
Childhood Malignant Brain Tumors: Balancing the Bench and Bedside


1
Children’s Brain Tumour Research Centre, Department of Paediatric Oncology, Royal Manchester Children’s Hospital, Manchester University NHS Foundation Trust, Manchester M13 9WL, UK
2
The Centre for Paediatric, Teenage and Young Adult Cancer, Institute of Cancer Sciences, The University of Manchester, Manchester M20 4BX, UK
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(23), 6099; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236099
Submission received: 8 November 2021
/
Revised: 29 November 2021
/
Accepted: 29 November 2021
/
Published: 3 December 2021
(This article belongs to the Special Issue Pediatric Cancers)
Abstract
:Simple Summary
Brain tumors remain the most common childhood solid tumors, accounting for approximately 25% of all pediatric cancers. They also represent the most common cause of cancer-related illness and death in this age group. Recent years have witnessed an evolution in our understanding of the biological underpinnings of many childhood brain tumors, potentially improving survival through both improved risk group allocation for patients to provide appropriate treatment intensity, and novel therapeutic breakthroughs. This review aims to summarize the molecular landscape, current trial-based standards of care, novel treatments being explored and future challenges for the three most common childhood malignant brain tumors—medulloblastomas, high-grade gliomas and ependymomas.
Abstract
Brain tumors are the leading cause of childhood cancer deaths in developed countries. They also represent the most common solid tumor in this age group, accounting for approximately one-quarter of all pediatric cancers. Developments in neuro-imaging, neurosurgical techniques, adjuvant therapy and supportive care have improved survival rates for certain tumors, allowing a future focus on optimizing cure, whilst minimizing long-term adverse effects. Recent times have witnessed a rapid evolution in the molecular characterization of several of the common pediatric brain tumors, allowing unique clinical and biological patient subgroups to be identified. However, a resulting paradigm shift in both translational therapy and subsequent survival for many of these tumors remains elusive, while recurrence remains a great clinical challenge. This review will provide an insight into the key molecular developments and global co-operative trial results for the most common malignant pediatric brain tumors (medulloblastoma, high-grade gliomas and ependymoma), highlighting potential future directions for management, including novel therapeutic options, and critical challenges that remain unsolved.
1. Introduction
Brain tumors are the most common solid tumors of childhood, accounting for approximately 25% of all pediatric malignancies, and represent the leading cause of cancer-induced morbidity and mortality in this age group [1]. With an incidence of approximately 6 per 100,000 children in industrialized society [2], these tumors represent a spectrum of clinically, pathologically and biologically diverse subtypes which can pose significant challenges in conducting research and clinical trials, necessitating international collaboration.
Over recent decades, cure rates for selected pediatric brain tumors (most notably medulloblastoma) have improved [3], predominantly as a consequence of advances in multiparametric neuro-imaging, neurosurgical techniques, radiation therapy and multiagent chemotherapy, together with improved supportive care. However, such survival advances are typically offset by a therapy-induced toxicity burden for the patient, with wide-reaching consequences for the child, their family and society. Moreover, for the majority of brain tumors, prognosis has remained static for over 30 years despite these technological improvements.
To overcome this impasse, the pediatric neuro-oncology community has shifted focus to develop risk-stratified treatment protocols that aim to reduce iatrogenic morbidity while maintaining outcomes for favorable-risk lesions, and improve cure rates for tumors refractory to conventional therapy, either through intensification or novel agents. This strategy has been supplemented by an evolution in our understanding of the molecular pathogenesis of almost all pediatric brain tumors.
Such molecular advances have identified potential cells of origin, and led to the identification of multiple biologically distinct subgroups within most brain tumor entities, therein allowing accurate risk stratification for affected children when incorporated with clinical, histological and survival data. In addition, oncogenic biological pathways amenable to manipulation using novel targeted agents have been identified.
This article will provide a summary of the most common malignant pediatric brain tumors (medulloblastoma, high-grade gliomas and ependymoma) with particular focus on inherent molecular advancements and potential future directions for management, including novel therapeutic options.
2. Medulloblastoma
2.1. Background
Medulloblastoma (MB) represents the most common malignant brain tumor in children, accounting for approximately 20% of all central nervous system (CNS) tumors [2,4]. It also comprises over 60% of intracranial embryonal tumors, a recently characterized entity consisting of atypical teratoid rhabdoid tumors (ATRTs), embryonal tumors with multilayer rosettes (ETMRs), CNS neuroblastoma with FOX2 alteration and malignant neuroepithelial tumors with BCOR alteration [5].
Arising within the cerebellum, MBs are observed across all age categories but are most frequently identified at a median age of five years [6]. Demographic, histological and prognostic heterogeneity embody MB, while it represents the first brain tumor where revolutionary global initiatives (such as the Medulloblastoma Advanced Genomics International Consortium (MAGIC)) have transformed our understanding of the molecular underpinnings of MB pathogenesis, enabling improved patient risk stratification to potentially influence clinical outcome [7].
2.2. Histopathology
MBs share a primitive embryonal phenotype comprising malignant cells of stereotypic histological patterns, dominated by neuronal antigen expression [8]. World Health Organization (WHO) pathological classification systems have historically divided MB into a classic subtype accounting for 72% of all cases, a desmoplastic/nodular variant of which medulloblastoma with extensive nodularity (MBEN) is a subgroup and a large cell/anaplastic variant which has historically been assigned an adverse prognostic association [5,9].
2.3. Molecular Classification
In the past decade, seminal transcriptomic MB analyses led to a global consensus establishing the identification of four discrete molecular subgroups, likely arising from distinct cells of origin—wingless-activated (WNT), sonic hedgehog (SHH), Group 3 and Group 4 MB [10,11]. Further molecular scrutiny of these four groups has now identified somatic mutations targeting chromatin modification as the leading driver for MB heterogeneity via epigenetic dysregulation [12]; further subdivisions have now been established [13,14,15,16] (Figure 1).
2.3.1. WNT Activated (WNT)
WNT MBs account for approximately 10% of all MBs, and often arise in older children with equal gender distribution [11]. Typically occurring in the midline, they frequently invade the lateral recess of the brainstem through the foramen of Luschka, due to a lower rhombic lip cell of origin [17,18]. They rarely metastasize and morphology is typically of the classic variant [8].
Somatic activating mutations in exon 3 of CTTNB1, which encodes B-catenin, are found in 80–90% of WNT MB, with 85–90% displaying monosomy 6 [19,20,21,22,23]. Mutations in the adenomatous polyposis coli (APC) gene are common in WNT tumors lacking CTTNB1 mutations [15,24]. Less frequently occurring mutations include TP53, SMARCA4, KMT2D and DDX3X [11,15,25,26]. TP53 mutation occurs only in a minority of WNT MB, and is not prognostic, unlike the SHH subtype [27].
2.3.2. Sonic Hedgehog-Activated-Activated (SHH)
SHH MB represents approximately 30% of all cases, presenting predominantly in a bimodal age distribution; below three years and in young adults [5,8,10]. Originating from granule progenitor cells SHH MBs localize almost exclusively within cerebellar hemispheres [17,28]. All nodular desmoplastic MBs belong to the SHH subgroup, although other histologies can be observed [21,29]. They are most commonly localized at diagnosis and morphology frequently correlates with underlying genetic abnormalities.
SHH MBs are characterized by activation of the SHH pathway as a result of somatic or germline mutations in a number of genes including SMO, PTCH1 and SUFU [30]. While PTCH1 mutations are seen across 30–50% of SHH MBs, SUFU and SMO mutations are typically seen in infant and adult SHH MBs, respectively [30]. TP53 mutations typically arise in childhood SHH MBs [27]. Recent epigenomic profiling has identified a further four clinically distinct granular molecular subclasses of SHH MB, alpha, beta, gamma and delta [13]. SHH-alpha MBs predominate in children, whereas infants are most commonly associated with SHH-beta and SHH-gamma, and SHH-delta is typically observed in adult patients [8].
2.3.3. Group 3
Group 3 tumors account for 25% of all MB cases, predominate in males and occur most frequently in younger children between the ages of 2 and 5 years [8]. Thought to arise from neural stem cell origin [31], Group 3 MBs have a short symptom interval and are frequently metastatic at diagnosis with small primary tumors [11,28,32].
As with Group 4 MB, Group 3 tumors are not characterized by a signature oncogenic pathway. Nevertheless, Group 3 MBs can be associated with activation of GABAergic and photoreceptor pathways [33,34]. Broad genomic aberrations are a feature, while recurrent somatic nucleotide variants are infrequent [7,12,26]. MYC amplification is the most common finding (in approximately 17% of cases) commonly occurring within a complex chromosomal rearrangement at the 8q24 locus, resulting in MYC–PVT1 fusion [7,12,13,34]. The presence of isochromosome 17q, activation of growth factor proto-oncogenes GFI1 and GFI1B, and amplification of transcription factor OTX2 are also observed [13,15,35].
2.3.4. Group 4
Group 4 tumors represent 35% of all MBs, have a male predisposition and are the dominant molecular subgroup in children of 3 to 16 years of age [8,36]. Similarly to Group 3 MB, they arise in the fourth ventricle and are frequently metastatic at diagnosis, but have a longer symptom interval [11,32].
Genetic abnormalities seen in Group 4 tumors include inactivating mutations of the histone demethylase KDMS6A and histone modulator PRDM6, tandem duplications of SNCAIP and amplifications of CDK6 and MYCN [7,12,25,26,33]. Chromosomal copy number variations include deletion of chromosome 8, 11 or 18p, gain of chromosome 1 or 17q and isochromosome 17q, the most common cytogenetic abnormality in the subgroup [37].
2.4. Prognostic Factors
Typical risk-stratification systems for MB incorporate age, extent of tumor resection, and metastatic status to define standard and high-risk cohorts, in turn determining therapy administered. Standard-risk patients are older than 3 years, have undergone gross or near total excision (below 1.5 cm2 of residual tumor) with localized disease while remaining patients are classified as high risk. However, these and historical prognostic markers (such as anaplastic morphology) may indeed be surrogates for the underlying MB molecular subgroup, suggesting future stratifications require further refinement.
Pediatric patients with standard-risk WNT-activated MB have an excellent prognosis with a 5 year progression-free survival above 90% following standard therapy. SHH MB demonstrates a range of outcomes. Infant SHH MBs beta and gamma have disparate outcomes, with beta conferring a poor prognosis, and gamma good outcomes [38,39]. TP53 germline positive SHH MBs confer a poor prognosis with a post-therapy 5 year survival of just 30–40%, particularly when associated with MYCN and GLI2 amplification [40], whereas wildtype SHH MB are associated with a favorable outcome with a 5 year survival of approximately 80% [8,27,30].
Group 3 and 4 MBs also demonstrate variable outcomes, influenced by inherent molecular heterogeneity spanning both groups [14]. For example, Group 3 MB generally carry a poor prognosis, particularly MYC amplified cases which are often refractory to conventional therapy [41,42,43], while Group 4 MBs demonstrate a variable prognosis, incorporating favorable-risk MBs harboring chromosome 11 loss or chromosome 17 gain [14]. Infantile Group 4 MBs are infrequent but carry a poor prognosis [44].
2.5. Current Management/Clinical Trials
The sequential trial-based addition of adjuvant craniospinal radiotherapy and combination chemotherapy to maximal safe tumor resection has improved survival rates for standard-risk patients immeasurably over the last 50 years and is now the accepted standard of care (Table 1). However, such improved cure rates are achieved at a significant burden to the survivor, with most experiencing chronic neurocognitive and neuroendocrine morbidities [45,46]. While standard-risk patients have benefited from a trial-validated reduction in craniospinal radiotherapy intensity [47] (Table 1), high-risk patients continue to require high-dose radiotherapy (36 Gy) and intensified chemotherapy regimens to maintain a 5 year progression-free survival (PFS) of up to 70% [48,49] (Table 1).
Current trial designs utilize refined patient risk stratifications which incorporate the additional knowledge of molecular MB subgroups. Open standard-risk studies including the Children’s Oncology Group (COG) ACNS1422 (NCT02724579), the North American SJMB12 (NCT01878617) and the European SIOP PNET5 trial (NCT02066220) are assessing whether treatment intensity can be reduced without compromising survival rates for favorable-risk MBs (particularly WNT-activated MBs).
Caution regarding de-escalation of therapy for WNT-activated MBs is evident from the premature termination of trial NCT02212574 which abandoned craniospinal irradiation for these patients, and a recent retrospective analysis of 93 WNT-activated MBs where relapse was associated with a reduction in the cumulative dosing of maintenance chemotherapy [61].
The PNET5 trial is also assessing the radio-sensitizing effect of carboplatin for non-WNT MB, while SJMB12 is the addition of targeted drug therapy in conjunction with conventional agents for specific molecular subgroups (SHH and high-risk Group 3 and 4 MBs). A European biomarker-driven phase III trial for newly stratified high-risk MB opened to recruitment in 2021. Of interest, post-operative residual tumor is not considered a high-risk feature in this study. The trial incorporates a double-randomized design, comparing the efficacy of hyper-fractionated radiotherapy and additional high-dose chemotherapy against standard radiotherapy, followed by a comparison of multimodal continuation chemotherapy versus single agent temozolomide (EudraCT Number: 2018-004250-17).
