
Identification of a Distinct miRNA Regulatory Network in the Tumor Microenvironment of Transformed Mycosis Fungoides

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Samples
2.2. RNA Sequencing and Library Preparation
2.3. Sequencing Data Analysis
2.4. Quantitative Real Time (qRT)-PCR Validation
2.5. Functional Gene Set Enrichment Analysis
2.6. Multiplex Immunofluorescence Staining
2.7. Statistical Analysis
3. Results
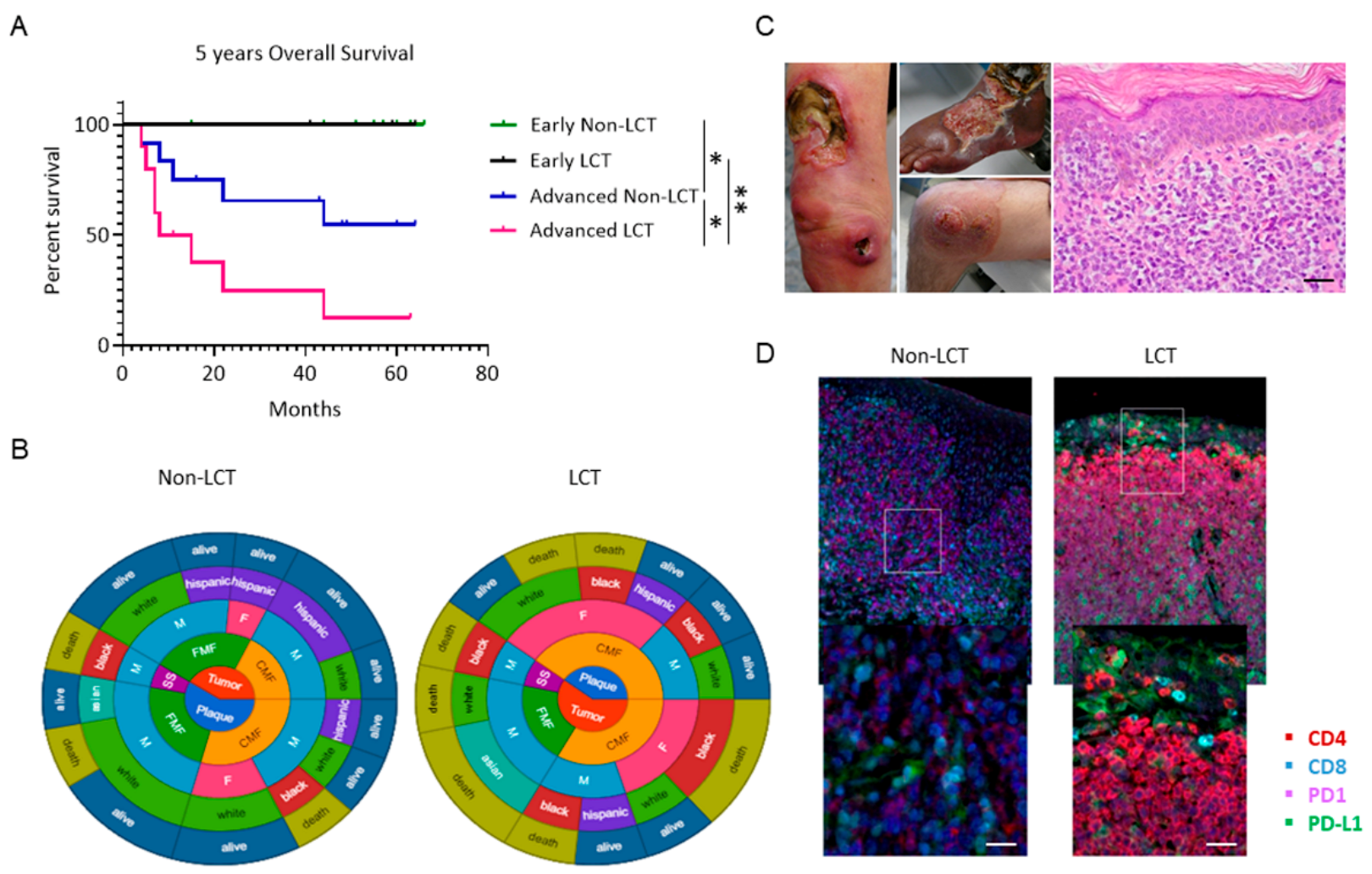
3.1. Outcomes for Patients with LCT-MF Are Discouraging
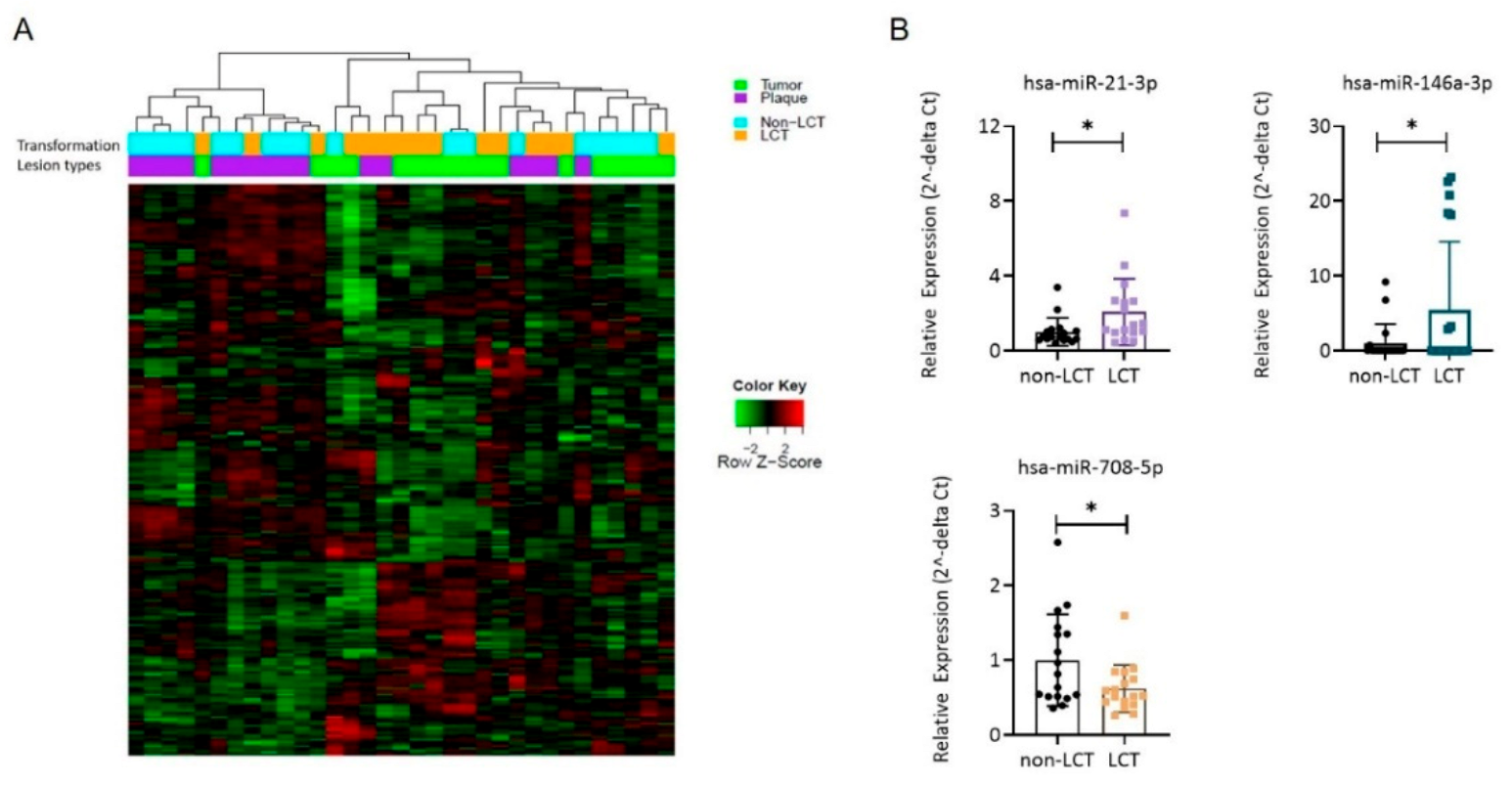
3.2. The miR Expression Profile in LCT-MF Is Distinct from That of Non-LCT
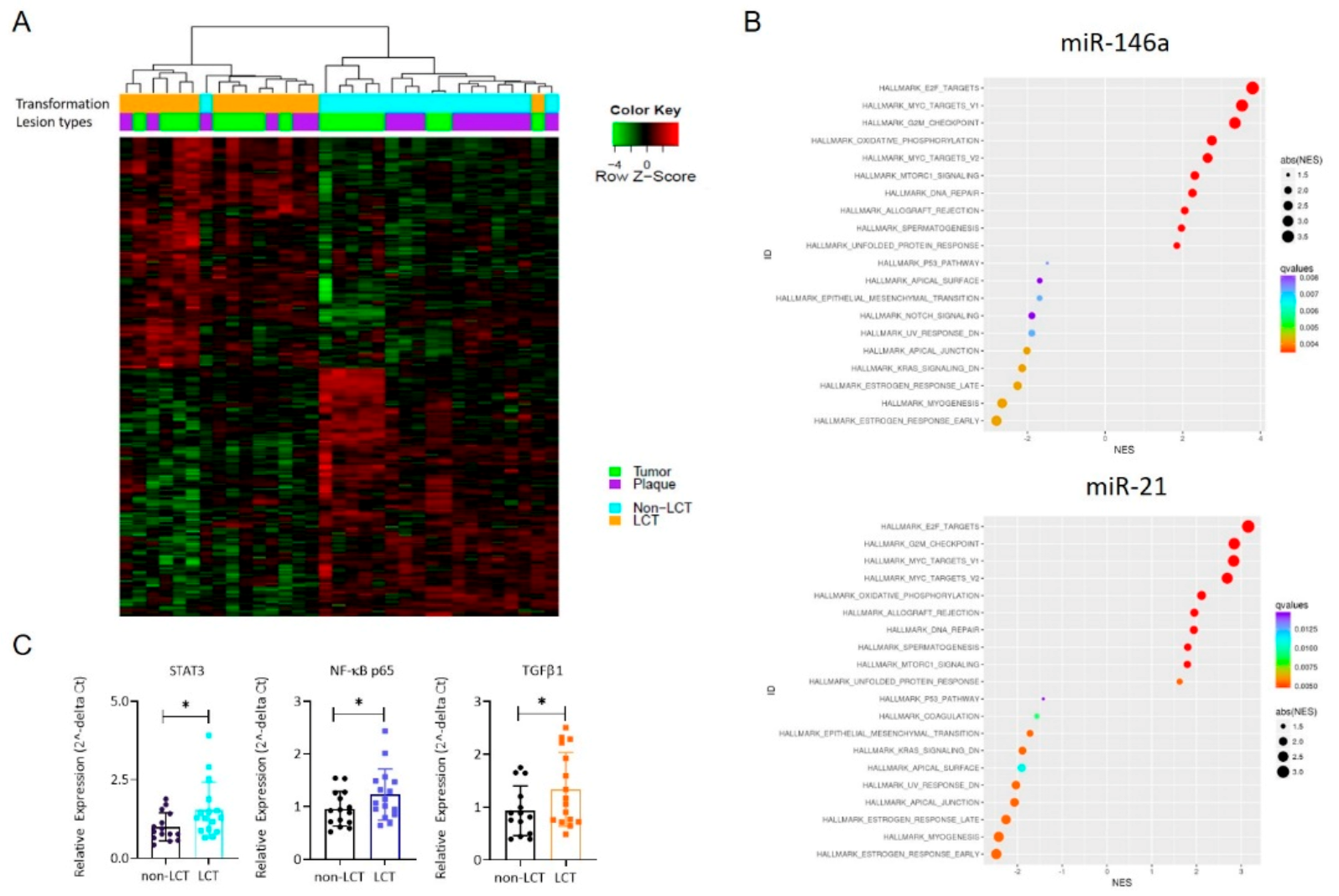
3.3. MRNA Expression Profiling Distinguishes LCT-MF from Non-LCT MF
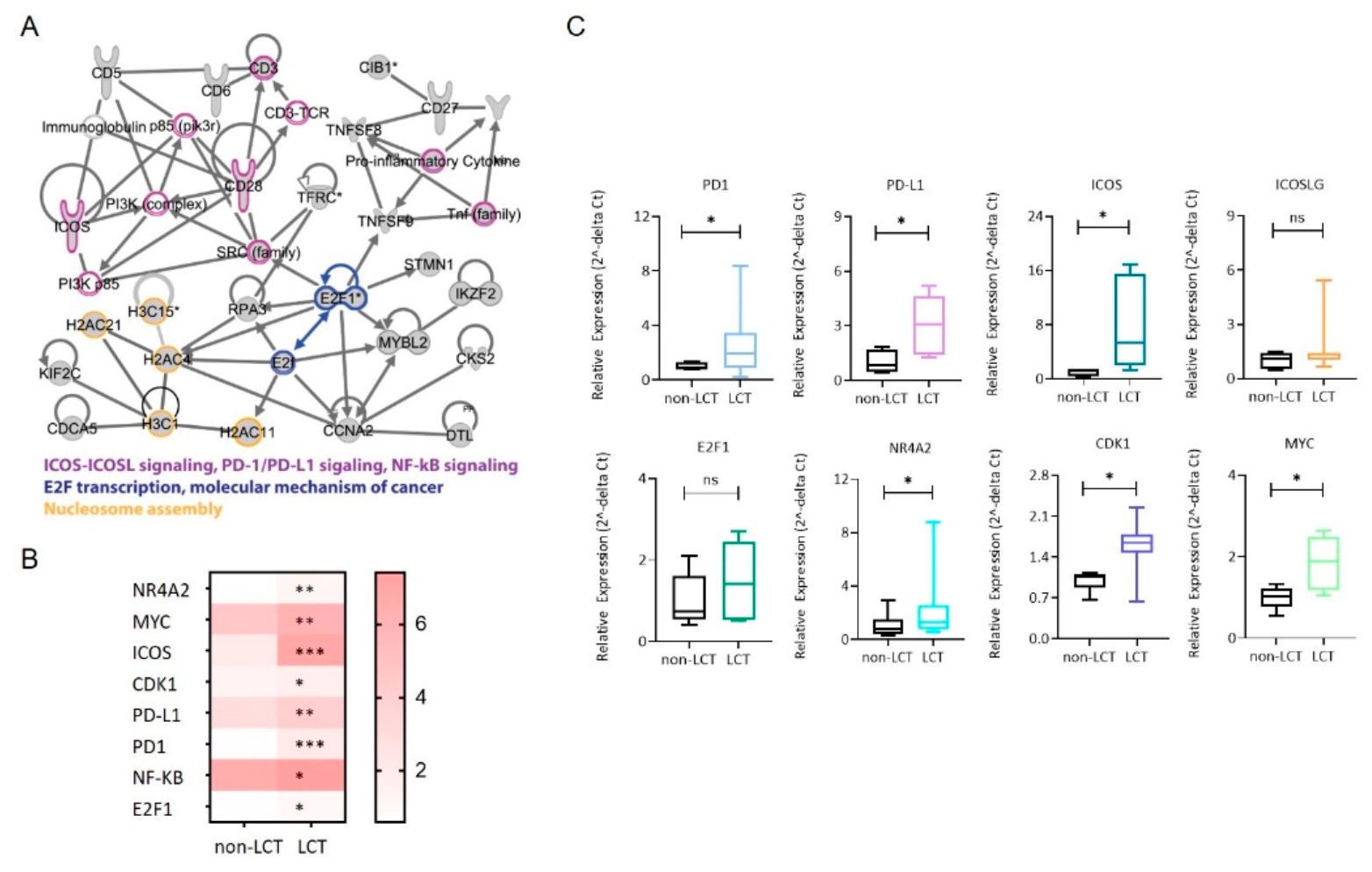
3.4. Identification of a Functional Interaction Network of Upregulated Genes in LCT-MF That Positively Correlated with miR-146a and miR-21
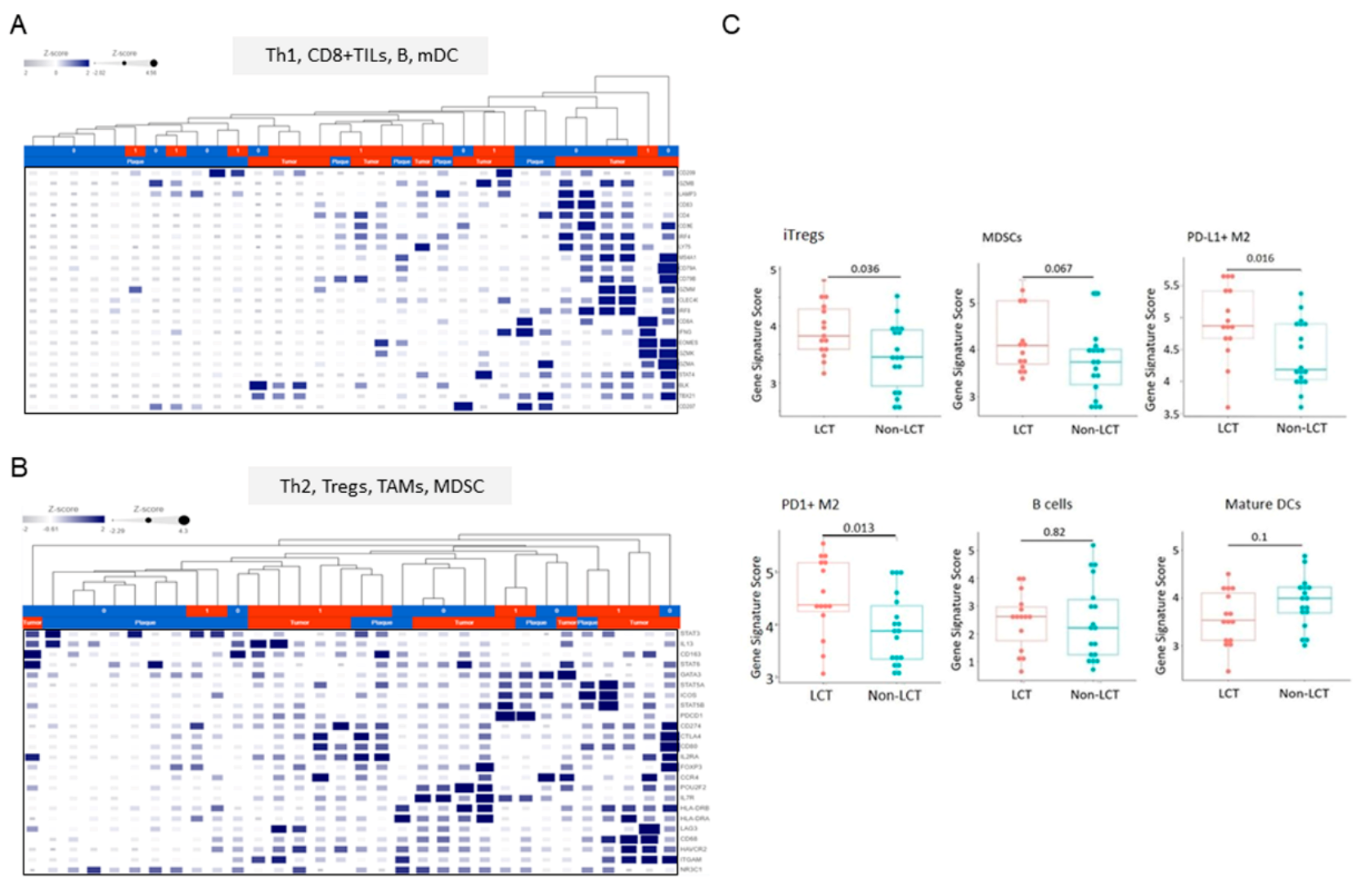
3.5. Distinct Microenvironmental Immune Cell Populations in LCT-MF
3.6. MiR Expression Correlates with the Immune Cell Gene Score in LCT-MF
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jawed, S.I.; Myskowski, P.L.; Horwitz, S.; Moskowitz, A.; Querfeld, C. Primary cutaneous T-cell lymphoma (mycosis fungoides and Sézary syndrome). J. Am. Acad. Dermatol. 2014, 70, 205.e1–205.e16. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Agar, N.S.; Wedgeworth, E.; Crichton, S.; Mitchell, T.; Cox, M.; Ferreira, S.; Robson, A.; Calonje, E.; Stefanato, C.M.; Wain, E.M.; et al. Survival Outcomes and Prognostic Factors in Mycosis Fungoides/Sézary Syndrome: Validation of the Revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer Staging Proposal. J. Clin. Oncol. 2010, 28, 4730–4739. [Google Scholar] [CrossRef] [PubMed]
