver function in portal hypertensive cirrhotic rats after administration of B. pseudocatenulatum CECT 7765. Eur. J. Nutr. 2019, 58, 1647–1658. [Google Scholar] [CrossRef]
- Tandon, P.; Moncrief, K.; Madsen, K.; Arrieta, M.C.; Owen, R.J.; Bain, V.G.; Wong, W.W.; Ma, M.M. Effects of probiotic therapy on portal pressure in patients with cirrhosis: A pilot study. Liver Int. 2009, 29, 1110–1115. [Google Scholar] [CrossRef]
- Jayakumar, S.; Carbonneau, M.; Hotte, N.; Befus, A.D.; St. Laurent, C.; Owen, R.; Mccarthy, M.; Madsen, K.; Bailey, R.J.; Ma, M.; et al. VSL#3® probiotic therapy does not reduce portal pressures in patients with decompensated cirrhosis. Liver Int. 2013, 33, 1470–1477. [Google Scholar] [CrossRef]
- Farzaei, M.H.; Zobeiri, M.; Parvizi, F.; El-Senduny, F.F.; Marmouzi, I.; Coy-Barrera, E.; Naseri, R.; Nabavi, S.M.; Rahimi, R.; Abdollahi, M. Curcumin in liver diseases: A systematic review of the cellular mechanisms of oxidative stress and clinical perspective. Nutrients 2018, 10, 855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pascoli, M.; Diví, M.; Rodríguez-Vilarrupla, A.; Rosado, E.; Gracia-Sancho, J.; Vilaseca, M.; Bosch, J.; García-Pagán, J.C. Resveratrol improves intrahepatic endothelial dysfunction and reduces hepatic fibrosis and portal pressure in cirrhotic rats. J. Hepatol. 2013, 58, 904–910. [Google Scholar] [CrossRef] [PubMed]
- Sacerdoti, D.; Pesce, P.; Di Pascoli, M.; Brocco, S.; Cecchetto, L.; Bolognesi, M. Arachidonic acid metabolites and endothelial dysfunction of portal hypertension. Prostaglandins Other Lipid Mediat. 2015, 120, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiest, R. The paradox of nitric oxide in cirrhosis and portal hypertension: Too much, not enough. Hepatology 2002, 35, 478–491. [Google Scholar] [CrossRef]
- Iwakiri, Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver Int. 2012, 32, 199–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buys, E.; Sips, P. New insights into the role of soluble guanylate cyclase in blood pressure regulation. Curr. Opin. Nephrol. Hypertens. 2014, 23, 135–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracia-Sancho, J.; Laviña, B.; Rodríguez-Vilarrupla, A.; García-Calderó, H.; Fernández, M.; Bosch, J.; García-Pagán, J.-C. Increased oxidative stress in cirrhotic rat livers: A potential mechanism contributing to reduced nitric oxide bioavailability. Hepatology 2008, 47, 1248–1256. [Google Scholar] [CrossRef]
- Bosch, J.; Gracia-Sancho, J.; Abraldes, J.G. Cirrhosis as new indication for statins. Gut 2020, 69, 953–962. [Google Scholar] [CrossRef]
- Mohanty, A.; Tate, J.P.; Garcia-Tsao, G. Statins Are Associated With a Decreased Risk of Decompensation and Death in Veterans With Hepatitis C–Related Compensated Cirrhosis. Gastroenterology 2016, 150, 430–440.e1. [Google Scholar] [CrossRef] [Green Version]
- Bosch, J.; Forns, X. Therapy: Statins and liver disease: From concern to “wonder” drugs? Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 320–321. [Google Scholar] [CrossRef] [PubMed]
- Hide, D.; Gil, M.; Andrade, F.; Rafael, D.; Raurell, I.; Bravo, M.; Barberá, A.; Gracia-Sancho, J.; Vargas, V.; Augustin, S.; et al. Simvastatin-loaded polymeric micelles are more effective and less toxic than conventional statins in a pre-clinical model of advanced chronic liver disease. Nanomed. Nanotechnol. Biol. Med. 2020, 29, 102267. [Google Scholar] [CrossRef]
- Kullak-Ublick, G.A.; Stieger, B.; Meier, P.J. Enterohepatic Bile Salt Transporters in Normal Physiology and Liver Disease. Gastroenterology 2004, 126, 322–342. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.A.; Kowdley, K.V. Obeticholic acid for the treatment of nonalcoholic steatohepatitis. Expert Rev. Gastroenterol. Hepatol. 2020, 14, 311–321. [Google Scholar] [CrossRef]
- US National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02308111 (accessed on 1 June 2021).
- US National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03439254 (accessed on 1 June 2021).