Infant MB represents a distinct, intensive chemotherapy-only treatment group [29]. Outcomes for infants with nodular desmoplastic SHH MB can be excellent, although it appears that this requires the inclusion of intrathecal methotrexate in addition to systemic therapy [38,39,62,63]. The COG ACNS0334 study of non-nodular desmoplastic MBs, incorporating both induction and high-dose tandem consolidation cycles of chemotherapy reported 100% survival for metastatic SHH MBs and a survival advantage for the incorporation of methotrexate at induction in Group 3MBs [64].
2.6. Novel Therapies
Advances in molecular understanding of MB pathogenesis have also provided the opportunity for the application of subgroup-specific novel targeted therapeutics, notably for SHH MBs. Vismodegib and sonidegib are SMO inhibitors that have shown objective responses in pediatric recurrent SHH MB [65,66,67,68,69,70,71]. For most patients, such responses were not sustained, as a result of mutations downstream from SMO re-activating the pathway [30]. Another important consideration of this therapy is the association with premature growth plate fusions which has led to modification of the current SJMB12 study [70,71,72]. Agents such as silmitasertib, targeting SMO downstream mutations in the SHH pathway, are under evaluation in relapsed SHH MB (NCT03904862). GLI inhibition by arsenic trioxide is another area of drug development in SHH MB and early phase pediatric tumor trial data are awaited (NCT00024258).
For non-SHH tumors, the aforementioned SJMB12 study is evaluating the addition of pemetrexed and gemcitabine to conventional chemotherapeutic agents for high-risk Group 3 and 4 MBs (large cell anaplastic histology, metastatic disease or MYC/MYCN upregulation) after promising high throughput in vitro drug assay analysis [73]. The CDK4/6-cyclin D-Rb pathway was identified as a potential therapeutic target in xenograft models for non-WNT MB [74]. Other proposed approaches include HDAC inhibitors, PI3K inhibition and BET-bromodomain inhibition to downregulate MYC expression in Group 3 MBs, and LSD1 inhibition of GFI1/GFI1B overexpression when present in Group 3 and 4 MBs [75,76,77,78].
Finally, despite the challenge posed by the lack of immunogenic targets in CNS tumors, immunotherapy has been proposed as a potential treatment option in relapsed/refractory MB [79]. Anti-EPHA2, HER2 and IL-13Rα2 chimeric antigen receptor T-cell (CAR-T) therapy has been shown to successfully treat murine Group 3 MBs [80] and early phase trials in children have commenced (NCT03500991, NCT04661384).
3. High-Grade Gliomas
3.1. Background
This group encapsulates all malignant lesions of glial origin. Alongside embryonal tumors, pediatric high-grade gliomas (pHGGs) are one of the most common malignant tumor groups of the childhood central nervous system, with a collective incidence of 1.1 per 100,000 children [2]. Despite a paradigm shift in our understanding of pHGG molecular subgrouping being distinct from adult counterparts, and some therapeutic successes for particular entities (such as infant HGG), little progress has been made over recent decades to improve the dismal prognosis; pHGGs account for over 40% of all childhood brain tumor deaths [81]. As a result, they remain the focus of several experimental therapeutic research teams.
3.2. Histopathology
The vast majority of pHGGs can be classified as anaplastic astrocytomas (WHO Grade III), or glioblastoma (Grade IV). Historically, a minority of diffuse intrinsic pontine gliomas (DIPGs) were morphologically consistent with diffuse astrocytoma (Grade II), likely resulting from sampling bias. However, the identification of pathognomonic oncogenic mutations in DIPG (particularly in histones 3.1 and 3.3), together with established malignant clinical characteristics, resulted in an amendment to current WHO nomenclature, with DIPGs now classified as diffuse midline gliomas with H3K27 mutation (Grade IV) [5].
3.3. Molecular Classification
Clear biological distinctions between pHGGs and adult counterparts are now established [82,83], providing a rationale for the failure of many novel therapies derived from adult tumor research. Molecular heterogeneity within pHGGs is also well described [84,85,86,87,88,89,90,91]. The largest molecular meta-analysis of pHGGs published to date, incorporating genomic, epigenomic and transcriptomic profiling has now identified at least nine pHGG subgroups with inherent biological and/or clinical characteristics such as age, tumor location and prognosis [90]. These subgroups express recurrent signature aberrations, which may lead to further refinement of subdivisions in the future (Figure 2).
The predominant pHGG subgroups express mutations of histones HIST1H3B (H3.1) at position K27, H3.2 (rarely) and H3F3A (H3.3) at positions K27 and G34 [90,92]. H3K27M pHGGs are characterized biologically by aberrant expression resulting from loss of trimethylation at lysine 27 on Histone 3 [93,94], and clinically by their midline location (pons, midbrain, thalamus, spina cord) and younger patient age [90,91]. H3.3 G34 subgroup pHGGs are typically located in hemispheric locations, impacting adolescent and older age groups [90,91,95]. The midline location may contribute to the significantly poorer prognosis reported in K27 pHGGs versus G34 counterparts [85,90,91,95], although the mutations alone have been reported as independent prognostic markers in multivariate analysis [90]. Secondary aberrations within the pHGG histone subgroups have also been identified. TOP3A, CCND2, PDGFRA, PPM1D, TP53 and FGFR1 mutations are more frequently identified in H3.3K27 pHGGs, while H3.1K27 tumors often demonstrate PI3K and ACVR1 mutations and H3.3 G34 pHGGs typically contain TP53 and ATRX mutations [90].
Other subgroups include the IDH mutant pHGGs, associated with a frontal location, an adolescent age range and improved prognosis, hypermutant pHGGs as seen in DNA replication repair deficiency disorders, infant HGGs characterized by NTRK mutations and pleomorphic xanthoastrocytoma-like pHGGs and BRAF mutated pHGGs, which may represent low-grade lesions that have undergone malignant transformation [90,91,95]. The latter two subgroups may be amenable to novel targeted inhibitor agents and often demonstrate good responses to therapy and improved survival outcomes. A final ‘wild-type’ subgroup comprises pHGGs harboring mutations in genes such as NF1, MYCN, EGFR, and CDK6 [90].
3.4. Prognostic Factors
Prior to the advent of molecular subclassification as described above, the two leading clinical prognostic factors were the extent of surgical resection and tumor histological grade with incomplete resection and Grade IV HGGs conferring a dismal prognosis [96,97]; this continues to be the case today but is supplemented by molecular stratification also. Some studies have also reported a prognostic influence of methylguanine-DNA-methyltransferase (MGMT) expression in the efficacy of temozolomide therapy and patient outcome [54,98].
3.5. Current Management/Clinical Trials
The global standard of care for pHGG, the Stupp regimen, stems from adult glioblastoma trial work, which demonstrated that the addition of the alkylating agent temozolomide alongside and after focal radiotherapy, improved progression-free and overall patient survival [99]. Given the molecular disparity between adult HGG and their childhood counterparts, it is therefore unsurprising that temozolomide in a Children’s Oncology Group (COG) pHGG trial analysis (ACNS0126) did not improve outcome compared with previous trials using varied adjuvant chemotherapies [54] (Table 1). However, it remains the standard of care because of the relatively low toxicity profile in comparison to alternative regimens.
The COG ACNS0423 trial noted a marginal outcome benefit for the addition of lomustine with temozolomide [55]; however, it was unclear if this was specific to certain molecular subgroups, while the myelosuppressive toxicity of the regime often proved restrictive. The German Hirntumor (HIT) co-operative group have also reported an improved survival rate for a subset of children with glioblastoma achieving gross total resection compared to historical controls, using an intensified chemotherapy regime alongside and after RT [100].
No definitive therapeutic breakthrough has been made in the treatment of DIPG (now diffuse midline glioma, H3K27 mutant), such that the standard therapy remains radiotherapy alone (Table 1). Modern, multinational collaborative trials, such as the Innovative Therapies for Children with Cancer (ITCC) BIOMEDE study, are developing a more nuanced approach alongside focal RT, utilizing novel inhibitor therapy to target corresponding molecular aberrations present in the lesion (dasatanib, everolimus, and erlotinib) (NCT02233049). Interim overall survival analysis of 193/250 randomized patients concluded that a preferential agent was unlikely to be demonstrated, with survival rates comparable with RT alone, albeit everolimus had the most favorable toxicity profile [58].
3.6. Novel Therapies
The paradigm shift in understanding of the molecular heterogeneity of pHGG, together with the failure of conventional therapeutics to significantly improve outcomes for several years, has shifted focus towards developing novel agents that manipulate the epigenetic and genomic aberrations inherent in pHGG molecular subgroups, immunotherapies, and the development of alternative drug administration routes to penetrate the blood–brain barrier such as convection enhanced delivery for diffuse midline glioma H3K27 mutant/DIPG [92,101,102,103,104,105].
Success of mutational target inhibition in specific pHGG subgroups gives credence to this new therapeutic standpoint. For pHGGs with BRAF V600E mutations, BRAF inhibitor (BRAFi) activity has been demonstrated as salvage therapy [106,107,108,109]; international co-operative studies are recruiting (NCT03919071). Similar findings of efficacy have been made with neurotrophic tyrosine receptor kinase inhibitor agents for infant HGGs [110,111] and immune checkpoint inhibition in hypermutant pHGGs resulting from replication repair deficiency disorders [112,113,114,115]. Follow-up co-operative early phase trials are now open (NCT04267146, NCT04323046 and NCT04655404).
With respect to the other main subgroups, targeting histone modification is a therapeutic research focus for the H3.1–3.3 pHGG subgroup. Histone deacetylase inhibitors (HDACi) such as panobinostat, vorinostat and valproic acid have been postulated to improve the therapeutic landscape for this subgroup following successful HDACi in vitro pHGG studies, but translational results to date have proved disappointing [116,117,118,119]. Other agents being looked at for this subgroup include ACVR1/ALK inhibitors [120,121] and the imipridones incorporating agents such as ONC201 [122,123,124].
For the IDH mutant pHGG subgroup, blood–brain barrier penetrant IDH inhibitors have been developed for glioma trials (NCT02273739, NCT03343197, NCT02073994 and NCT04056910). These may be specific to IDH-1 (ivosedinib), IDH-2 (enasidenib) or both (vorasidenib). In addition, the use of PARP (poly-adenosine 50-diphosphate-ribose) inhibitors alongside temozolomide as a radiosensitizer is being explored [125].
Immunotherapeutic strategies other than checkpoint blockade are also being evaluated in pHGGs, including cancer peptide vaccine therapy with antigens such as Ephrin A2 (EphA2), interleukin 13 receptor alpha 2 (IL13Ra2), survivin and HLA-A2 (NCT01130077) [126,127,128], autologous dendritic cell vaccine therapy [129], and chimeric antigen receptor (CAR)-T therapy where studies are recruiting (anti-IL13aR2; NCT02208362, anti-GD2; NCT04196413, anti-B7 H3; NCT04185038).
4. Ependymoma
4.1. Background
Ependymoma is the second most common malignant brain tumor entity in children, after medulloblastoma, representing approximately 10% of all childhood CNS tumors [130]. Most cases present in patients aged below five years and have a male predominance (male: female ratio 0.23: 0.17) [130,131]. Although able to arise anywhere in the neuraxis, over 90% of pediatric ependymomas are intracranial (IC) in origin. Of these, two-thirds occur in the posterior fossa (PF), with the remaining one-third located in the supratentorial (ST) compartment [132]. Leptomeningeal metastasis is uncommon, reported in 2–20% of cases [133,134].
No inherited disorders are consistently reported to predispose to IC pediatric ependymomas. Neurofibromatosis type 2 appears to be associated with the development of spinal ependymomas but typically in the adult population [135].
4.2. Histopathology
Current histological classification of ependymoma remains according to the current WHO grading scheme, resulting in four main histological subgroups: subependymoma and myxopapillary ependymoma (grade I), classic (grade II) and anaplastic (grade III) [120]. Subependymoma typically arise in the ventricles of adults, while myxopapillary ependymoma occur exclusively in the spine [5,136]. Consequently, classic and anaplastic variants typically account for all pediatric IC ependymomas. Morphologically they are both characterized by the tumor cell formation into true rosettes (around a canal) or pseudorosettes (around a blood vessel) while anaplasia is signified by increased mitotic figures, necrosis, microvascular proliferation, and an increased an increased cellular nucleus/cytoplasmic ratio [5]. Common immunohistochemical findings include positive staining for glial fibrillary acid protein (GFAP), expression of EMA, S100 and vimentin [5,137].
The utilization of histological grading as a prognostic marker has failed to consistently be of value, in part due to the subjective nature of grade assignation and tumor heterogeneity. These factors, alongside improved understanding of the genomic landscape of pediatric ependymoma, has led to the Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxonomy (cIMPACT) to recommend that the WHO adopt a new, integrated histological/biological classification system for ependymomas [138].
4.3. Molecular Classification
Genomic and methylomic profiling of ependymoma has revealed nine distinct molecular subtypes, four of which account for most pediatric IC ependymoma across the PF (PF-A and PF-B) and ST (ST-ZFTA and ST-YAP) compartments [139] (Figure 3).