- Vergier, B.; De Muret, A.; Beylot-Barry, M.; Vaillant, L.; Ekouevi, D.; Chene, G.; Carlotti, A.; Franck, N.; Dechelotte, P.; Souteyrand, P.; et al. Transformation of mycosis fungoides: Clinicopathological and prognostic features of 45 cases. French Study Group of Cutaneious Lymphomas. Blood 2000, 95, 2212–2218. [Google Scholar]
- Pulitzer, M.; Myskowski, P.L.; Horwitz, S.M.; Querfeld, C.; Connolly, B.; Li, J.; Murali, R. Mycosis fungoides with large cell transformation: Clinicopathological features and prognostic factors. Pathology 2014, 46, 610–616. [Google Scholar] [CrossRef] [Green Version]
- Kadin, M.E.; Hughey, L.C.; Wood, G.S. Large-cell transformation of mycosis fungoides–differential diagnosis with implications for clinical management: A consensus statement of the US Cutaneous Lymphoma Consortium. J. Am. Acad. Dermatol. 2014, 70, 374–376. [Google Scholar] [CrossRef] [PubMed]
- Scarisbrick, J.J.; Prince, H.M.; Vermeer, M.; Quaglino, P.; Horwitz, S.; Porcu, P.; Stadler, R.; Wood, G.S.; Beylot-Barry, M.; Pham-Ledard, A.; et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sézary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J. Clin. Oncol. 2015, 33, 3766–3773. [Google Scholar] [CrossRef]
- Mao, X.; Orchard, G.; Lillington, D.; Child, F.; Vonderheid, E.; Nowell, P.; Bagot, M.; Bensussan, A.; Russell-Jones, R.; Young, B.; et al. BCL2 and JUNB abnormalities in primary cutaneous lymphomas. Br. J. Dermatol. 2004, 151, 546–556. [Google Scholar] [CrossRef]
- Mao, X.; Orchard, G.; Lillington, D.M.; Russell-Jones, R.; Young, B.D.; Whittaker, S.J. Amplification and overexpression of JUNB is associated with primary cutaneous T-cell lymphomas. Blood 2003, 101, 1513–1519. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.; Kaestel, C.G.; Eriksen, K.W.; Woetmann, A.; Stokkedal, T.; Kaltoft, K.; Geisler, C.; Röpke, C.; Ødum, N. Inhibition of constitutively activated Stat3 correlates with altered Bcl-2/Bax expression and induction of apoptosis in mycosis fungoides tumor cells. Leukemia 1999, 13, 735–738. [Google Scholar] [CrossRef] [Green Version]
- Navas, I.C.; Ortiz-Romero, P.L.; Villuendas, R.; Martínez, P.; García, C.; Gómez, E.; Rodriguez, J.L.; García, D.; Vanaclocha, F.; Iglesias, L.; et al. p16INK4a Gene Alterations Are Frequent in Lesions of Mycosis Fungoides. Am. J. Pathol. 2000, 156, 1565–1572. [Google Scholar] [CrossRef] [Green Version]
- Hrovatin, K.; Kunej, T. Classification of miRNA-related sequence variations. Epigenomics 2018, 10, 463–481. [Google Scholar] [CrossRef]
- Shen, X.; Wang, B.; Li, K.; Wang, L.; Zhao, X.; Xue, F.; Shi, R.; Zheng, J. MicroRNA Signatures in Diagnosis and Prognosis of Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2018, 138, 2024–2032. [Google Scholar] [CrossRef] [Green Version]
- Ralfkiaer, U.; Hagedorn, P.H.; Bangsgaard, N.; Løvendorf, M.; Ahler, C.B.; Svensson, L.; Kopp, K.L.; Vennegaard, M.T.; Lauenborg, B.; Zibert, J.R.; et al. Diagnostic microRNA profiling in cutaneous T-cell lymphoma (CTCL). Blood 2011, 118, 5891–5900. [Google Scholar] [CrossRef]
- Querfeld, C.; Foss, F.M.; Kim, Y.H.; Pinter-Brown, L.; William, B.M.; Porcu, P.; Pacheco, T.; Haverkos, B.M.; DeSimone, J.; Guitart, J.; et al. Phase 1 Trial of Cobomarsen, an Inhibitor of Mir-155, in Cutaneous T Cell Lymphoma. Blood 2018, 132, 2903. [Google Scholar] [CrossRef]
- Querfeld, C. The emerging role of micro RNA s in the molecular diagnosis of mycosis fungoides. Br. J. Dermatol. 2019, 180, 984–985. [Google Scholar] [CrossRef]