- Tripodi, A.; Primignani, M.; Chantarangkul, V.; Dell’Era, A.; Clerici, M.; de Franchis, R.; Colombo, M.; Mannucci, P.M. An Imbalance of Pro- vs Anti-Coagulation Factors in Plasma From Patients With Cirrhosis. Gastroenterology 2009, 137, 2105–2111. [Google Scholar] [CrossRef]
- Fernández, M.; Semela, D.; Bruix, J.; Colle, I.; Pinzani, M.; Bosch, J. Angiogenesis in liver disease. J. Hepatol. 2009, 50, 604–620. [Google Scholar] [CrossRef]
- Fortea, J.I.; Zipprich, A.; Fernandez-Mena, C.; Puerto, M.; Bosoi, C.R.; Almagro, J.; Hollenbach, M.; Bañares, J.; Rodríguez-Sánchez, B.; Cercenado, E.; et al. Enoxaparin does not ameliorate liver fibrosis or portal hypertension in rats with advanced cirrhosis. Liver Int. 2018, 38, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Woolbright, B.L.; Ding, W.X.; Jaeschke, H. Caspase inhibitors for the treatment of liver disease: Friend or foe? Expert Rev. Gastroenterol. Hepatol. 2017, 11, 397–399. [Google Scholar] [CrossRef] [Green Version]
- Liss, K.H.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef] [Green Version]
- US National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04849728?term=lanifibranor&cond=Liver+Diseases&draw=2&rank=2 (accessed on 1 June 2021).
- US National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02784444?term=MSDC-0602k&cond=Liver+Diseases&draw=2&rank=1 (accessed on 1 June 2021).
- US National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02704403?term=elafibranor&cond=Liver+Diseases&draw=2&rank=5 (accessed on 1 June 2021).
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.I.; Mateva, L.; et al. The panppar agonist lanifibranor induces both resolution of NASH and regression of fibrosis after 24 weeks of treatment in non-cirrhotic NASH: Results of the NATIVE phase 2b trial. Hepatology 2020, 72, 9A–10A. [Google Scholar] [CrossRef]
- Hutchinson, J.H.; Rowbottom, M.W.; Lonergan, D.; Darlington, J.; Prodanovich, P.; King, C.D.; Evans, J.F.; Bain, G. Small Molecule Lysyl Oxidase-like 2 (LOXL2) Inhibitors: The Identification of an Inhibitor Selective for LOXL2 over LOX. ACS Med. Chem. Lett. 2017, 8, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Yang, A.; Jia, J.; Popov, Y.V.; Schuppan, D.; You, H. Lysyl oxidase (LOX) family members: Rationale and their potential as therapeutic targets for liver fibrosis. Hepatology 2020, 72, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J.; Harrison, S.A.; Ratziu, V.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Aguilar Schall, R.; Jia, C.; McColgan, B.; et al. The Natural History of Advanced Fibrosis Due to Nonalcoholic Steatohepatitis: Data From the Simtuzumab Trials. Hepatology 2019, 70, 1913–1927. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.A.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duarte, S.M.B.; Stefano, J.T.; Oliveira, C.P. Microbiota and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis (NAFLD/NASH). Ann. Hepatol. 2019, 18, 416–421. [Google Scholar] [CrossRef]
- Bajaj, J.S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 235–246. [Google Scholar] [CrossRef]
- Li, Y.; Tang, R.; Leung, P.S.C.; Gershwin, M.E.; Ma, X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun. Rev. 2017, 16, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Möller, N.P.; Scholz-Ahrens, K.E.; Roos, N.; Schrezenmeir, J. Bioactive peptides and proteins from foods: Indication for health effects. Eur. J. Nutr. 2008, 47, 171–182. [Google Scholar] [CrossRef]
- Romero-Gómez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, M.C.P.; Aghemo, A.; Lleo, A.; Bodini, G.; Furnari, M.; Marabotto, E.; Miele, L.; Giannini, E.G. Mediterranean diet and NAFLD: What we know and questions that still need to be answered. Nutrients 2019, 11, 2971. [Google Scholar] [CrossRef] [Green Version]