PF-A ependymomas are biologically characterized by epigenetic dysregulation of DNA methylation and histone modification, often accompanying lack of H3K27 trimethylation [147,149,150]. With the exception of some genomic imbalances, namely 1q gain and 6q loss, they typically demonstrate a balanced genome [139,147,149]. They are most common in infants and young children, have a tendency towards infiltration, dissemination and consequent poor prognosis [153]. Due to their predominant lateral location and inherent invasiveness, gross total resection (GTR) is often difficult to achieve and therefore relapse rates are high [154]. PF-B ependymomas are characteristically enriched with numerous cytogenetic abnormalities and are more common in adolescents and young adults [139,152,155]. They originate in the midline yet are often amenable to surgical resection, have a low metastatic potential and therefore have a superior outcome to PF-A tumors [139,152,155]. Recent methylation profiling work to further categories these two PF subgroups have reported two major subgroups, nine minor PF-A subtypes and five PF-B subgroups displaying variable clinical and genetic heterogeneity [140,156].
Greater than 70% ST ependymomas contain a zinc finger translocation associated (ZFTA, previously C11Orf95) gene fusion, most commonly RELA-ZFTA and are termed ST-RELA or, more recently ST-ZFTA [139,143,151]. This subtype is found in children and adults, but rarely infants and is often located in frontal or parietal lobes, often with intra-tumoral hemorrhage, cysts or necrosis [157]. ST-YAP is the remaining molecular subgroup, characterized by the fusion of the YAP1 oncogene with MAMLD1 [142]. ST-YAP tumors typically arise in ventricular or periventricular locations among infants [142]. Up to 15% of ST ependymoma may not harbor a RELA or YAP1 fusion [158].
4.4. Prognostic Factors
Interest remains in identifying prognostic markers to aid patient risk stratification for future ependymoma trial design to improve upon the relative poor long-term outcomes that exist. Akin to medulloblastoma, several clinical and histological putative markers (location, age, tumor grade) have been rendered obsolete by the identification of molecular subgrouping.
The most consistent clinical marker is the extent of surgical resection, with some studies reporting a 60% difference in survival between cases of complete and incomplete tumor resection [59,120,132,159,160,161,162]. The positive prognostic effect of complete excision is maintained across molecular subgroups [120,139].
The infiltrative nature, localization and predisposition to metastasis suggests PF-A ependymomas should exhibit a poorer prognosis when compared with PF-B counterparts, an assumption supported by a retrospective analysis 820 patients with PF ependymoma across four independent cohorts [161]. The recent prospective Children’s Oncology Group (COG) ACN0121 clinical trial, however, found no difference between PF-A and PF-B patient survival, although likely reflecting a paucity of PF-B cases [59]. The study did identify an adverse association with 1q gain in PF-A cases, with survival as low as 30% despite tumor resection and radiotherapy administration [59]. As stated above, tumor gain of chromosome 1q and loss of chromosome 6q are the most commonly observed chromosomal imbalances in ependymoma and appear adverse prognostic factors [59,120,139,141,144,145,146,148,152]. A recent retrospective molecular profiling study of 212 primary PF-B ependymomas identified loss of 13q as a potential novel adverse marker [140].
A retrospective cohort study of 122 ST ependymomas identified ZFTA/RELA fusion as a poor prognostic marker, regardless of the attainment of resection status, with 10 year PFS and overall survival (OS) of approximately 20% and 50%, respectively [139]. The same study conversely identified excellent ST-YAP1 survival rates of 100% [139]. Nevertheless, data from the ACNS0121 clinical trial failed to show any adverse prognostic implication for ST molecular subgroups, again potentially influenced by the case numbers involved [59].
4.5. Current Management/Clinical Trials
The globally accepted standard for pediatric IC ependymomas is maximal, safe surgical resection followed by involved field adjuvant radiotherapy (RT), dosed at 54–59.4 Gy, founded from a 2009 St Jude’s Children’s Research Hospital single-center study of 107 children, demonstrating a 7 year PFS of 77% and OS of 85% [160]. Exceptions to this are in metastatic cases where craniospinal radiotherapy is typically utilized for older children, and infant IC ependymomas, where a chemotherapy only strategy is reserved in order to avoid or delay radiotherapy to the developing brain, with eligibility thresholds of 12 to 18 months for PF tumors and up to 3 years for ST tumors.
Concerns regarding radiotherapy-induced neurotoxicity in young children have resulted in IC ependymoma being the most common pediatric tumor treated with proton beam radiotherapy. By reducing radiation exposure to healthy tissue while delivering therapeutic doses, this modality delivers comparable disease control to modern photon radiotherapy without unexpected toxicity [163,164,165]. Data continue to be collated on latent toxicity [164].
Recent, large international co-operative IC ependymoma trials have been designed to validate the findings of the 2009 St. Jude’s study, evaluate the utility of an aggressive surgical approach, and verify a therapeutic role for chemotherapy either pre or post-radiotherapy (Table 1), since historical data have proven contradictory and inconclusive. The North American CCG-9924 study reported a PFS benefit from immediate post-operative chemotherapy prior to radiotherapy in patients where over 90% of the tumor has been resected [166]; however, this approach has been rebutted by other trial groups [167]. Similarly, outcomes from chemotherapeutic, radiation-sparing strategies for infants have been inconsistent and ultimately disappointing for the majority of children, with only a minority ultimately sparing radiation [168,169,170,171].
The COG ACNS0121 trial confirmed the efficacy of an aggressive surgical approach followed by immediate post-operative radiotherapy, even for children below 3 years of age when compared to historical controls [59]. Long-term follow-up of these younger patients is eagerly awaited. The impact of post-operative chemotherapy to facilitate second-look surgery could not be determined. The COG ACNS0831 study followed on from ACNS0121, with the randomized addition of continuation chemotherapy (vincristine, cisplatin, cyclophosphamide and etoposide) for children treated with adjuvant focal RT following a complete or near total resection [60]. An interim “as treated” analysis of patients was undertaken due to significant non-compliance in patients randomized to receive chemotherapy. This reported a survival advantage for patients receiving chemotherapy (3 year EFS 80% vs. 71%; 1-sided p-value = 0.0121) [60].
The open phase II/III SIOP-Europe Ependymoma II trial (NCT02265770) has design similarities with the COG studies, making compliance with post-irradiation chemotherapy randomization imperative to validate the findings from ACNS 0831. Through patient allocation to three strata, the trial also attempts to evaluate the value of pre-radiotherapy chemotherapy and a 8 Gy radiotherapy boost in cases of incomplete resection, and the addition of a of a histone de-acetylase (HDAC) inhibitor, sodium valproate, for infants receiving one year of conventional multiagent chemotherapy.
4.6. Novel Therapies
Several biological models and patient derived xenografts have been developed to recapitulate ependymoma subgroups in order to identify new therapeutic targets and test novel therapies [172,173,174]. High throughput drug screening in murine models of ZFTA fusion-negative supratentorial ependymoma, characterized by the Ephb2 oncogene identified 5-fluoracil (5-FU) as a potential active drug against this subtype [174,175]. Fibroblast growth factor receptor inhibitors have also been shown to have activity against patient derived ependymoma cell models and demonstrate efficacy in the clinic [176]. As detailed above, the use of histone deacetylase inhibitors as differentiation therapy is currently under evaluation in the current SIOP-Europe trial, following in vitro analyses [177,178]. Similarly, the phase I/Ib COZMOS trial is evaluating the DNA methyltransferase inhibitor 5′Azacitidine in combination with carboplatin, on the premise that inhibition of aberrant DNA methylation will have therapeutic benefit (NCT03206021). Other novel therapies being explored include chimeric antigen receptor T-Cells (HER2; NCT03500991), based on encouraging pre-clinical murine work [80] and metronomic antiangiogenic therapy [179,180].
5. Conclusions
This review exposes the need for the pediatric neuro-oncology community to address the disparity that has developed between advances at the bench compared to the bedside. The potential for an era of biology driven patient care clearly exists yet, at present, international clinical trials struggle to keep pace with the scientific progress made to date. Indeed, many are being rendered outdated before they open to recruitment when evaluated against current molecular advances. This challenge is not unsurmountable and indeed should be embraced as recent years have demonstrated a paradigm shift in our understanding of the molecular pathogenesis across principal malignant brain tumor groups, therein serving as the foundation for developing both risk stratification systems and novel agents as part of the next generation of clinical trials. Nevertheless, results from the review highlight that the statistical design, regulatory infrastructure and ultimately funding of such studies will need urgent consideration to achieve these objectives.
5.1. Clinical Trials and Therapeutic Protocols
We have shown that for pediatric medulloblastoma, the four established intrinsic molecular subgroups have now been superseded by the identification of up to 14 subtypes, each demonstrating a disparate corresponding clinical profile. In contrast, most treatment protocols over the past 20 years have continued to treat MBs with the historical backbone of craniospinal radiotherapy and multiagent chemotherapy, only recently tailoring therapy intensity according to WNT/non-WNT subgrouping, without particular focus on the three other subgroups. Encouragingly, open international trials are now attempting to stratify patients and adapt therapy according to molecular diversity. For example, the SIOP-Europe PNET5 study is following a risk-adapted treatment stratification according to low and high-risk WNT subgroups, the SHH-alpha MB subtype (which demonstrate TP53 mutations), standard-risk biological profiles (including MYCN amplified Group 4 MB) and children with a germline mutational profile (NCT02066220). The SJMB12 trial, in addition to evaluating treatment de-escalation for WNT-subgroup patients, is assessing the addition of smoothened inhibitor Vismodegib for SHH MB, and the incorporation of gemcitabine and pemetrexed for high-risk Group 3 and 4 MB patients (NCT01878617). Finally, the SIOP-Europe high-risk medulloblastoma trial is using molecular screening to identify appropriate cases for increased-intensity treatments, including MYC/MYCN amplification (excluding MYCN amplified Group 4 MB) and SHH-alpha MB (EudraCT Number: 2018-004250-17).
Attempts to integrate molecular pathogenesis to inform on therapeutic stratification for most childhood high-grade gliomas or pediatric intracranial ependymoma unfortunately lag significantly behind the progress observed with medulloblastoma. As shown in this review, there is now compelling evidence that molecular subgrouping alone is an independent survival marker for childhood ependymoma, while prognostic adversity is further conferred by the presence of genomic aberrations including chromosome 1q gain and 6q loss in PF-A ependymomas, and potentially 13q loss in PF-B ependymomas. Despite this, international ependymoma clinical trials continue to risk stratify children according to the clinical parameters of patient age and resection status alone; an omission that will require addressing in future clinical trial strategies. With the exception of BIOMEDE 1, large-scale international pediatric HGG trials have also not incorporated biologically derived therapeutic stratification systems, principally because the finding that HGGs encompass an array of discrete subtypes is a relatively recent discovery.
As with medulloblastoma, the observation of up to 14 discrete molecular subtypes of PF ependymoma, at least 3 subtypes of ST ependymoma and up to 10 pediatric HGG subtypes clearly presents a challenge for future trial design. As can be seen from Table 1 of this article, the duration of an international pediatric brain tumor trial can take up to 10 years to complete patient accrual, and even longer to publish data. In order to tailor therapeutic intensity or introduce novel agents against the array of specific tumor subtypes now published in this review, future trials will require novel statistical designs that embrace truly global collaboration to generate timely, rigorous results as increasing molecular subcategorization will lead to significantly smaller patient subpopulations from which statistically sound conclusions must be drawn. Such collaborative efforts may also support less affluent countries to provide equity in diagnostic and therapeutic approaches. Duration of follow-up for specific patient populations will also need to be considered, as evidenced by the high proportion of late relapses in Group 3/4, subtype VIII MB and some non PF-A subgroups of ependymoma.
5.2. Conventional and Novel Therapies
While advances in adjuvant therapy have undoubtedly improved the survival of children with malignant brain tumors, the ‘one-therapy-fits-all’ paradigm fails to reflect and tailor to the diverse molecular landscape now apparent. As highlighted by this review, integrating clinical and biological data to generate risk-adapted treatment stratifications can potentially modify conventional therapy intensity and enable the introduction of novel agents.
De-escalation of radiotherapy dosing is being evaluated in several of the current international medulloblastoma clinical trials highlighted in the review. However, such an approach could also be considered for other molecularly-defined tumor entities including Group 4 (often subtype IV) medulloblastomas with chromosome 11 loss, completely resected ST-YAP1 ependymomas, completely resected PF-B ependymomas without 13q loss, and ‘infant’ or ‘LGG-like’ pediatric HGGs. Clearly, any de-escalation of therapy must be approached with extreme caution, as evidenced by the failure of trial NCT02212574 for WNT-activated MB, where a post-operative chemotherapy only strategy led to unacceptable relapse rates.
For some unfavorable-risk tumors, the option of increasing treatment intensity is a possibility as evidenced by current high-risk medulloblastoma trial strategies; however, any trial adopting this approach should consider incorporating disability or health status outcome measures, as they will help determine the quality of potential survivorship afforded [181]. The efficacy of chemotherapy in pediatric ependymoma remains contentious but a potential option to explore for escalation of therapy in certain cases (for instance PF-A tumors with chromosome 1q gain or 6q loss). The interim analysis results of the COG ACNS0831 trial suggested a potential survival advantage for children receiving continuation chemotherapy following tumor excision and post-operative irradiation, yet this requires validation ideally by the open phase II/III SIOP-Europe Ependymoma II trial. The administration of conventional chemotherapy agents and novel agents by alternative means, such as convection enhanced delivery to overcome the blood–brain barrier in diffuse midline glioma, H3K27M pediatric HGGs is also under consideration.