- Lindahl, L.M.; Besenbacher, S.; Rittig, A.H.; Celis, P.; Willerslev-Olsen, A.; Gjerdrum, L.M.R.; Krejsgaard, T.; Johansen, C.; Litman, T.; Woetmann, A.; et al. Prognostic miRNA classifier in early-stage mycosis fungoides: Development and validation in a Danish nationwide study. Blood 2018, 131, 759–770. [Google Scholar] [CrossRef] [Green Version]
- Manso, R.; Martínez-Magunacelaya, N.; Eraña-Tomás, I.; Monsálvez, V.; Rodríguez-Peralto, J.L.; Ortiz-Romero, P.-L.; Santonja, C.; Cristóbal, I.; A Piris, M.; Rodriguez-Pinilla, S.M. Mycosis fungoides progression could be regulated by microRNAs. PLoS ONE 2018, 13, e0198477. [Google Scholar] [CrossRef]
- Olsen, E.; Vonderheid, E.; Pimpinelli, N.; Willemze, R.; Kim, Y.; Knobler, R.; Zackheim, H.; Duvic, M.; Estrach, T.; Lamberg, S.; et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: A proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007, 110, 1713–1722. [Google Scholar] [CrossRef] [Green Version]
- Querfeld, C.; Leung, S.; Myskowski, P.L.; Curran, S.A.; Goldman, D.A.; Heller, G.; Wu, X.; Kil, S.H.; Sharma, S.; Finn, K.J.; et al. Primary T Cells from Cutaneous T-cell Lymphoma Skin Explants Display an Exhausted Immune Checkpoint Profile. Cancer Immunol. Res. 2018, 6, 900–909. [Google Scholar] [CrossRef] [Green Version]
- Benner, M.F.; Jansen, P.M.; Vermeer, M.; Willemze, R. Prognostic factors in transformed mycosis fungoides: A retrospective analysis of 100 cases. Blood 2012, 119, 1643–1649. [Google Scholar] [CrossRef] [Green Version]
- Duvic, M.; Olsen, E.A.; Breneman, D.; Pacheco, T.R.; Parker, S.; Vonderheid, E.C.; Abuav, R.; Ricker, J.L.; Rizvi, S.; Chen, C.; et al. Evaluation of the Long-Term Tolerability and Clinical Benefit of Vorinostat in Patients With Advanced Cutaneous T-Cell Lymphoma. Clin. Lymphoma Myeloma 2009, 9, 412–416. [Google Scholar] [CrossRef]