- Anania, C.; Massimo Perla, F.; Olivero, F.; Pacifico, L.; Chiesa, C. Mediterranean diet and nonalcoholic fatty liver disease. World J. Gastroenterol. 2018, 24, 2083–2094. [Google Scholar] [CrossRef]
- Suárez, M.; Boqué, N.; del Bas, J.M.; Mayneris-Perxachs, J.; Arola, L.; Caimari, A. Mediterranean diet and multi-ingredient-based interventions for the management of non-alcoholic fatty liver disease. Nutrients 2017, 9, 1052. [Google Scholar] [CrossRef] [Green Version]
- Faghihzadeh, F.; Hekmatdoost, A.; Adibi, P. Resveratrol and liver: A systematic review. J. Res. Med. Sci. 2015, 20, 797–810. [Google Scholar] [CrossRef] [PubMed]
- Szkudelski, T.; Szkudelska, K. Potential of resveratrol in mitigating metabolic disturbances induced by ethanol. Biomed. Pharmacother. 2018, 101, 579–584. [Google Scholar] [CrossRef]
- Boyer-Diaz, Z.; Morata, P.; Aristu-Zabalza, P.; Gibert-Ramos, A.; Bosch, J.; Gracia-Sancho, J. Oxidative Stress in Chronic Liver Disease and Portal Hypertension: Potential of DHA as Nutraceutical. Nutrients 2020, 12, 2627. [Google Scholar] [CrossRef] [PubMed]
- Salomone, F.; Godos, J.; Zelber-Sagi, S. Natural antioxidants for non-alcoholic fatty liver disease: Molecular targets and clinical perspectives. Liver Int. 2016, 36, 5–20. [Google Scholar] [CrossRef]
- Van De Wier, B.; Koek, G.H.; Bast, A.; Haenen, G.R.M.M. The potential of flavonoids in the treatment of non-alcoholic fatty liver disease. Crit. Rev. Food Sci. Nutr. 2017, 57, 834–855. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Yin, L.; Tang, C.; Yin, C. Multifunctional polymeric nanoparticles for oral delivery of TNF-α siRNA to macrophages. Biomaterials 2013, 34, 2843–2854. [Google Scholar] [CrossRef]
- Hassan, R.; Tammam, S.N.; El Safy, S.; Abdel-Halim, M.; Asimakopoulou, A.; Weiskirchen, R.; Mansour, S. Prevention of hepatic stellate cell activation using JQ1- and atorvastatin-loaded chitosan nanoparticles as a promising approach in therapy of liver fibrosis. Eur. J. Pharm. Biopharm. 2019, 134, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.T.T.; Dong, Z.; Su, L.; Boyer, C.; George, J.; Davis, T.P.; Wang, J. The Use of Nanoparticles to Deliver Nitric Oxide to Hepatic Stellate Cells for Treating Liver Fibrosis and Portal Hypertension. Small 2015, 11, 2291–2304. [Google Scholar] [CrossRef]
- Dropmann, A.; Dooley, S.; Dewidar, B.; Hammad, S.; Dediulia, T.; Werle, J.; Hartwig, V.; Ghafoory, S.; Woelfl, S.; Korhonen, H.; et al. TGF-β2 silencing to target biliary-derived liver diseases. Gut 2020, 69, 1677–1690. [Google Scholar] [CrossRef]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef]
- Bent, E.H.; Wehrenberg-Klee, E.; Koay, E.J.; Goyal, L.; Wo, J.Y. Integration of Systemic and Liver-Directed Therapies for Locally Advanced Hepatocellular Cancer: Harnessing Potential Synergy for New Therapeutic Horizons. J. Natl. Compr. Canc. Netw. 2021, 19, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma review-article. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, O.; Kaleli, H.N.; Ozer, E. Molecular Pathogenesis of Nonalcoholic Steatohepatitis-(NASH-) Related Hepatocellular Carcinoma. Can. J. Gastroenterol. Hepatol. 2018, 2018, 8547363. [Google Scholar] [CrossRef] [Green Version]
- Kostallari, E.; Shah, V.H. Angiocrine signaling in the hepatic sinusoids in health and disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G246–G251. [Google Scholar] [CrossRef] [Green Version]
- Benedicto, A.; Herrero, A.; Romayor, I.; Marquez, J.; Smedsrød, B.; Olaso, E.; Arteta, B. Liver sinusoidal endothelial cell ICAM-1 mediated tumor/endothelial crosstalk drives the development of liver metastasis by initiating inflammatory and angiogenic responses. Sci. Rep. 2019, 9, 13111. [Google Scholar] [CrossRef] [Green Version]
- Lefere, S.; Degroote, H.; Van Vlierberghe, H.; Devisscher, L. Unveiling the depletion of Kupffer cells in experimental hepatocarcinogenesis through liver macrophage subtype-specific markers. J. Hepatol. 2019, 71, 631–633. [Google Scholar] [CrossRef] [Green Version]