Parallel to modifying the intensity or administration of conventional therapies for childhood malignant brain tumors, much hope rests on establishing novel agents to target aberrant molecular aberrations underpinning tumorigenesis. This review highlights many of the developments in this field across medulloblastoma, pediatric high-grade gliomas and ependymomas. International trial outcomes are awaited for medulloblastoma subgroup-targeted therapy in SJMB12 and combination HDACi therapy across infants in the SIOP Ependymoma II study, while the success of BRAFi and NTRKi in certain pHGG subtypes and the evolving array of targeted primary treatment options for pediatric low-grade glioma give cause for optimism.
While encouraging, challenges nevertheless remain. As described in this review, novel agents against malignant brain tumors are being evaluated in early-phase pediatric studies, yet few successful candidates targeting the spectrum of molecular subtypes that now exist have been identified. One explanation for this is that many early-phase neuro-oncology trials in children assess novel agents in the relapse setting, rather than as primary therapy. In turn, this could potentially generate misleading results on drug efficacy, as evidenced by pre-clinical relapsed medulloblastoma work implicating clonal selection as a potential cause for the disappearance of targetable aberrations between patient-matched primary and relapsed tumors [182,183]. However, the paucity of effective novel agents also reflects the ongoing need for improved pre-clinical models that accurately replicate the specific human disease subtype interrogated, including appropriate immunocompetent murine models to test potential immunotherapies. A further explanation is that many pediatric malignant brain tumors appear driven by epigenetic dysregulation such that tumors rarely harbor immediately actionable mutations, or display significant molecular heterogeneity making resistance to single agent targeted therapy anticipated, as is described for SHH-activated medulloblastoma [30]. Consequently, it is presumed that combination therapy, utilizing novel agents alongside conventional modalities, will better enable local and disseminated disease control rather than a single agent approach in future studies.
5.3. Future Challenges
This review highlights the molecular heterogeneity across the most common pediatric malignant brain tumors, together with its relevance to current diagnostic and therapeutic protocols, and strategies to correct the consequent imbalance that arises from bench to bedside. The tumor groups discussed in this review have key clinical challenges that now warrant focus, including intensification or novel combination therapy for unfavorable-risk tumors, de-escalation of intensity for favorable-risk lesions, the treatment of relapse, and a reduction in morbidity, disability and late effects (Table 2). It is now incumbent on the neuro-oncology community to meet and overcome these challenges; in an age of digital technology and social media, where the latest global scientific breakthroughs are acknowledged promptly in the public domain, the families of our patients are demanding this of us.
Author Contributions
Conceptualization, J.-P.K., C.T.; Writing-original draft preparation, J.-P.K., C.T.; Writing-review and editing, J.-P.K., C.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no active funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Heath, J.A.; Zacharoulis, S.; Kieran, M.W. Pediatric Neuro Oncol.: Current status and future directions. Asia Pac. J. Clin. Oncol. 2012, 8, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro Oncol. 2021, 23, iii1–iii105. [Google Scholar] [CrossRef] [PubMed]
- Packer, R.J.; Gajjar, A.; Vezina, G.; Rorke-Adams, L.; Burger, P.C.; Robertson, P.L.; Bayer, L.; LaFond, D.; Donahue, B.R.; Marymont, M.H.; et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J. Clin. Oncol. 2006, 24, 4202–4208. [Google Scholar] [CrossRef]
- Khanna, V.; Achey, R.L.; Ostrom, Q.T.; Block-Beach, H.; Kruchko, C.; Barnholtz-Sloan, J.S.; de Blank, P.M. Incidence and survival trends for medulloblastomas in the United States from 2001 to 2013. J. Neurooncol. 2017, 135, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizer, B.L.; Clifford, S.C. The potential impact of tumour biology on improved clinical practice for medulloblastoma: Progress towards biologically driven clinical trials. Br. J. Neurosurg. 2009, 23, 364–375. [Google Scholar] [CrossRef]
- Northcott, P.A.; Shih, D.J.; Peacock, J.; Garzia, L.; Morrissy, A.S.; Zichner, T.; Stutz, A.M.; Korshunov, A.; Reimand, J.; Schumacher, S.E.; et al. Subgroup-specific structural variation across 1000 medulloblastoma genomes. Nature 2012, 488, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.A. Pathology, diagnostics, and classification of medulloblastoma. Brain Pathol. 2020, 30, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, C.G.; Kepner, J.L.; Goldthwaite, P.T.; Kun, L.E.; Duffner, P.K.; Friedman, H.S.; Strother, D.R.; Burger, P.C. Histopathologic grading of medulloblastomas: A Pediatric Oncology Group study. Cancer 2002, 94, 552–560. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Bailey, S.; Clifford, S.C.; Doz, F.; Kool, M.; Dufour, C.; Vassal, G.; Milde, T.; et al. Risk stratification of childhood medulloblastoma in the molecular era: The current consensus. Acta Neuropathol. 2016, 131, 821–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northcott, P.A.; Jones, D.T.; Kool, M.; Robinson, G.W.; Gilbertson, R.J.; Cho, Y.J.; Pomeroy, S.L.; Korshunov, A.; Lichter, P.; Taylor, M.D.; et al. Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 2012, 12, 818–834. [Google Scholar] [CrossRef] [Green Version]
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e736. [Google Scholar] [CrossRef] [Green Version]
- Sharma, T.; Schwalbe, E.C.; Williamson, D.; Sill, M.; Hovestadt, V.; Mynarek, M.; Rutkowski, S.; Robinson, G.W.; Gajjar, A.; Cavalli, F.; et al. Second-generation molecular subgrouping of medulloblastoma: An international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol. 2019, 138, 309–326. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Grobner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Gibson, P.; Tong, Y.; Robinson, G.; Thompson, M.C.; Currle, D.S.; Eden, C.; Kranenburg, T.A.; Hogg, T.; Poppleton, H.; Martin, J.; et al. Subtypes of medulloblastoma have distinct developmental origins. Nature 2010, 468, 1095–1099. [Google Scholar] [CrossRef]
- Patay, Z.; DeSain, L.A.; Hwang, S.N.; Coan, A.; Li, Y.; Ellison, D.W. MR Imaging Characteristics of Wingless-Type-Subgroup Pediatric Medulloblastoma. AJNR Am. J. Neuroradiol. 2015, 36, 2386–2393. [Google Scholar] [CrossRef] [Green Version]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Jager, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.J.; Pugh, T.J.; Hovestadt, V.; Stutz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef]
- Kool, M.; Korshunov, A.; Remke, M.; Jones, D.T.; Schlanstein, M.; Northcott, P.A.; Cho, Y.J.; Koster, J.; Schouten-van Meeteren, A.; van Vuurden, D.; et al. Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012, 123, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korshunov, A.; Sahm, F.; Zheludkova, O.; Golanov, A.; Stichel, D.; Schrimpf, D.; Ryzhova, M.; Potapov, A.; Habel, A.; Meyer, J.; et al. DNA methylation profiling is a method of choice for molecular verification of pediatric WNT-activated medulloblastomas. Neuro Oncol. 2019, 21, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Robinson, G.W.; Kratz, C.P.; Mabbott, D.J.; Pomeroy, S.L.; Clifford, S.C.; Rutkowski, S.; Ellison, D.W.; Malkin, D.; Taylor, M.D.; et al. Medulloblastoma. Nat. Rev. Dis. Primers 2019, 5, 11. [Google Scholar] [CrossRef]
- Waszak, S.M.; Northcott, P.A.; Buchhalter, I.; Robinson, G.W.; Sutter, C.; Groebner, S.; Grund, K.B.; Brugieres, L.; Jones, D.T.W.; Pajtler, K.W.; et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: A retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018, 19, 785–798. [Google Scholar] [CrossRef]
- Pugh, T.J.; Weeraratne, S.D.; Archer, T.C.; Pomeranz Krummel, D.A.; Auclair, D.; Bochicchio, J.; Carneiro, M.O.; Carter, S.L.; Cibulskis, K.; Erlich, R.L.; et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature 2012, 488, 106–110. [Google Scholar] [CrossRef]
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel mutations target distinct subgroups of medulloblastoma. Nature 2012, 488, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Zhukova, N.; Ramaswamy, V.; Remke, M.; Pfaff, E.; Shih, D.J.; Martin, D.C.; Castelo-Branco, P.; Baskin, B.; Ray, P.N.; Bouffet, E.; et al. Subgroup-specific prognostic implications of TP53 mutation in medulloblastoma. J. Clin. Oncol. 2013, 31, 2927–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raybaud, C.; Ramaswamy, V.; Taylor, M.D.; Laughlin, S. Posterior fossa tumors in children: Developmental anatomy and diagnostic imaging. Childs Nerv. Syst. 2015, 31, 1661–1676. [Google Scholar] [CrossRef]
- Lafay-Cousin, L.; Smith, A.; Chi, S.N.; Wells, E.; Madden, J.; Margol, A.; Ramaswamy, V.; Finlay, J.; Taylor, M.D.; Dhall, G.; et al. Clinical, Pathological, and Molecular Characterization of Infant Medulloblastomas Treated with Sequential High-Dose Chemotherapy. Pediatr. Blood Cancer 2016, 63, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Jones, D.T.; Jager, N.; Northcott, P.A.; Pugh, T.J.; Hovestadt, V.; Piro, R.M.; Esparza, L.A.; Markant, S.L.; Remke, M.; et al. Genome sequencing of SHH medulloblastoma predicts genotype-related response to smoothened inhibition. Cancer Cell 2014, 25, 393–405. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.H.; Xu, Q.F.; Cui, Y.H.; Li, N.; Bian, X.W.; Lv, S.Q. Medulloblastoma stem cells: Promising targets in medulloblastoma therapy. Cancer Sci. 2016, 107, 583–589. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Shih, D.; Wang, X.; Northcott, P.A.; Faria, C.C.; Raybaud, C.; Tabori, U.; Hawkins, C.; Rutka, J.; et al. Duration of the pre-diagnostic interval in medulloblastoma is subgroup dependent. Pediatr. Blood Cancer 2014, 61, 1190–1194. [Google Scholar] [CrossRef]
- Cho, Y.J.; Tsherniak, A.; Tamayo, P.; Santagata, S.; Ligon, A.; Greulich, H.; Berhoukim, R.; Amani, V.; Goumnerova, L.; Eberhart, C.G.; et al. Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J. Clin. Oncol. 2011, 29, 1424–1430. [Google Scholar] [CrossRef]
- Northcott, P.A.; Korshunov, A.; Witt, H.; Hielscher, T.; Eberhart, C.G.; Mack, S.; Bouffet, E.; Clifford, S.C.; Hawkins, C.E.; French, P.; et al. Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 2011, 29, 1408–1414. [Google Scholar] [CrossRef]
- Northcott, P.A.; Lee, C.; Zichner, T.; Stutz, A.M.; Erkek, S.; Kawauchi, D.; Shih, D.J.; Hovestadt, V.; Zapatka, M.; Sturm, D.; et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature 2014, 511, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Maier, H.; Dalianis, T.; Kostopoulou, O.N. New Approaches in Targeted Therapy for Medulloblastoma in Children. Anticancer. Res. 2021, 41, 1715–1726. [Google Scholar] [CrossRef]