- Han, Z.; Estephan, R.J.; Wu, X.; Su, C.; Yuan, Y.C.; Qin, H.; Kil, S.H.; Morales, C.; Schmolze, D.; Sanchez, J.F.; et al. MiRNA regulation of T cell exhaustion in cutaneous T cell lymphoma. J. Investig. Dermatol. 2021, in press. [Google Scholar] [CrossRef]
- Moyal, L.; Barzilai, A.; Gorovitz, B.; Hirshberg, A.; Amariglio, N.; Jacob-Hirsch, J.; Maron, L.; Feinmesser, M.; Hodak, E. miR-155 is involved in tumor progression of mycosis fungoides. Exp. Dermatol. 2013, 22, 431–433. [Google Scholar] [CrossRef] [Green Version]
- Labbaye, C.; Testa, U. The emerging role of MIR-146A in the control of hematopoiesis, immune function and cancer. J. Hematol. Oncol. 2012, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Testa, U.; Pelosi, E.; Castelli, G.; Labbaye, C. miR-146 and miR-155: Two Key Modulators of Immune Response and Tumor Development. Non-Coding RNA 2017, 3, 22. [Google Scholar] [CrossRef]
- Ralfkiaer, U.; Lindahl, L.M.; Lindal, L.; Litman, T.; Gjerdrum, L.M.R.; Ahler, C.B.; Gniadecki, R.; Marstrand, T.; Fredholm, S.; Iversen, L.; et al. MicroRNA expression in early mycosis fungoides is distinctly different from atopic dermatitis and advanced cutaneous T-cell lymphoma. Anticancer. Res. 2014, 34, 7207–7217. [Google Scholar]
- Lindahl, L.M.; Fredholm, S.; Joseph, C.; Nielsen, B.S.; Jønson, L.; Willerslev-Olsen, A.; Gluud, M.; Blümel, E.; Petersen, D.L.; Sibbesen, N.; et al. STAT5 induces miR-21 expression in cutaneous T cell lymphoma. Oncotarget 2016, 7, 45730–45744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasham, A.; Samuel, W.; Cao, H.; Patel, R.; Mehta, R.; Stern, J.L.; Reid, G.; Woolley, A.G.; Miller, L.D.; A Black, M.; et al. YB-1, the E2F Pathway, and Regulation of Tumor Cell Growth. J. Natl. Cancer