- Kietzmann, T. Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol. 2017, 11, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.L.; Qurashi, M.; Shetty, S. The Role of Sinusoidal Endothelial Cells in the Axis of Inflammation and Cancer Within the Liver. Front. Physiol. 2020, 11, 990. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, J.; Tsui, Y.M.; Shi, C.; Wang, Y.; Zhang, X.; Yan, Q.; Chen, M.; Jiang, C.; Yuan, Y.F.; et al. RALYL increases hepatocellular carcinoma stemness by sustaining the mRNA stability of TGF-β2. Nat. Commun. 2021, 12, 1518. [Google Scholar] [CrossRef]
- Geraud, C.; Mogler, C.; Runge, A.; Evdokimov, K.; Lu, S.; Schledzewski, K.; Arnold, B.; Hammerling, G.; Koch, P.S.; Breuhahn, K.; et al. Endothelial transdifferentiation in hepatocellular carcinoma: Loss of Stabilin-2 expression in peri-tumourous liver correlates with increased survival. Liver Int. 2013, 33, 1428–1440. [Google Scholar] [CrossRef]
- Faillaci, F.; Marzi, L.; Critelli, R.; Milosa, F.; Schepis, F.; Turola, E.; Andreani, S.; Vandelli, G.; Bernabucci, V.; Lei, B.; et al. Liver Angiopoietin-2 Is a Key Predictor of De Novo or Recurrent Hepatocellular Cancer After Hepatitis C Virus Direct-Acting Antivirals. Hepatology 2018, 68, 1010–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism | Drug | Administration Method | Experimental Model | Structural Effects | Hemodynamic Effects | Direct Cellular Effects | Reference |
|---|---|---|---|---|---|---|---|
| Inhibition of vasoconstriction | SC-560 (COX-1 selective inhibitor) and SQ 29,548 (TXA2 receptor antagonist) | 5 µM during 15 min before the hemodynamic study. Preincubation of the liver. | Male Wistar CCl4-cirrhotic rats | - | ↑ Vasodilatory response to acetylcholine, ↓TXB2 | - | [152] |
| Terutroban (TP-receptor antagonist) | 30 mg/kg once a day for 2 weeks after development of ascites | Male Wistar CCl4-cirrhotic rats | ↓Fibrosis, ↓collagen I and TGF-β mRNA, ↓α-SMA protein | ↓ PP, ↓ HVR | - | [153] | |
| COX-1 siRNA | Intravenous injections with 0.6 mg/kg every other day at 8 weeks after starting CCl4-cirrhosis induction | Male C57BL/6 CCl4-cirrhotic mice | - | ↓ PP | Inhibition of the COX-1/TXA2 pathway | [154] | |
| Ifetroban sodium (TX receptor antagonist) or CGS 12970 (TX synthase inhibitor) | 3 mg/kg or 10 mg/kg, respectively, every day, starting the last 2 weeks of alcohol treatment | Male Wistar alcohol and fat-induced cirrhotic rats | ↓ Necrosis, ↓ inflammation and ↓ fibrosis ↓NF-kB activation, ↓TNFα, COX-2 and TGF-β1 expression. | - | - | [155] | |
| Nitroflurbiprofen or flurbiprofen | In vivo: 45 mg/kg or 30 mg/kg, respectively, intraperitoneally injected 24 h and 1 h prior to the hemodynamic and perfusion experiments. In vitro: From 25 to 250 umol/L | Male Wistar TAA-cirrhotic rats | - | ↓ PP, ↓ splanchnic hyperemia, ↓HVR, ↑ vasodilation, ↓TXB2 | In vitro: ↓ HSC contraction | [156] | |
| ET-A (ABT-627), ET-B (A-192621) or a mixed ET receptor antagonist (A-182086) | 50 mg/kg, 40 mg/kg, or 30 mg/kg, respectively, once a day for 8 weeks during cirrhosis induction | Male BALB/c CCl4-cirrhotic mice | ↓Fibrosis, ↓α-SMA protein and collagen I mRNA | ↓ PP | - | [157] | |
| BQ-123 (ET-A antagonist) | 10 nmol/min infused via a catheter in the mesenteric vein for 10 min | Healthy male Wistar rats | - | ↓ PP | ↑ Number and diameter of fenestrae | [158] | |
| BQ-123 (ET-A antagonist) | 1000 and 3000 nmol/min infused for 20 min | 16 CH patients with PH | - | ↓ MAP and pulmonary vascular resistance index. No effects on HVPG. | - | [159] | |
| BQ-123 or Ambrisentan (ET-A antagonists) | 300, 500, 1000 and 2000 nmol/L of BQ-123 infused through the hepatic artery. 5 or 10 mg single oral administration of ambrisentan | 26 CH patients | - | BQ123: Vasodilation of the hepatic artery, ↓ HVPG. Ambrisentan: ↓ HVPG | - | [160] | |