- Szalontay, L.; Khakoo, Y. Medulloblastoma: An Old Diagnosis with New Promises. Curr. Oncol. Rep. 2020, 22, 90. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Rudneva, V.A.; Buchhalter, I.; Billups, C.A.; Waszak, S.M.; Smith, K.S.; Bowers, D.C.; Bendel, A.; Fisher, P.G.; Partap, S.; et al. Risk-adapted therapy for young children with medulloblastoma (SJYC07): Therapeutic and molecular outcomes from a multicentre, phase 2 trial. Lancet Oncol. 2018, 19, 768–784. [Google Scholar] [CrossRef]
- Lafay-Cousin, L.; Bouffet, E.; Strother, D.; Rudneva, V.; Hawkins, C.; Eberhart, C.; Horbinski, C.; Heier, L.; Souweidane, M.; Williams-Hughes, C.; et al. Phase II Study of Nonmetastatic Desmoplastic Medulloblastoma in Children Younger Than 4 Years of Age: A Report of the Children’s Oncology Group (ACNS1221). J. Clin. Oncol. 2020, 38, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, A.; Remke, M.; Kool, M.; Hielscher, T.; Northcott, P.A.; Williamson, D.; Pfaff, E.; Witt, H.; Jones, D.T.; Ryzhova, M.; et al. Biological and clinical heterogeneity of MYCN-amplified medulloblastoma. Acta Neuropathol. 2012, 123, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, T.; Schmidt, R.; Remke, M.; Korshunov, A.; Hovestadt, V.; Jones, D.T.; Felsberg, J.; Kaulich, K.; Goschzik, T.; Kool, M.; et al. Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol. 2014, 128, 137–149. [Google Scholar] [CrossRef] [Green Version]
- von Bueren, A.O.; Kortmann, R.D.; von Hoff, K.; Friedrich, C.; Mynarek, M.; Muller, K.; Goschzik, T.; Zur Muhlen, A.; Gerber, N.; Warmuth-Metz, M.; et al. Treatment of Children and Adolescents with Metastatic Medulloblastoma and Prognostic Relevance of Clinical and Biologic Parameters. J. Clin. Oncol. 2016, 34, 4151–4160. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Remke, M.; Adamski, J.; Bartels, U.; Tabori, U.; Wang, X.; Huang, A.; Hawkins, C.; Mabbott, D.; Laperriere, N.; et al. Medulloblastoma subgroup-specific outcomes in irradiated children: Who are the true high-risk patients? Neuro Oncol. 2016, 18, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, D.J.; Northcott, P.A.; Remke, M.; Korshunov, A.; Ramaswamy, V.; Kool, M.; Luu, B.; Yao, Y.; Wang, X.; Dubuc, A.M.; et al. Cytogenetic prognostication within medulloblastoma subgroups. J. Clin. Oncol. 2014, 32, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Laughton, S.J.; Merchant, T.E.; Sklar, C.A.; Kun, L.E.; Fouladi, M.; Broniscer, A.; Morris, E.B.; Sanders, R.P.; Krasin, M.J.; Shelso, J.; et al. Endocrine outcomes for children with embryonal brain tumors after risk-adapted craniospinal and conformal primary-site irradiation and high-dose chemotherapy with stem-cell rescue on the SJMB-96 trial. J. Clin. Oncol. 2008, 26, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Mulhern, R.K.; Merchant, T.E.; Gajjar, A.; Reddick, W.E.; Kun, L.E. Late neurocognitive sequelae in survivors of brain tumours in childhood. Lancet Oncol. 2004, 5, 399–408. [Google Scholar] [CrossRef]
- Packer, R.J.; Goldwein, J.; Nicholson, H.S.; Vezina, L.G.; Allen, J.C.; Ris, M.D.; Muraszko, K.; Rorke, L.B.; Wara, W.M.; Cohen, B.H.; et al. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: A Children’s Cancer Group Study. J. Clin. Oncol. 1999, 17, 2127–2136. [Google Scholar] [CrossRef] [PubMed]
- Gajjar, A.; Chintagumpala, M.; Ashley, D.; Kellie, S.; Kun, L.E.; Merchant, T.E.; Woo, S.; Wheeler, G.; Ahern, V.; Krasin, M.J.; et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): Long-term results from a prospective, multicentre trial. Lancet Oncol. 2006, 7, 813–820. [Google Scholar] [CrossRef]
- Tarbell, N.J.; Friedman, H.; Polkinghorn, W.R.; Yock, T.; Zhou, T.; Chen, Z.; Burger, P.; Barnes, P.; Kun, L. High-risk medulloblastoma: A pediatric oncology group randomized trial of chemotherapy before or after radiation therapy (POG 9031). J. Clin. Oncol. 2013, 31, 2936–2941. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.E.; Bailey, C.C.; Robinson, K.; Weston, C.L.; Ellison, D.; Ironside, J.; Lucraft, H.; Gilbertson, R.; Tait, D.M.; Walker, D.A.; et al. Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: The International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J. Clin. Oncol. 2003, 21, 1581–1591. [Google Scholar] [CrossRef]
- Lannering, B.; Rutkowski, S.; Doz, F.; Pizer, B.; Gustafsson, G.; Navajas, A.; Massimino, M.; Reddingius, R.; Benesch, M.; Carrie, C.; et al. Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: Results from the randomized multicenter HIT-SIOP PNET 4 trial. J. Clin. Oncol. 2012, 30, 3187–3193. [Google Scholar] [CrossRef] [PubMed]
- Michalski, J.M.; Janss, A.; Vezina, G.; Gajjar, A.; Pollack, I.; Merchant, T.E.; Fitzgerald, T.J.; Booth, T.; Tarbell, N.J.; Li, Y.; et al. Results of COG ACNS0331: A Phase III Trial of Involved-Field Radiotherapy (IFRT) and Low Dose Craniospinal Irradiation (LD-CSI) with Chemotherapy in Average-Risk Medulloblastoma: A Report from the Children’s Oncology Group. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 937–938. [Google Scholar] [CrossRef]
- Leary, S.E.S.; Packer, R.J.; Li, Y.; Billups, C.A.; Smith, K.S.; Jaju, A.; Heier, L.; Burger, P.; Walsh, K.; Han, Y.; et al. Efficacy of Carboplatin and Isotretinoin in Children with High-risk Medulloblastoma: A Randomized Clinical Trial from the Children’s Oncology Group. JAMA Oncol. 2021, 7, 1313–1321. [Google Scholar] [CrossRef]
- Cohen, K.J.; Heideman, R.L.; Zhou, T.; Holmes, E.J.; Lavey, R.S.; Bouffet, E.; Pollack, I.F. Temozolomide in the treatment of children with newly diagnosed diffuse intrinsic pontine gliomas: A report from the Children’s Oncology Group. Neuro Oncol. 2011, 13, 410–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakacki, R.I.; Cohen, K.J.; Buxton, A.; Krailo, M.D.; Burger, P.C.; Rosenblum, M.K.; Brat, D.J.; Hamilton, R.L.; Eckel, S.P.; Zhou, T.; et al. Phase 2 study of concurrent radiotherapy and temozolomide followed by temozolomide and lomustine in the treatment of children with high-grade glioma: A report of the Children’s Oncology Group ACNS0423 study. Neuro Oncol. 2016, 18, 1442–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, K.A.; Zhou, T.; McNall-Knapp, R.Y.; Jakacki, R.I.; Levy, A.S.; Vezina, G.; Pollack, I.F. Motexafin-gadolinium and involved field radiation therapy for intrinsic pontine glioma of childhood: A children’s oncology group phase 2 study. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, e55–e60. [Google Scholar] [CrossRef] [Green Version]
- Grill, J.; Massimino, M.; Bouffet, E.; Azizi, A.A.; McCowage, G.; Canete, A.; Saran, F.; Le Deley, M.C.; Varlet, P.; Morgan, P.S.; et al. Phase II, Open-Label, Randomized, Multicenter Trial (HERBY) of Bevacizumab in Pediatric Patients with Newly Diagnosed High-Grade Glioma. J. Clin. Oncol. 2018, 36, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Grill, J.; Le Teuff, G.; Nysom, K.; Blomgren, K.; Hargrave, D.; MacCowage, G.; Bautista, F.; Van Vuurden, D.; Dangouloff-Ros, V.; Puget, S.; et al. Biological medicine for diffuse intrinsic pontine glioma (DIPG) eradication: Results of the three arm biomarker-driven randomized BIOMEDE 1.0 trial. Neuro Oncol. 2020, 22 (Suppl. S3), iii293. [Google Scholar] [CrossRef]
- Merchant, T.E.; Bendel, A.E.; Sabin, N.D.; Burger, P.C.; Shaw, D.W.; Chang, E.; Wu, S.; Zhou, T.; Eisenstat, D.D.; Foreman, N.K.; et al. Conformal Radiation Therapy for Pediatric Ependymoma, Chemotherapy for Incompletely Resected Ependymoma, and Observation for Completely Resected, Supratentorial Ependymoma. J. Clin. Oncol. 2019, 37, 974–983. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.; Onar-Thomas, A.; Ellison, D.; Owens-Pickle, E.; Wu, S.; Leary, S.; Fouladi, M. ACNS0831, Phase III Rrandomized trial of post-radiation chemotherapy in patients with newly diagnosed ependymoma ages 1 to 21 years. Neuro Oncol. 2021, 22, iii318. [Google Scholar] [CrossRef]
- Nobre, L.; Zapotocky, M.; Khan, S.; Fukuoka, K.; Fonseca, A.; McKeown, T.; Sumerauer, D.; Vicha, A.; Grajkowska, W.A.; Trubicka, J.; et al. Pattern of Relapse and Treatment Response in WNT-Activated Medulloblastoma. Cell Rep. Med. 2020, 1, 100038. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, S.; Bode, U.; Deinlein, F.; Ottensmeier, H.; Warmuth-Metz, M.; Soerensen, N.; Graf, N.; Emser, A.; Pietsch, T.; Wolff, J.E.; et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N. Engl. J. Med. 2005, 352, 978–986. [Google Scholar] [CrossRef] [Green Version]
- von Bueren, A.O.; von Hoff, K.; Pietsch, T.; Gerber, N.U.; Warmuth-Metz, M.; Deinlein, F.; Zwiener, I.; Faldum, A.; Fleischhack, G.; Benesch, M.; et al. Treatment of young children with localized medulloblastoma by chemotherapy alone: Results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro Oncol. 2011, 13, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Mazewski, C.; Kang, G.; Kellie, S.; Gossett, J.; Leary, S.; Li, B.; Arigides, P.; Hayes, L.; Reddy, A.; Shaw, D.; et al. Efficacy of methotrexate (MTX) according to molecular sub-type in young children with medulloblastoma (MB): A report from Children’s Oncology Group Phase III Trial ACNS0334. Neuro Oncol. 2020, 22, iii396. [Google Scholar] [CrossRef]
- Bautista, F.; Fioravantti, V.; de Rojas, T.; Carceller, F.; Madero, L.; Lassaletta, A.; Moreno, L. Medulloblastoma in children and adolescents: A systematic review of contemporary phase I and II clinical trials and biology update. Cancer Med. 2017, 6, 2606–2624. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [Green Version]
- Robinson, G.W.; Orr, B.A.; Wu, G.; Gururangan, S.; Lin, T.; Qaddoumi, I.; Packer, R.J.; Goldman, S.; Prados, M.D.; Desjardins, A.; et al. Vismodegib Exerts Targeted Efficacy Against Recurrent Sonic Hedgehog-Subgroup Medulloblastoma: Results from Phase II Pediatric Brain Tumor Consortium Studies PBTC-025B and PBTC-032. J. Clin. Oncol. 2015, 33, 2646–2654. [Google Scholar] [CrossRef]
- Li, Y.; Song, Q.; Day, B.W. Phase I and phase II sonidegib and vismodegib clinical trials for the treatment of paediatric and adult MB patients: A systemic review and meta-analysis. Acta Neuropathol. Commun. 2019, 7, 123. [Google Scholar] [CrossRef]
- Gajjar, A.; Packer, R.J.; Foreman, N.K.; Cohen, K.; Haas-Kogan, D.; Merchant, T.E.; Committee, C.O.G.B.T. Children’s Oncology Group’s 2013 blueprint for research: Central nervous system tumors. Pediatr. Blood Cancer 2013, 60, 1022–1026. [Google Scholar] [CrossRef] [Green Version]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef] [Green Version]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 2017, 19, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Robinson, G.W.; Kaste, S.C.; Chemaitilly, W.; Bowers, D.C.; Laughton, S.; Smith, A.; Gottardo, N.G.; Partap, S.; Bendel, A.; Wright, K.D.; et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget 2017, 8, 69295–69302. [Google Scholar] [CrossRef] [Green Version]