Inst. 2012, 104, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lummus, S.C.; Donson, A.M.; Gowan, K.; Jones, K.L.; Vibhakar, R.; Foreman, N.K.; Kleinschmidt-DeMasters, B.K. p16Loss and E2F/cell cycle deregulation in infant posterior fossa ependymoma. Pediatr. Blood Cancer 2017, 64, e26656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteleone, N.; Lutz, C.S. miR-708-5p: A microRNA with emerging roles in cancer. Oncotarget 2017, 8, 71292–71316. [Google Scholar] [CrossRef] [Green Version]
- Han, B.-W.; Feng, D.-D.; Li, Z.-G.; Luo, X.-Q.; Zhang, H.; Li, X.-J.; Zhang, X.-J.; Zheng, L.-L.; Zeng, C.-W.; Lin, K.-Y.; et al. A set of miRNAs that involve in the pathways of drug resistance and leukemic stem-cell differentiation is associated with the risk of relapse and glucocorticoid response in childhood ALL. Hum. Mol. Genet. 2011, 20, 4903–4915. [Google Scholar] [CrossRef]
- Huang, W.; Wang, W.-T.; Fang, K.; Chen, Z.-H.; Sun, Y.-M.; Han, C.; Sun, L.-Y.; Luo, X.-Q.; Chen, Y.-Q. MIR-708 promotes phagocytosis to eradicate T-ALL cells by targeting CD47. Mol. Cancer 2018, 17, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Nirmal, A.J.; Regan, T.; Shih, B.B.; Hume, D.A.; Sims, A.H.; Freeman, T.C. Immune Cell Gene Signatures for Profiling the Microenvironment of Solid Tumors. Cancer Immunol. Res. 2018, 6, 1388–1400. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, B.R.; Zain, J.; Rosen, S.T.; Querfeld, C. Tumor microenvironment in mycosis fungoides and Sézary syndrome. Curr. Opin. Oncol. 2016, 28, 88–96. [Google Scholar] [CrossRef]