| Ambrisentan (ET-A antagonist) | 2 or 30 mg/kg/day for 2 weeks alone or in combination with 10 mg/kg/day of atorvastatin. | Male Sprague-Dawley rats with NASH induced by a HFGFD. | ↓ NAS score | ↓ PP, ↓ HVR | Improved markers of microvascular dysfunction, ↓ HSC contraction. | [161] | |
| Induction of vasodilation | BAY 60-2770 (sGC activator) | 0.3 mg/kg daily for 1 week | LSEC and HSC isolated from healthy and TAA-cirrhotic male Sprague-Dawley rats | ↓ Fibrosis, ↓ Cirrhosis | - | Restoration of LSEC phenotype and quiescence of HSC | [127] |
| Riociguat (sGC stimulator) | 1 mg/kg daily, for 1 to 3 weeks | Male Sprague Dawley cBDL or CCl4-cirrhotic rats | ↓ Fibrosis, ↓ Inflammation | ↓ PP, ↑ vasodilation pathways | ↓ HSC α-SMA expression | [162] | |
| Praliciguat (sGC stimulator) | STAM/HC: 3 or 10 mg/kg/day for 6 weeks during cirrhosis induction. TAA: 1, 3 or 10 mg/kg/day for 4 weeks during cirrhosis induction. CCl4: 1, 3 or 10 mg/kg/day for 6 weeks during cirrhosis induction. | Male C57/B6 mouse model with steatosis and metabolism with high cholesterol (STAM/HC), and TAA or CCl4-induced cirrhotic Sprague-Dawley rats | ↓ Fibrosis, ↓ Inflammation | ↓ MAP | ↓ TGF-β-induced HSC activation, ↓TGF-β and PDGF-b, ↓ Macrophage infiltration | [163] | |
| Sildenafil (PDE5 inhibitor) | 0.25 mg/kg twice a day for 1 week, starting 3 weeks after the surgery | Male Sprague-Dawley rats with cBDL-induced cirrhosis | ↑ BH4, total hepatic biopterin and GTPCH-I activity | ↑ sinusoid area, volumetric flow and perfused sinusoids ↓ PP, ↓ PPP | ↑ NO bioavailability, ↑ phosphorilation of eNOS and Akt, ↑ NOx production | [164] | |
| Udenafil (PDE5 inhibitor) | Series A: 1, 5 or 25 mg/kg/day, starting 1 week after the cBDL surgery and continued for 3 weeks. Series B: Single dose of 5 or 25 mg/kg 4 weeks after surgery | Male Sprague-Dawley cBDL-cirrhotic rats | - | ↓ PP | ↓ HSC mRNA expression of procollagen type I and α-SMA | [165] | |
| Udenafil | 1 mg/kg and 5 mg/kg for 60 min | Male Sprague Dawley cBDL or CCl4-cirrhotic rats | - | ↓ PP, ↓ HVR, ↑ intrahepatic vasodilation | ↑ eNOS protein, ↑ cGMP | [166] | |
| Vardenafil (PDE5 inhibitor) | A single dose of 10 mg | 18 CH patients | - | ↑ Portal blood flow, ↓ PP | - | [167] | |
| Udenafil | 12.5, 25, 50, 75 and 100 mg daily for one week | 35 patients with compensated liver cirrhosis and HVPG ≥12 mmHg | - | ↓ HVPG, MAP | - | [168] | |
| Tempol (SOD mimetic) | In vivo: 180 µmol for 30 min during the hemodynamic study. In vitro: 50 µM for 6 h. | In vivo: Male Wistar CCl4 cirrhotic rats. In vitro: LSEC isolated from treated Wistar rats incubated for 6 h with a superoxide dismutase inhibitor. | In vivo: ↓ oxidative stress, ↑ cGMP. | ↓ PP, ↑ portal blood flow, ↓ vascular resistance, ↓ MAP | In vitro: ↓ oxidative stress, ↑ NO. | [169] | |
| rMnSOD (recombinant manganese SOD) | Healthy rats: 15 µg/kg 2 h before the experiment. Cirrhotic rats: 15 µg/kg daily for 7 days. In vitro: 1 µM overnight | Male Wistar healty, CCl4 and cBDL-cirrhotic rats. In vitro: LX2 cells | ↓ oxidative stress, ↓ deposition of fibrillar collagen | ↓ PP, ↓ HVR, ↑ vasorelaxation | In vitro: ↓ oxidative stress, ↓ α-SMA and collagen I gene expression | [170] | |
| Simvastatin | One time dose of 40 mg, 30 and/or 60 min before the study | 30 CH patients with HVPG ≥ 12 mm | - | ↑ Hepatic blood flow, ↓ HVR | ↑ NO levels | [171] | |
| Simvastatin | 20 mg/day for 15 days, and 40 mg/day the following 15 days | 59 patients with CH and PH | - | ↓ HVPG, ↑ liver perfusion and function | - | [172] | |
| Atorvastatin, mevastatin, simvastatin or lovastatin | 0.1, 1 or 10 µM for 24 h | LSEC isolated from male Wistar CCl4-cirrhotic rats | - | - | ↑ KLF2, eNOS and thrombomodulin mRNA expression | [76] | |