- Morfouace, M.; Shelat, A.; Jacus, M.; Freeman, B.B.; Turner, D.; Robinson, S.; Zindy, F.; Wang, Y.D.; Finkelstein, D.; Ayrault, O.; et al. Pemetrexed and gemcitabine as combination therapy for the treatment of Group3 medulloblastoma. Cancer Cell 2014, 25, 516–529. [Google Scholar] [CrossRef] [Green Version]
- Sangar, M.L.C.; Genovesi, L.A.; Nakamoto, M.W.; Davis, M.J.; Knobluagh, S.E.; Ji, P.; Millar, A.; Wainwright, B.J.; Olson, J.M. Inhibition of CDK4/6 by Palbociclib Significantly Extends Survival in Medulloblastoma Patient-Derived Xenograft Mouse Models. Clin. Cancer Res. 2017, 23, 5802–5813. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhayay, P.; Bergthold, G.; Nguyen, B.; Schubert, S.; Gholamin, S.; Tang, Y.; Bolin, S.; Schumacher, S.E.; Zeid, R.; Masoud, S.; et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014, 20, 912–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Rudneva, V.A.; Erkek, S.; Zapatka, M.; Chau, L.Q.; Tacheva-Grigorova, S.K.; Garancher, A.; Rusert, J.M.; Aksoy, O.; Lea, R.; et al. Lsd1 as a therapeutic target in Gfi1-activated medulloblastoma. Nat. Commun. 2019, 10, 332. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Liu, K.W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [Green Version]
- Sayour, E.J.; Mitchell, D.A. Immunotherapy for Pediatric Brain Tumors. Brain Sci. 2017, 7, 137. [Google Scholar] [CrossRef] [Green Version]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; de Blank, P.M.; Kruchko, C.; Petersen, C.M.; Liao, P.; Finlay, J.L.; Stearns, D.S.; Wolff, J.E.; Wolinsky, Y.; Letterio, J.J.; et al. Alex’s Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro Oncol. 2015, 16 (Suppl. S10), x1–x36. [Google Scholar] [CrossRef]
- Diaz, A.K.; Baker, S.J. The genetic signatures of pediatric high-grade glioma: No longer a one-act play. Semin. Radiat. Oncol. 2014, 24, 240–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tonjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [Green Version]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturm, D.; Bender, S.; Jones, D.T.; Lichter, P.; Grill, J.; Becher, O.; Hawkins, C.; Majewski, J.; Jones, C.; Costello, J.F.; et al. Paediatric and adult glioblastoma: Multiform (epi)genomic culprits emerge. Nat. Rev. Cancer 2014, 14, 92–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatwin, H.V.; Cruz Cruz, J.; Green, A.L. Pediatric high-grade glioma: Moving toward subtype-specific multimodal therapy. FEBS J. 2021, 288, 6127–6141. [Google Scholar] [CrossRef]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pages, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Lewis, P.W.; Muller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [Green Version]
- Korshunov, A.; Ryzhova, M.; Hovestadt, V.; Bender, S.; Sturm, D.; Capper, D.; Meyer, J.; Schrimpf, D.; Kool, M.; Northcott, P.A.; et al. Integrated analysis of pediatric glioblastoma reveals a subset of biologically favorable tumors with associated molecular prognostic markers. Acta Neuropathol. 2015, 129, 669–678. [Google Scholar] [CrossRef]
- Finlay, J.L.; Boyett, J.M.; Yates, A.J.; Wisoff, J.H.; Milstein, J.M.; Geyer, J.R.; Bertolone, S.J.; McGuire, P.; Cherlow, J.M.; Tefft, M.; et al. Randomized phase III trial in childhood high-grade astrocytoma comparing vincristine, lomustine, and prednisone with the eight-drugs-in-1-day regimen. Childrens Cancer Group. J. Clin. Oncol. 1995, 13, 112–123. [Google Scholar] [CrossRef]
- Pollack, I.F.; Boyett, J.M.; Yates, A.J.; Burger, P.C.; Gilles, F.H.; Davis, R.L.; Finlay, J.L.; Children’s Cancer, G. The influence of central review on outcome associations in childhood malignant gliomas: Results from the CCG-945 experience. Neuro Oncol. 2003, 5, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Pollack, I.F.; Hamilton, R.L.; Sobol, R.W.; Burnham, J.; Yates, A.J.; Holmes, E.J.; Zhou, T.; Finlay, J.L. O6-methylguanine-DNA methyltransferase expression strongly correlates with outcome in childhood malignant gliomas: Results from the CCG-945 Cohort. J. Clin. Oncol. 2006, 24, 3431–3437. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Wolff, J.E.; Driever, P.H.; Erdlenbruch, B.; Kortmann, R.D.; Rutkowski, S.; Pietsch, T.; Parker, C.; Metz, M.W.; Gnekow, A.; Kramm, C.M. Intensive chemotherapy improves survival in pediatric high-grade glioma after gross total resection: Results of the HIT-GBM-C protocol. Cancer 2010, 116, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.C.; Kennedy, B.; Yanes, C.L.; Garvin, J.; Needle, M.; Canoll, P.; Feldstein, N.A.; Bruce, J.N. Convection-enhanced delivery of topotecan into diffuse intrinsic brainstem tumors in children. J. Neurosurg Pediatr. 2013, 11, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Souweidane, M.M.; Kramer, K.; Pandit-Taskar, N.; Zhou, Z.; Haque, S.; Zanzonico, P.; Carrasquillo, J.A.; Lyashchenko, S.K.; Thakur, S.B.; Donzelli, M.; et al. Convection-enhanced delivery for diffuse intrinsic pontine glioma: A single-centre, dose-escalation, phase 1 trial. Lancet Oncol. 2018, 19, 1040–1050. [Google Scholar] [CrossRef]
- Tosi, U.; Souweidane, M. Convection Enhanced Delivery for Diffuse Intrinsic Pontine Glioma: Review of a Single Institution Experience. Pharmaceutics 2020, 12, 660. [Google Scholar] [CrossRef]
- Saito, R.; Kanamori, M.; Sonoda, Y.; Yamashita, Y.; Nagamatsu, K.; Murata, T.; Mugikura, S.; Kumabe, T.; Wembacher-Schroder, E.; Thomson, R.; et al. Phase I trial of convection-enhanced delivery of nimustine hydrochloride (ACNU) for brainstem recurrent glioma. Neurooncol. Adv. 2020, 2, vdaa033. [Google Scholar] [CrossRef]
- Abedalthagafi, M.; Mobark, N.; Al-Rashed, M.; AlHarbi, M. Epigenomics and immunotherapeutic advances in pediatric brain tumors. NPJ Precis. Oncol. 2021, 5, 34. [Google Scholar] [CrossRef]
- Robinson, G.W.; Orr, B.A.; Gajjar, A. Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer 2014, 14, 258. [Google Scholar] [CrossRef] [Green Version]
- Bautista, F.; Paci, A.; Minard-Colin, V.; Dufour, C.; Grill, J.; Lacroix, L.; Varlet, P.; Valteau-Couanet, D.; Geoerger, B. Vemurafenib in pediatric patients with BRAFV600E mutated high-grade gliomas. Pediatr. Blood Cancer 2014, 61, 1101–1103. [Google Scholar] [CrossRef]
- Ceccon, G.; Werner, J.M.; Dunkl, V.; Tscherpel, C.; Stoffels, G.; Brunn, A.; Deckert, M.; Fink, G.R.; Galldiks, N. Dabrafenib Treatment in a Patient with an Epithelioid Glioblastoma and BRAF V600E Mutation. Int. J. Mol. Sci. 2018, 19, 1090. [Google Scholar] [CrossRef] [Green Version]
- Toll, S.A.; Tran, H.N.; Cotter, J.; Judkins, A.R.; Tamrazi, B.; Biegel, J.A.; Dhall, G.; Robison, N.J.; Waters, K.; Patel, P.; et al. Sustained response of three pediatric BRAF(V600E) mutated high-grade gliomas to combined BRAF and MEK inhibitor therapy. Oncotarget 2019, 10, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, D.S.; Wong, M.; Mayoh, C.; Kumar, A.; Tsoli, M.; Mould, E.; Tyrrell, V.; Khuong-Quang, D.A.; Pinese, M.; Gayevskiy, V.; et al. Brief Report: Potent clinical and radiological response to larotrectinib in TRK fusion-driven high-grade glioma. Br. J. Cancer 2018, 119, 693–696. [Google Scholar] [CrossRef] [Green Version]
- Alharbi, M.; Mobark, N.A.; Balbaid, A.A.O.; Alanazi, F.A.; Aljabarat, W.A.R.; Bakhsh, E.A.; Ramkissoon, S.H.; Abedalthagafi, M. Regression of ETV6-NTRK3 Infantile Glioblastoma after First-Line Treatment with Larotrectinib. JCO Precis. Oncol. 2020, 4, 796–800. [Google Scholar] [CrossRef]
- AlHarbi, M.; Ali Mobark, N.; AlMubarak, L.; Aljelaify, R.; AlSaeed, M.; Almutairi, A.; Alqubaishi, F.; Hussain, M.E.; Balbaid, A.A.O.; Said Marie, A.; et al. Durable Response to Nivolumab in a Pediatric Patient with Refractory Glioblastoma and Constitutional Biallelic Mismatch Repair Deficiency. Oncologist 2018, 23, 1401–1406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting from Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larouche, V.; Atkinson, J.; Albrecht, S.; Laframboise, R.; Jabado, N.; Tabori, U.; Bouffet, E. Sustained complete response of recurrent glioblastoma to combined checkpoint inhibition in a young patient with constitutional mismatch repair deficiency. Pediatr. Blood Cancer 2018, 65, e27389. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Hummel, T.R.; Wagner, L.; Ahern, C.; Fouladi, M.; Reid, J.M.; McGovern, R.M.; Ames, M.M.; Gilbertson, R.J.; Horton, T.; Ingle, A.M.; et al. A pediatric phase 1 trial of vorinostat and temozolomide in relapsed or refractory primary brain or spinal cord tumors: A Children’s Oncology Group phase 1 consortium study. Pediatr. Blood Cancer 2013, 60, 1452–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.A.; Tolstrup, M.; Moller, H.J.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Laursen, A.L.; Ostergaard, L.; Sogaard, O.S. Activation of latent human immunodeficiency virus by the histone deacetylase inhibitor panobinostat: A pilot study to assess effects on the central nervous system. Open Forum Infect. Dis. 2015, 2, ofv037. [Google Scholar] [CrossRef] [Green Version]
- Muscal, J.A.; Thompson, P.A.; Horton, T.M.; Ingle, A.M.; Ahern, C.H.; McGovern, R.M.; Reid, J.M.; Ames, M.M.; Espinoza-Delgado, I.; Weigel, B.J.; et al. A phase I trial of vorinostat and bortezomib in children with refractory or recurrent solid tumors: A Children’s Oncology Group phase I consortium study (ADVL0916). Pediatr. Blood Cancer 2013, 60, 390–395. [Google Scholar] [CrossRef] [Green Version]
- Su, J.M.; Li, X.N.; Thompson, P.; Ou, C.N.; Ingle, A.M.; Russell, H.; Lau, C.C.; Adamson, P.C.; Blaney, S.M. Phase 1 study of valproic acid in pediatric patients with refractory solid or CNS tumors: A children’s oncology group report. Clin. Cancer Res. 2011, 17, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Pajtler, K.W.; Mack, S.C.; Ramaswamy, V.; Smith, C.A.; Witt, H.; Smith, A.; Hansford, J.R.; von Hoff, K.; Wright, K.D.; Hwang, E.; et al. The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol. 2017, 133, 5–12. [Google Scholar] [CrossRef]
- Carvalho, D.; Taylor, K.R.; Olaciregui, N.G.; Molinari, V.; Clarke, M.; Mackay, A.; Ruddle, R.; Henley, A.; Valenti, M.; Hayes, A.; et al. ALK2 inhibitors display beneficial effects in preclinical models of ACVR1 mutant diffuse intrinsic pontine glioma. Commun. Biol. 2019, 2, 156. [Google Scholar] [CrossRef] [PubMed]
- Arrillaga-Romany, I.; Chi, A.S.; Allen, J.E.; Oster, W.; Wen, P.Y.; Batchelor, T.T. A phase 2 study of the first imipridone ONC201, a selective DRD2 antagonist for oncology, administered every three weeks in recurrent glioblastoma. Oncotarget 2017, 8, 79298–79304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, A.S.; Tarapore, R.S.; Hall, M.D.; Shonka, N.; Gardner, S.; Umemura, Y.; Sumrall, A.; Khatib, Z.; Mueller, S.; Kline, C.; et al. Pediatric and adult H3 K27M-mutant diffuse midline glioma treated with the selective DRD2 antagonist ONC201. J. Neurooncol. 2019, 145, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.D.; Odia, Y.; Allen, J.E.; Tarapore, R.; Khatib, Z.; Niazi, T.N.; Daghistani, D.; Schalop, L.; Chi, A.S.; Oster, W.; et al. First clinical experience with DRD2/3 antagonist ONC201 in H3 K27M-mutant pediatric diffuse intrinsic pontine glioma: A case report. J. Neurosurg Pediatr. 2019, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lesueur, P.; Lequesne, J.; Grellard, J.M.; Dugue, A.; Coquan, E.; Brachet, P.E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef]