- Schwingshackl, P.; Obermoser, G.; Nguyen, V.; Fritsch, P.; Sepp, N.; Romani, N. Distribution and Maturation of Skin Dendritic Cell Subsets in Two Forms of Cutaneous T-Cell Lymphoma: Mycosis Fungoides and Sézary Syndrome. Acta Derm. Venereol. 2012, 92, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Luftl, M.; Feng, A.; Licha, E.; Schuler, G. Dendritic cells and apoptosis in mycosis fungoides. Br. J. Dermatol. 2002, 147, 1171–1179. [Google Scholar] [CrossRef]
- Wu, X.; Schulte, B.C.; Zhou, Y.; Haribhai, D.; Mackinnon, A.; Plaza, J.A.; Williams, C.B.; Hwang, S.T. Depletion of M2-Like Tumor-Associated Macrophages Delays Cutaneous T-Cell Lymphoma Development In Vivo. J. Investig. Dermatol. 2014, 134, 2814–2822. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, P.; Wang, X.-F. Microenvironmental regulation of cancer metastasis by miRNAs. Trends Cell Biol. 2014, 24, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Shahi, P.; Werb, Z. microRNA-mediated regulation of the tumor microenvironment. Cell Cycle 2013, 12, 3262–3271. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Non-LCT | LCT |
|---|---|---|
| Patients/Skin biopsy specimens | 17/20 | 11/14 |
| Age (years), median (range) | 58.4 (30.8–85.4) | 68 (41.3–82.8) |
| Gender | ||
| 14 (82.4%) | 6 (54.5%) |
| 3 (17.6%) | 5 (45.5%) |
| Race/Ethnicity | ||
| 8 (47.1%) | 3 (27.3%) |
| 2 (11.8%) | 6 (54.5%) |
| 5 (29.4%) | 1 (9.1%) |
| 2 (11.8%) | 1 (9.1%) |
| Clinical stage | ||
| 3 (17.6%) | 0 |
| 5 (29.4%) | 3 (27.3%) |
| 8 (47.1%) | 5 (45.5%) |
| 1 (5.9%) | 3 (27.3%) |
| MF histologic subtype | ||
| 11 (64.7%) | 9 (81.8%) |
| 6 (35.3%) | 2 (18.2%) |
| Genes | Log FC | Log CPM | F | p Value | lFDR | Non-LCT | LCT | Status |
|---|---|---|---|---|---|---|---|---|
| hsa-miR-146a-3p | 1.71 | 4.97 | 11.81 | 0.001717 | 0.174221 | 16 | 49 | Up |
| hsa-miR-21-3p | 1.05 | 7.56 | 10.12 | 0.003353 | 0.202974 | 105 | 288 | Up |
| hsa-miR-136-5p | 0.89 | 5.21 | 9.07 | 0.005179 | 0.227844 | 32 | 42 | Up |