| Simvastatin | LX-2 cells: 0.1, 1 and 10 µM for 24 and 72 h. HSC: 10 uM | LX-2 cells and primary HSC | - | - | ↑ KLF2 mRNA expression, ↓ α-SMA mRNA and protein expression | [136] | |
| Atorvastatin | 15 mg/kg once per day for 1 week | Male Sprague-Dawley BDL-cirrhotic rats | - | ↓ PP, ↓ HVR, ↓ shunting | ↑ eNOS mRNA, protein expression, ↑ PKG activity, ↓ HSC contraction | [173] | |
| Atorvastatin | 15 mg/kg daily. Prophylaxis group: 1, 2, 3 or 5 weeks of treatment after BDL. Therapy group: 1 week of treatment at different time points after BDL. | Male Sprague-Dawley BDL-cirrhotic rats | Prophylaxis: ↓ fibrosis, Therapy: ↓ fibrosis, apoptosis. | - | Prophylaxis: ↓ ECM and HSC activation. Therapy: ↓ ECM, ↓ HSC activation, proliferation and apoptosis. | [174] | |
| Simvastatin | 25 mg/kg/day for 3 days | Male Wistar CCl4-cirrhotic rats | ↑ eNOS activity | - | ↑ LSEC function | [175] | |
| Simvastatin | Chronic treatment: 20 mg/kg/day by gavage. Acute treatment: incubation of the portal-systemic collateral vascular bed for 25 min with 10 µM | Male Sprague-Dawley with portal hypertension induced by partial portal vein ligation | - | ↓ PP, ↓ collateral vascular resistance | ↑ SRS eNOS, COX-2 and TXA2 mRNA expression | [176] | |
| Simvastatin | 20 mg/kg by gavage from 2 days prior to ligation until 7 days after the operation | Male Sprague-Dawley with portal hypertension induced by partial portal vein ligation | - | ↓ PP, ↓ collateral vascular resistance | - | [177] | |
| Simvastatin, Atorvastatin | 10 mg/kg/day of one drug for 2 weeks | Male Sprague-Dawley OFA rats with NASH induced by a HFGFD | ↓ Steatosis, ballooning and inflammation, ↓ histological NASH | ↓ PP | ↑ eNOS and AKT phosphorylation, restoration of LSEC phenotype and quiescence of HSC | [178] | |
| Simvastatin | In vivo: 4 mg/kg/day for 8 weeks. In vitro: 10 µM for 24 h | In vivo: male Wistar rats with NASH induced by HFD. In vitro: LX-2 cell line activated with TGF-β. | In vivo:↓ liver inflammatory cells infiltration, ↓ steatosis, ↑ mRNA and protein eNOS ↓ iNOS and collagen I. | - | In vivo: ↓ liver inflammatory cells infiltration. In vitro: ↓ LX-2 activation, ↓ mRNA and protein α-SMA and collagen I. | [179] | |
| Simvastatin | 25 mg/kg given 3 and 23 h after LPS challenge, or 25 mg/kg/day, from 3 days before LPS injection | Male Wistar rats administered with LPS and evaluated 6 and 24 h later | ↓ Inflammation, leukocyte infiltration | ↓ PP | ↓ Sinusoidal endothelial dysfunction, ↑ eNOS phosphorylation | [180] | |
| Simvastatin | 25 mg/kg/day in CCl4 and TAA-induced ACLD animals, 5 mg/kg/day in BDL-induced animals for 3 days and a last dose 30 min before the LPS injection | ACLD rats (CCl4, BDL or TAA) subjected to ACLF challenge with an injection of 1 mg/kg of LPS before the study | ↓ ACLF-derived complications, ↑ survival, ↓ inflammation | ↓ PP | ↑ Sinusoidal function, ↓ LPS-mediated activation of HSC | [181] | |
| Simvastatin | 5 mg/kg/day starting 3 days before the experiments | Male Sprague-Dawley cBDL-cirrhotic rats subject to hemorrhage/resuscitation | ↓ ALT, AST, ↓ RNA expression of inflammatory genes | ↓ Vasoconstriction | - | [182] | |
| Simvastatin | 5 mg/kg/day for 15 days by gavage | Male Wistar rats (16 months old) with ACLD (CCl4). | ↓ Fibrosis, | ↓ Hepatic microvascular dysfunction, | ↑ Fenestration, ↑ markers of hepatocyte function, ↓ markers of HSC activation | [183] | |
| INT-747 (FXR receptor agonist) | TAA-cirrhotic rats received two doses of 30 mg/kg 24 and 4 h before the experiments. Another grup of TAA and BDL-cirrhotic rats received 30 mg daily and 5 mg/kg every 2 days for 10 days, respectively. | Male Wistar TAA or cBDL-cirrhotic rats | - | ↓ PP, ↓ HVR, ↑ hepatic microvascular function | ↑ NO | [184] | |