- Okada, H.; Low, K.L.; Kohanbash, G.; McDonald, H.A.; Hamilton, R.L.; Pollack, I.F. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J. Neurooncol. 2008, 88, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Potter, D.M.; Connelly, A.K.; Dibridge, S.A.; Whiteside, T.L.; Okada, H. Antigen-specific immune responses and clinical outcome after vaccination with glioma-associated antigen peptides and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in children with newly diagnosed malignant brainstem and nonbrainstem gliomas. J. Clin. Oncol. 2014, 32, 2050–2058. [Google Scholar] [CrossRef] [Green Version]
- Chheda, Z.S.; Kohanbash, G.; Okada, K.; Jahan, N.; Sidney, J.; Pecoraro, M.; Yang, X.; Carrera, D.A.; Downey, K.M.; Shrivastav, S.; et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J. Exp. Med. 2018, 215, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Benitez-Ribas, D.; Cabezon, R.; Florez-Grau, G.; Molero, M.C.; Puerta, P.; Guillen, A.; Gonzalez-Navarro, E.A.; Paco, S.; Carcaboso, A.M.; Santa-Maria Lopez, V.; et al. Corrigendum: Immune Response Generated with the Administration of Autologous Dendritic Cells Pulsed with an Allogenic Tumoral Cell-Lines Lysate in Patients with Newly Diagnosed Diffuse Intrinsic Pontine Glioma. Front. Oncol. 2018, 8, 201. [Google Scholar] [CrossRef]
- McGuire, C.S.; Sainani, K.L.; Fisher, P.G. Incidence patterns for ependymoma: A surveillance, epidemiology, and end results study. J. Neurosurg. 2009, 110, 725–729. [Google Scholar] [CrossRef]
- Amirian, E.S.; Armstrong, T.S.; Aldape, K.D.; Gilbert, M.R.; Scheurer, M.E. Predictors of survival among pediatric and adult ependymoma cases: A study using Surveillance, Epidemiology, and End Results data from 1973 to 2007. Neuroepidemiology 2012, 39, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Kilday, J.P.; Rahman, R.; Dyer, S.; Ridley, L.; Lowe, J.; Coyle, B.; Grundy, R. Pediatric ependymoma: Biological perspectives. Mol. Cancer Res. 2009, 7, 765–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benesch, M.; Mynarek, M.; Witt, H.; Warmuth-Metz, M.; Pietsch, T.; Bison, B.; Pfister, S.M.; Pajtler, K.W.; Kool, M.; Schuller, U.; et al. Newly Diagnosed Metastatic Intracranial Ependymoma in Children: Frequency, Molecular Characteristics, Treatment, and Outcome in the Prospective HIT Series. Oncologist 2019, 24, e921–e929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharoulis, S.; Ji, L.; Pollack, I.F.; Duffner, P.; Geyer, R.; Grill, J.; Schild, S.; Jaing, T.H.; Massimino, M.; Finlay, J.; et al. Metastatic ependymoma: A multi-institutional retrospective analysis of prognostic factors. Pediatr. Blood Cancer 2008, 50, 231–235. [Google Scholar] [CrossRef]
- Plotkin, S.R.; O’Donnell, C.C.; Curry, W.T.; Bove, C.M.; MacCollin, M.; Nunes, F.P. Spinal ependymomas in neurofibromatosis Type 2: A retrospective analysis of 55 patients. J. Neurosurg. Spine 2011, 14, 543–547. [Google Scholar] [CrossRef]
- Bandopadhayay, P.; Silvera, V.M.; Ciarlini, P.; Malkin, H.; Bi, W.L.; Bergthold, G.; Faisal, A.M.; Ullrich, N.J.; Marcus, K.; Scott, R.M.; et al. Myxopapillary ependymomas in children: Imaging, treatment and outcomes. J. Neurooncol. 2016, 126, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Goldwein, J.W.; Leahy, J.M.; Packer, R.J.; Sutton, L.N.; Curran, W.J.; Rorke, L.B.; Schut, L.; Littman, P.S.; D’Angio, G.J. Intracranial ependymomas in children. Int. J. Radiat. Oncol. Biol. Phys. 1990, 19, 1497–1502. [Google Scholar] [CrossRef]
- Ellison, D.W.; Aldape, K.D.; Capper, D.; Fouladi, M.; Gilbert, M.R.; Gilbertson, R.J.; Hawkins, C.; Merchant, T.E.; Pajtler, K.; Venneti, S.; et al. cIMPACT-NOW update 7: Advancing the molecular classification of ependymal tumors. Brain Pathol. 2020, 30, 863–866. [Google Scholar] [CrossRef]
- Pajtler, K.W.; Witt, H.; Sill, M.; Jones, D.T.; Hovestadt, V.; Kratochwil, F.; Wani, K.; Tatevossian, R.; Punchihewa, C.; Johann, P.; et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015, 27, 728–743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalli, F.M.G.; Hubner, J.M.; Sharma, T.; Luu, B.; Sill, M.; Zapotocky, M.; Mack, S.C.; Witt, H.; Lin, T.; Shih, D.J.H.; et al. Heterogeneity within the PF-EPN-B ependymoma subgroup. Acta Neuropathol. 2018, 136, 227–237. [Google Scholar] [CrossRef]
- Andreiuolo, F.; Le Teuff, G.; Bayar, M.A.; Kilday, J.P.; Pietsch, T.; von Bueren, A.O.; Witt, H.; Korshunov, A.; Modena, P.; Pfister, S.M.; et al. Integrating Tenascin-C protein expression and 1q25 copy number status in pediatric intracranial ependymoma prognostication: A new model for risk stratification. PLoS ONE 2017, 12, e0178351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreiuolo, F.; Varlet, P.; Tauziede-Espariat, A.; Junger, S.T.; Dorner, E.; Dreschmann, V.; Kuchelmeister, K.; Waha, A.; Haberler, C.; Slavc, I.; et al. Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: An entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol. 2019, 29, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Arabzade, A.; Zhao, Y.; Varadharajan, S.; Chen, H.C.; Jessa, S.; Rivas, B.; Stuckert, A.J.; Solis, M.; Kardian, A.; Tlais, D.; et al. ZFTA-RELA Dictates Oncogenic Transcriptional Programs to Drive Aggressive Supratentorial Ependymoma. Cancer Discov. 2021, 11, 2200–2215. [Google Scholar] [CrossRef] [PubMed]
- Araki, A.; Chocholous, M.; Gojo, J.; Dorfer, C.; Czech, T.; Heinzl, H.; Dieckmann, K.; Ambros, I.M.; Ambros, P.F.; Slavc, I.; et al. Chromosome 1q gain and tenascin-C expression are candidate markers to define different risk groups in pediatric posterior fossa ependymoma. Acta Neuropathol. Commun. 2016, 4, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baroni, L.; Sundaresan, L.; Heled, A.; Coltin, H.; Pajtler, K.W.; Lin, T.; Merchant, T.E.; McLendon, R.; Faria, C.; Buntine, M.; et al. Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol. 2021, 23, 1360–1370. [Google Scholar] [CrossRef] [PubMed]
- Kilday, J.P.; Mitra, B.; Domerg, C.; Ward, J.; Andreiuolo, F.; Osteso-Ibanez, T.; Mauguen, A.; Varlet, P.; Le Deley, M.C.; Lowe, J.; et al. Copy number gain of 1q25 predicts poor progression-free survival for pediatric intracranial ependymomas and enables patient risk stratification: A prospective European clinical trial cohort analysis on behalf of the Children’s Cancer Leukaemia Group (CCLG), Societe Francaise d’Oncologie Pediatrique (SFOP), and International Society for Pediatric Oncology (SIOP). Clin. Cancer Res. 2012, 18, 2001–2011. [Google Scholar] [CrossRef] [Green Version]
- Mack, S.C.; Witt, H.; Piro, R.M.; Gu, L.; Zuyderduyn, S.; Stutz, A.M.; Wang, X.; Gallo, M.; Garzia, L.; Zayne, K.; et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014, 506, 445–450. [Google Scholar] [CrossRef]
- Mendrzyk, F.; Korshunov, A.; Benner, A.; Toedt, G.; Pfister, S.; Radlwimmer, B.; Lichter, P. Identification of gains on 1q and epidermal growth factor receptor overexpression as independent prognostic markers in intracranial ependymoma. Clin. Cancer Res. 2006, 12, 2070–2079. [Google Scholar] [CrossRef] [Green Version]
- Michealraj, K.A.; Kumar, S.A.; Kim, L.J.Y.; Cavalli, F.M.G.; Przelicki, D.; Wojcik, J.B.; Delaidelli, A.; Bajic, A.; Saulnier, O.; MacLeod, G.; et al. Metabolic Regulation of the Epigenome Drives Lethal Infantile Ependymoma. Cell 2020, 181, 1329–1345.e1324. [Google Scholar] [CrossRef] [PubMed]
- Panwalkar, P.; Clark, J.; Ramaswamy, V.; Hawes, D.; Yang, F.; Dunham, C.; Yip, S.; Hukin, J.; Sun, Y.; Schipper, M.J.; et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017, 134, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.; Mohankumar, K.M.; Punchihewa, C.; Weinlich, R.; Dalton, J.D.; Li, Y.; Lee, R.; Tatevossian, R.G.; Phoenix, T.N.; Thiruvenkatam, R.; et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 2014, 506, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witt, H.; Mack, S.C.; Ryzhova, M.; Bender, S.; Sill, M.; Isserlin, R.; Benner, A.; Hielscher, T.; Milde, T.; Remke, M.; et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 2011, 20, 143–157. [Google Scholar] [CrossRef] [Green Version]
- Zapotocky, M.; Beera, K.; Adamski, J.; Laperierre, N.; Guger, S.; Janzen, L.; Lassaletta, A.; Figueiredo Nobre, L.; Bartels, U.; Tabori, U.; et al. Survival and functional outcomes of molecularly defined childhood posterior fossa ependymoma: Cure at a cost. Cancer 2019, 125, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.K.; Foreman, N.K.; Gaspar, L.E.; Trinidad, E.; Handler, M.H. Maximally safe resection followed by hypofractionated re-irradiation for locally recurrent ependymoma in children. Pediatr. Blood Cancer 2009, 52, 804–807. [Google Scholar] [CrossRef]
- Ramaswamy, V.; Taylor, M.D. Treatment implications of posterior fossa ependymoma subgroups. Chin. J. Cancer 2016, 35, 93. [Google Scholar] [CrossRef] [Green Version]
- Pajtler, K.W.; Wen, J.; Sill, M.; Lin, T.; Orisme, W.; Tang, B.; Hubner, J.M.; Ramaswamy, V.; Jia, S.; Dalton, J.D.; et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 2018, 136, 211–226. [Google Scholar] [CrossRef]
- Nowak, J.; Junger, S.T.; Huflage, H.; Seidel, C.; Hohm, A.; Vandergrift, L.A.; von Hoff, K.; Rutkowski, S.; Pietsch, T.; Warmuth-Metz, M. MRI Phenotype of RELA-fused Pediatric Supratentorial Ependymoma. Clin. Neuroradiol. 2019, 29, 595–604. [Google Scholar] [CrossRef]
- Fukuoka, K.; Kanemura, Y.; Shofuda, T.; Fukushima, S.; Yamashita, S.; Narushima, D.; Kato, M.; Honda-Kitahara, M.; Ichikawa, H.; Kohno, T.; et al. Significance of molecular classification of ependymomas: C11orf95-RELA fusion-negative supratentorial ependymomas are a heterogeneous group of tumors. Acta Neuropathol. Commun. 2018, 6, 134. [Google Scholar] [CrossRef] [Green Version]
- Bouffet, E.; Perilongo, G.; Canete, A.; Massimino, M. Intracranial ependymomas in children: A critical review of prognostic factors and a plea for cooperation. Med. Pediatr. Oncol. 1998, 30, 319–329; discussion 329–331. [Google Scholar] [CrossRef]
- Merchant, T.E.; Li, C.; Xiong, X.; Kun, L.E.; Boop, F.A.; Sanford, R.A. Conformal radiotherapy after surgery for paediatric ependymoma: A prospective study. Lancet Oncol. 2009, 10, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, V.; Hielscher, T.; Mack, S.C.; Lassaletta, A.; Lin, T.; Pajtler, K.W.; Jones, D.T.; Luu, B.; Cavalli, F.M.; Aldape, K.; et al. Therapeutic Impact of Cytoreductive Surgery and Irradiation of Posterior Fossa Ependymoma in the Molecular Era: A Retrospective Multicohort Analysis. J. Clin. Oncol. 2016, 34, 2468–2477. [Google Scholar] [CrossRef] [PubMed]