| hsa-miR-889 | 0.88 | 6.39 | 8.56 | 0.006415 | 0.242032 | 72 | 96 | Up |
| hsa-miR-539-3p | 0.80 | 3.14 | 8.50 | 0.006581 | 0.243816 | 7 | 10 | Up |
| hsa-miR-708-5p | −1.45 | 8.83 | 12.76 | 0.001194 | 0.163057 | 592 | 290 | Down |
| hsa-miR-744-5p | −0.81 | 7.40 | 11.97 | 0.001618 | 0.172186 | 207 | 122 | Down |
| hsa-miR-3653 | −0.97 | 2.99 | 9.94 | 0.003613 | 0.206878 | 9 | 5 | Down |
| hsa-let-7g-5p | −0.64 | 13.97 | 9.91 | 0.003657 | 0.20753 | 18189 | 13575 | Down |
| hsa-let-7b-5p | −0.71 | 12.52 | 9.17 | 0.004964 | 0.225185 | 7078 | 4459 | Down |
| hsa-miR-664-5p | −0.87 | 3.25 | 8.85 | 0.005685 | 0.233856 | 11 | 7 | Down |
| hsa-miR-5701 | −1.02 | 3.59 | 8.42 | 0.006808 | 0.246205 | 13 | 9 | Down |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Raimondo, C.; Han, Z.; Su, C.; Wu, X.; Qin, H.; Sanchez, J.F.; Yuan, Y.-C.; Martinez, X.; Abdulla, F.; Zain, J.; et al. Identification of a Distinct miRNA Regulatory Network in the Tumor Microenvironment of Transformed Mycosis Fungoides. Cancers 2021, 13, 5854. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225854
Di Raimondo C, Han Z, Su C, Wu X, Qin H, Sanchez JF, Yuan Y-C, Martinez X, Abdulla F, Zain J, et al. Identification of a Distinct miRNA Regulatory Network in the Tumor Microenvironment of Transformed Mycosis Fungoides. Cancers. 2021; 13(22):5854. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225854
Chicago/Turabian StyleDi Raimondo, Cosimo, Zhen Han, Chingyu Su, Xiwei Wu, Hanjun Qin, James F. Sanchez, Yate-Ching Yuan, Xochiquetzal Martinez, Farah Abdulla, Jasmine Zain, and et al. 2021. "Identification of a Distinct miRNA Regulatory Network in the Tumor Microenvironment of Transformed Mycosis Fungoides" Cancers 13, no. 22: 5854. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13225854