| OCA (FXR agonist) | In vivo: 10 mg/kg either every 2 days during the last 4 weeks of the TAA intoxication or every 2 days for 4 weeks after cirrhosis development. In vitro: 0.1, 1 and 10 μM | In vivo: Male Wistar TAA-cirrhotic rats In vitro: hepatocytes, LSEC, HSC and Kupffer cells isolated from mice liver | In vivo: ↓ liver fibrosis, ↓inflammation | ↓ PP, ↓ HVR | ↓ LSEC activation, ↓ Kupffer cell activation, ↓HSC activation | [185] | |
| Targeting other vascular alterations | Enoxaparin (anticoagulant) | In vivo: 1.8 mg/kg. -Short-term: Daily for 1 week (CCl4) -Long-term: Daily for 3 weeks (CCl4 and TAA) -Preventive: Daily for the last 3 weeks of the induction of cirrhosis | In vivo: Male Wistar CCl4 or male Sprague-Dawley TAA-cirrhotic rats In vitro: primary HSCs isolated from CCl4-cirrhotic rats | In vivo: ↓ liver fibrosis, ↑liver function, ↓ liver microthrombosis | ↓ PP, ↓ HVR | In vivo: ↓ HSC activation, ↓ oxidative stress, In vitro: improved HSC phenotype | [186] |
| Rivaroxaban (Anticoagulant) | In vivo: 20 mg/kg/day for 2 weeks. In vitro: 25, 50, 100 ng/mL during 24 h | In vivo: Wistar CCl4 or Sprague-Dawley TAA-cirrhotic rats In vitro: primary HSCs isolated from CCl4-cirrhotic rats | No regression of fibrosis | ↓ PP | In vivo: ↑ NO (CCl4), ↑ LSEC phenotype, ↓HSC activation, ↓ liver microthrombosis. In vitro: ↑ HSC phenotype | [187] | |
| Sorafenib (multikinase inhibitor) | In vivo: 2 mg/kg/day for 2 weeks in PPVL rats. 1 mg/kg/day for 2 weeks in cBDL-cirrhotic rats. | Male Sprague-Dawley PPVL or cBDL-cirrhotic rats (PPVL, cBDL) | ↓ splanchnic neovascularization, ↓inflammation, ↓ liver damage, ↓ liver fibrosis, ↓ angiogenesis | ↓ PP (cBDL) | - | [188] | |
| Sorafenib, Imatinib or the combination of both (multikinase inhibitors) | 30 mg/kg or 50 mg/kg, respectively, five times/week for 3 weeks | Female Balb/c Concanavalin A-acute liver fibrosis mice | ↓ liver fibrosis | - | ↓ HSC activation | [189] | |
| Cell death and inflammation | Emricasan (Caspase inhibitor) | 10 mg/kg/day for 7 days, starting 1 week after the animals developed cirrhosis | Male Wistar CCl4-cirrhotic rats | ↓ AST, ↑ Bile, ↓ fibrosis, ↓ inflammation, ↑ hepatocyte phenotype | ↓ PP, ↓ PPP, ↑ vasodilation | ↓ Cell death, ↓ HSC activation and number, ↑ LSEC fenestrae, ↓ vWF, ↑ NO, improved HSC, LSEC and KC phenotype | [190] |
| Emricasan | 5, 25 or 50 mg daily for 24 weeks | 196 patients with NASH cirrhosis | - | No significant differences in HVPG. Small ↓ HVPG in the compensated subgroup. | - | [191] | |
| Seolnsertib (ASK1 inhibitor) | 18 mg or 6 mg daily for 48 weeks | Two phase III trials: 802 and 877 patients with NASH cirrhosis | No regression of fibrosis | - | - | [192] | |
| Fenofibrate (PPARα agonist) | 25 mg/kg daily for 7 days | Male Wistar CCl4-cirrhotic rats | ↓ liver fibrosis | ↓ PP, ↑ MAP | ↓ HSC activation, ↑ NO bioavailability, ↑ hepatic microvascular function | [193] | |
| Fenofibrate, Lanifibranor pioglitazone and GW501516 (PPAR agonists) | 100, 30, 30, 10 mg/kg/day via oral gavage for up to 6 weeks | Choline-deficient, amino acid-defined high fat diet and WD-fed CCl4-cirrhotic rats | ↓ liver fibrosis, ↓steatosis, ↓liver injury | - | ↓ HSC activation | [194] | |
| Lanifibranor (pan-PPAR agonist) | In vivo: 100 mg/kg/day for two weeks In vitro: 1, 3 or 10 µM for 24 h | In vivo: Male Sprague-Dawley TAA or cBDL-cirrhotic rats In vitro: Primary human liver cells from patients with cirrhosis | In vivo: ↓ liver fibrosis, ↑ liver function, ↓ inflammation | ↓ PP, ↓ HVR | In vivo: ↓ HSC activation, improved LSEC phenotype, ↑ hepatic microvascular function In vitro: improved HSC phenotype, ↓ HSC contraction capacity | [195] | |
| Liraglutide (GLP-1 analogue) | In vivo: 0.5 mg/kg/day for 15 days. In vitro: 50 µM for 72 h | In vivo: Male Wistar TAA-cirrhotic rats In vitro: Primary human HSC from patients with cirrhosis and Immortalized human-activated HSC LX-2 | In vivo: ↓ liver fibrosis | ↓ PP | In vivo: ↓ HSC activation, improved LSEC phenotype, ↑ hepatic microvascular function In vitro: ↓ HSC activation, ↓ HSC contraction capacity, ↓ inflammation | [196] | |
| Liraglutide | 1.8 mg/day for 48 weeks | 52 patients with NASH | ↓ hepatic steatosis, ↓ hepatocyte ballooning | - | - | [197] | |
| Semaglutide (GLP-1 analogue) | 0.4 mg/day for 72 weeks | 162 patients with NASH | ↑ NASH resolution. No improvement in fibrosis stage | - | - | [198] | |
| Metformin | 300 mg/kg/day for 1 week | In vivo: Male Wistar CCl4 or Sprague-Dawley cBDL-cirrhotic rats In vitro: LX-2 cell line | In vivo: ↓ liver fibrosis, ↓ inflammation | ↓ PP, ↓ HVR | In vivo: ↓ HSC activation, ↑ NO bioavailability, ↓ oxidative stress In vitro: ↓ markers of HSC activation | [199] | |
| ObR-Ab (Leptin receptor antagonist) | 8 µg/kg/day for 1 week | Male Wistar CCl4-cirrhotic rats | - | ↓ PP | ↑ GMPc, ↓ oxidative stress | [200] | |
| Lifestyle and dietary interventions | Docosahexaenoic acid | After cirrhosis induction, 500 mg/kg/day for two weeks | Male Sprague-Dawley TAA-cirrhotic rats | Recovery of normal fatty acid enzymatic activity and fatty acid composition, ↓ oxidative stress, ↓ inflammation, ↓ steatosis | ↓ PP | ↓ HSC activation, ↓ ECM accumulation | [201] |
| VSL#3 (probiotics) | 900 billion CFU per day for 6 weeks | 17 patients with cirrhosis | ↓ bacterial translocation, ↓ inflammation | ↓ HVPG | - | [202] | |
| VSL#3 | 900 billion CFU per day for 24 weeks | 130 patients with cirrhosis with a recent episode of hepatic encephalopathy | ↓ CTP and MELD scores, ↓plasma proinflammatory markers (TNFα, IL1β and IL6) | - | - | [203] | |
| VSL#3 | 900 billion CFU per day for 2 months | 94 patients with cirrhosis | ↓ TNFα | ↓ HVPG | - | [204] | |
| CECT7765 (Pseudocatenulatum) | 1 billion CFU daily for 1 week | Male Sprague-Dawley cBDL-cirrhotic rats | ↑ liver function: ↑ bilirubin and alkaline phosphatase. ↓ inflammation: ↓ TNFα, IL-6 and NO. ↑ FXR and eNOS gene expression. ↓ iNOS and COX-2 | ↓ portal vein area and portal flow | [205] | ||
| VSL#3 | 900 billion CFU twice daily for 2 months | 8 cirrhotic patients | - | No changes in HVPG | - | [206] | |
| VSL#3 | 900 billion CFU twice daily for 2 months | 17 patients with decompensated cirrhosis and HVPG ≥10 | - | No changes in HVPG | - | [207] | |
| Curcumin | 50 mg/kg suspended in 0.5% carboxymethyl cellulose daily for 6 weeks | Male Sprague-Dawley rats with NASH induced by HFD | ↓ inflammation, ↓ steatosis, ↓ insulin resistance | ↓ MDA, ↑ hepatic GSH content, ↑ SOD activity, ↑ HO-1 | [208] | ||
| Resveratrol | 10 and 20 mg/kg/day for 2 weeks | In vivo: Male Wistar CCl4-cirrhotic rats In vitro: LX-2 cell line | In vivo: ↓ liver fibrosis, ↓inflammation | ↓ PP | In vivo: ↑ hepatic microvascular function, ↓ oxidative damage, ↓ HSC activation. In vitro: ↓ HSC activation markers | [209] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibert-Ramos, A.; Sanfeliu-Redondo, D.; Aristu-Zabalza, P.; Martínez-Alcocer, A.; Gracia-Sancho, J.; Guixé-Muntet, S.; Fernández-Iglesias, A. The Hepatic Sinusoid in Chronic Liver Disease: The Optimal Milieu for Cancer. Cancers 2021, 13, 5719. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225719
Gibert-Ramos A, Sanfeliu-Redondo D, Aristu-Zabalza P, Martínez-Alcocer A, Gracia-Sancho J, Guixé-Muntet S, Fernández-Iglesias A. The Hepatic Sinusoid in Chronic Liver Disease: The Optimal Milieu for Cancer. Cancers. 2021; 13(22):5719. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225719
Chicago/Turabian StyleGibert-Ramos, Albert, David Sanfeliu-Redondo, Peio Aristu-Zabalza, Ana Martínez-Alcocer, Jordi Gracia-Sancho, Sergi Guixé-Muntet, and Anabel Fernández-Iglesias. 2021. "The Hepatic Sinusoid in Chronic Liver Disease: The Optimal Milieu for Cancer" Cancers 13, no. 22: 5719. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225719