- Thompson, Y.Y.; Ramaswamy, V.; Diamandis, P.; Daniels, C.; Taylor, M.D. Posterior fossa ependymoma: Current insights. Childs Nerv. Syst. 2015, 31, 1699–1706. [Google Scholar] [CrossRef]
- Indelicato, D.J.; Bradley, J.A.; Rotondo, R.L.; Nanda, R.H.; Logie, N.; Sandler, E.S.; Aldana, P.R.; Ranalli, N.J.; Beier, A.D.; Morris, C.G.; et al. Outcomes following proton therapy for pediatric ependymoma. Acta Oncol. 2018, 57, 644–648. [Google Scholar] [CrossRef]
- Indelicato, D.J.; Ioakeim-Ioannidou, M.; Bradley, J.A.; Mailhot-Vega, R.B.; Morris, C.G.; Tarbell, N.J.; Yock, T.; MacDonald, S.M. Proton Therapy for Pediatric Ependymoma: Mature Results from a Bicentric Study. Int. J. Radiat. Oncol. Biol. Phys. 2021, 110, 815–820. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, S.M.; Safai, S.; Trofimov, A.; Wolfgang, J.; Fullerton, B.; Yeap, B.Y.; Bortfeld, T.; Tarbell, N.J.; Yock, T. Proton radiotherapy for childhood ependymoma: Initial clinical outcomes and dose comparisons. Int. J. Radiat. Oncol. Biol. Phys. 2008, 71, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Garvin, J.H., Jr.; Selch, M.T.; Holmes, E.; Berger, M.S.; Finlay, J.L.; Flannery, A.; Goldwein, J.W.; Packer, R.J.; Rorke-Adams, L.B.; Shiminski-Maher, T.; et al. Phase II study of pre-irradiation chemotherapy for childhood intracranial ependymoma. Children’s Cancer Group protocol 9942: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2012, 59, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- von Hoff, K.; Kortmann, R.D.; Gerber, N.U.; Mynarek, M. Risk-adapted treatment for non-metastatic ependymoma: Preliminary results of the nonrandomized prospective phase II Clinical Trial Hit2000. Neuro Oncol. 2014, 16, i17. [Google Scholar]
- Duffner, P.K.; Krischer, J.P.; Sanford, R.A.; Horowitz, M.E.; Burger, P.C.; Cohen, M.E.; Friedman, H.S.; Kun, L.E. Prognostic factors in infants and very young children with intracranial ependymomas. Pediatr. Neurosurg. 1998, 28, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Grundy, R.G.; Wilne, S.H.; Robinson, K.J.; Ironside, J.W.; Cox, T.; Chong, W.K.; Michalski, A.; Campbell, R.H.; Bailey, C.C.; Thorp, N.; et al. Primary postoperative chemotherapy without radiotherapy for treatment of brain tumours other than ependymoma in children under 3 years: Results of the first UKCCSG/SIOP CNS 9204 trial. Eur. J. Cancer 2010, 46, 120–133. [Google Scholar] [CrossRef]
- Zacharoulis, S.; Levy, A.; Chi, S.N.; Gardner, S.; Rosenblum, M.; Miller, D.C.; Dunkel, I.; Diez, B.; Sposto, R.; Ji, L.; et al. Outcome for young children newly diagnosed with ependymoma, treated with intensive induction chemotherapy followed by myeloablative chemotherapy and autologous stem cell rescue. Pediatr. Blood Cancer 2007, 49, 34–40. [Google Scholar] [CrossRef]
- Grill, J.; Le Deley, M.C.; Gambarelli, D.; Raquin, M.A.; Couanet, D.; Pierre-Kahn, A.; Habrand, J.L.; Doz, F.; Frappaz, D.; Gentet, J.C.; et al. Postoperative chemotherapy without irradiation for ependymoma in children under 5 years of age: A multicenter trial of the French Society of Pediatric Oncology. J. Clin. Oncol. 2001, 19, 1288–1296. [Google Scholar] [CrossRef]
- Milde, T.; Hielscher, T.; Witt, H.; Kool, M.; Mack, S.C.; Deubzer, H.E.; Oehme, I.; Lodrini, M.; Benner, A.; Taylor, M.D.; et al. Nestin expression identifies ependymoma patients with poor outcome. Brain Pathol. 2012, 22, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Mohankumar, K.M.; Currle, D.S.; White, E.; Boulos, N.; Dapper, J.; Eden, C.; Nimmervoll, B.; Thiruvenkatam, R.; Connelly, M.; Kranenburg, T.A.; et al. An in vivo screen identifies ependymoma oncogenes and tumor-suppressor genes. Nat. Genet. 2015, 47, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, J.M.; Shelat, A.A.; Carcaboso, A.M.; Kranenburg, T.A.; Arnold, L.A.; Boulos, N.; Wright, K.; Johnson, R.A.; Poppleton, H.; Mohankumar, K.M.; et al. An integrated in vitro and in vivo high-throughput screen identifies treatment leads for ependymoma. Cancer Cell 2011, 20, 384–399. [Google Scholar] [CrossRef] [Green Version]
- Wright, K.D.; Daryani, V.M.; Turner, D.C.; Onar-Thomas, A.; Boulos, N.; Orr, B.A.; Gilbertson, R.J.; Stewart, C.F.; Gajjar, A. Phase I study of 5-fluorouracil in children and young adults with recurrent ependymoma. Neuro Oncol. 2015, 17, 1620–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotsch, D.; Kirchhofer, D.; Englinger, B.; Jiang, L.; Okonechnikov, K.; Senfter, D.; Laemmerer, A.; Gabler, L.; Pirker, C.; Donson, A.M.; et al. Targeting fibroblast growth factor receptors to combat aggressive ependymoma. Acta Neuropathol. 2021, 142, 339–360. [Google Scholar] [CrossRef]
- Mork, S.J.; Loken, A.C. Ependymoma: A follow-up study of 101 cases. Cancer 1977, 40, 907–915. [Google Scholar] [CrossRef]
- Rahman, R.; Osteso-Ibanez, T.; Hirst, R.A.; Levesley, J.; Kilday, J.P.; Quinn, S.; Peet, A.; O’Callaghan, C.; Coyle, B.; Grundy, R.G. Histone deacetylase inhibition attenuates cell growth with associated telomerase inhibition in high-grade childhood brain tumor cells. Mol. Cancer Ther. 2010, 9, 2568–2581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alderete, D.; Baroni, L.; Sampor, C.; Freytes, C.; Pennella, C. Antiangiogenic metronomic therapy for children with recurrent ependymoma. Neuro Oncol. 2018, 20, i74. [Google Scholar] [CrossRef] [Green Version]
- Gillan, E. Response of recurrent ependymoma to MEMMAT based metronomic antiangiogenic combination therapy utilizing tapered bevacizumab and maintenance therapy with celecoxib and fenofibrate. Neuro Oncol. 2020, 22, iii317. [Google Scholar] [CrossRef]
- Bull, K.S.; Hornsey, S.; Kennedy, C.R.; Darlington, A.E.; Grootenhuis, M.A.; Hargrave, D.; Liossi, C.; Shepherd, J.P.; Walker, D.A.; Morris, C. Systematic review: Measurement properties of patient-reported outcome measures evaluated with childhood brain tumor survivors or other acquired brain injury. Neurooncol. Pract. 2020, 7, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Morrissy, A.S.; Garzia, L.; Shih, D.J.; Zuyderduyn, S.; Huang, X.; Skowron, P.; Remke, M.; Cavalli, F.M.; Ramaswamy, V.; Lindsay, P.E.; et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature 2016, 529, 351–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Northcott, P.A.; Dubuc, A.; Dupuy, A.J.; Shih, D.J.; Witt, H.; Croul, S.; Bouffet, E.; Fults, D.W.; Eberhart, C.G.; et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature 2012, 482, 529–533. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Molecular subgroups and in-group subtypes of medulloblastoma; the four globally recognized molecular subgroups of medulloblastoma (WNT, SHH, Group 3 and Group 4) are shown, together with the current subtypes within WNT and SHH subgroups, as per [13], and Groups 3 and 4, in accordance with [14,15]. Two WNT-activated subtypes are reported, alongside 4 SHH subtypes. Groups 3 and 4 are likely now best considered as a spectrum of 8 different subtypes, each with biological and clinical characteristics. Age-related cartoons depict infant, young child (2–5 years), child (5–12 years), adolescent and older (12+ years). Key: OS = overall survival, DN = desmoplastic/nodular histology, LCA = large cell anaplastic histology, MBEN = medulloblastoma with extensive nodularity, amp. = amplification, mut. = mutation, del. = deletion, and actvn. = activation.
Figure 1.
Molecular subgroups and in-group subtypes of medulloblastoma; the four globally recognized molecular subgroups of medulloblastoma (WNT, SHH, Group 3 and Group 4) are shown, together with the current subtypes within WNT and SHH subgroups, as per [13], and Groups 3 and 4, in accordance with [14,15]. Two WNT-activated subtypes are reported, alongside 4 SHH subtypes. Groups 3 and 4 are likely now best considered as a spectrum of 8 different subtypes, each with biological and clinical characteristics. Age-related cartoons depict infant, young child (2–5 years), child (5–12 years), adolescent and older (12+ years). Key: OS = overall survival, DN = desmoplastic/nodular histology, LCA = large cell anaplastic histology, MBEN = medulloblastoma with extensive nodularity, amp. = amplification, mut. = mutation, del. = deletion, and actvn. = activation.

Figure 2.
Molecular subgroups of pediatric high-grade glioma. At least nine subgroups are thought to exist, with biological and clinical features highlighted in accordance with [84,90,91]. Age-related cartoons depict infant, young child (2–5 years), child (5–12 years), and adolescent/adult (12+ years). Key: amp. = amplification, mut. = mutation, del. = deletion, IDH = isocitrate dehydrogenase, and PXA, pleomorphic xanthoastrocytoma.
Figure 2.
Molecular subgroups of pediatric high-grade glioma. At least nine subgroups are thought to exist, with biological and clinical features highlighted in accordance with [84,90,91]. Age-related cartoons depict infant, young child (2–5 years), child (5–12 years), and adolescent/adult (12+ years). Key: amp. = amplification, mut. = mutation, del. = deletion, IDH = isocitrate dehydrogenase, and PXA, pleomorphic xanthoastrocytoma.

Figure 3.
Predominant molecular subtypes of pediatric intracranial ependymoma. Posterior fossa and supratentorial childhood ependymomas are shown, further categorized into four in-group subtypes; PF-A, PF-B, ZFTA-fused and YAP1-fused. The clinical and biological characteristics of these subtypes are shown, in accordance with [59,120,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153]. Nine molecular subtypes of ependymoma are reported but the remaining subtypes occur in either the spinal cord (spinal subependymoma, spinal myxopapillary ependymoma, spinal ependymoma) or the adult brain (subependymoma: PF and ST) so are not depicted in this figure. Age-related cartoons depict infant, young child (2–5 years), child (5–12 years), adolescent/adult (12+ years). Key: WHO = World Health Organization, CIN = chromosomal instability, GTR = gross total resection, IR = incomplete resection.
Figure 3.
Predominant molecular subtypes of pediatric intracranial ependymoma. Posterior fossa and supratentorial childhood ependymomas are shown, further categorized into four in-group subtypes; PF-A, PF-B, ZFTA-fused and YAP1-fused. The clinical and biological characteristics of these subtypes are shown, in accordance with [59,120,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153]. Nine molecular subtypes of ependymoma are reported but the remaining subtypes occur in either the spinal cord (spinal subependymoma, spinal myxopapillary ependymoma, spinal ependymoma) or the adult brain (subependymoma: PF and ST) so are not depicted in this figure. Age-related cartoons depict infant, young child (2–5 years), child (5–12 years), adolescent/adult (12+ years). Key: WHO = World Health Organization, CIN = chromosomal instability, GTR = gross total resection, IR = incomplete resection.


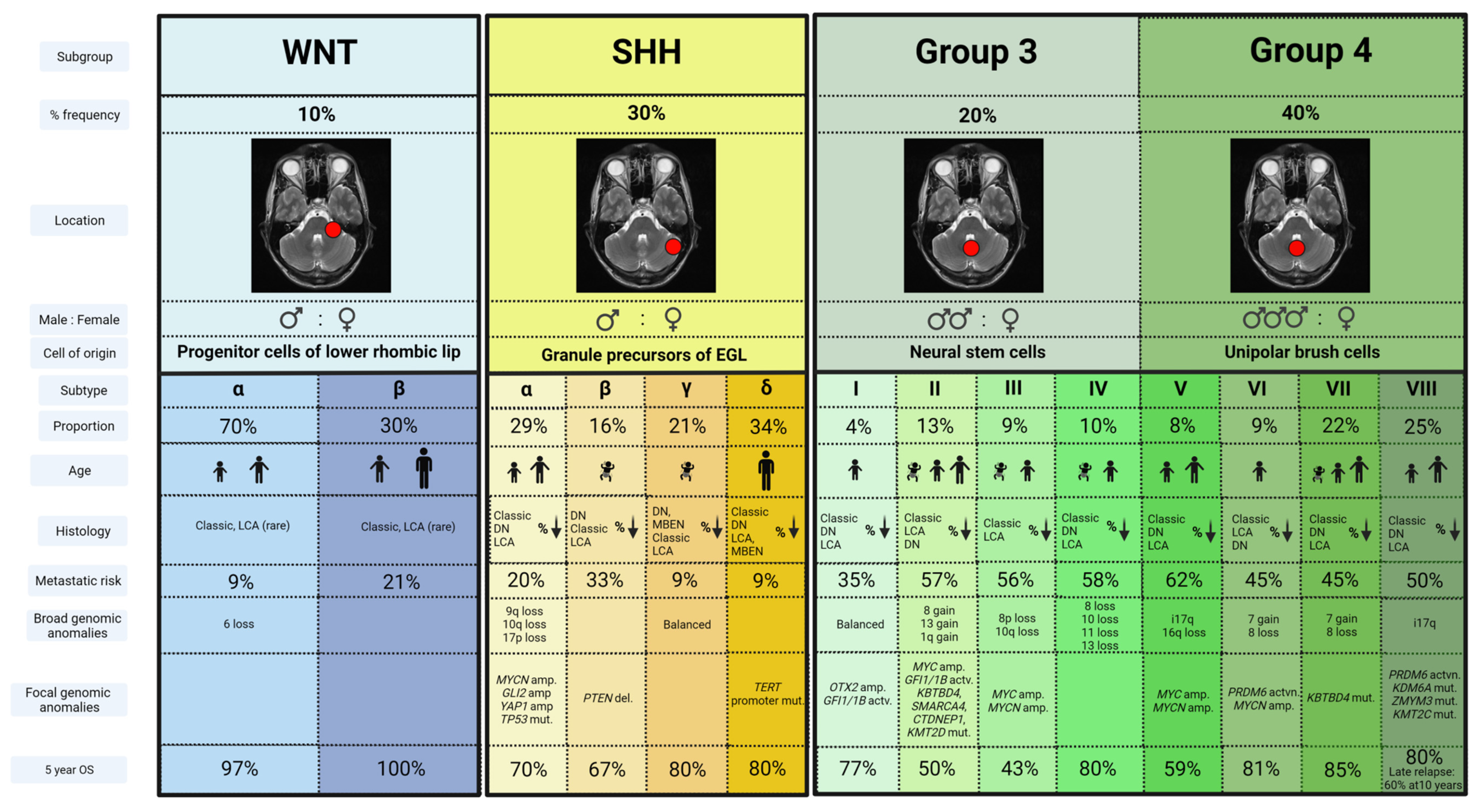{kind=link}
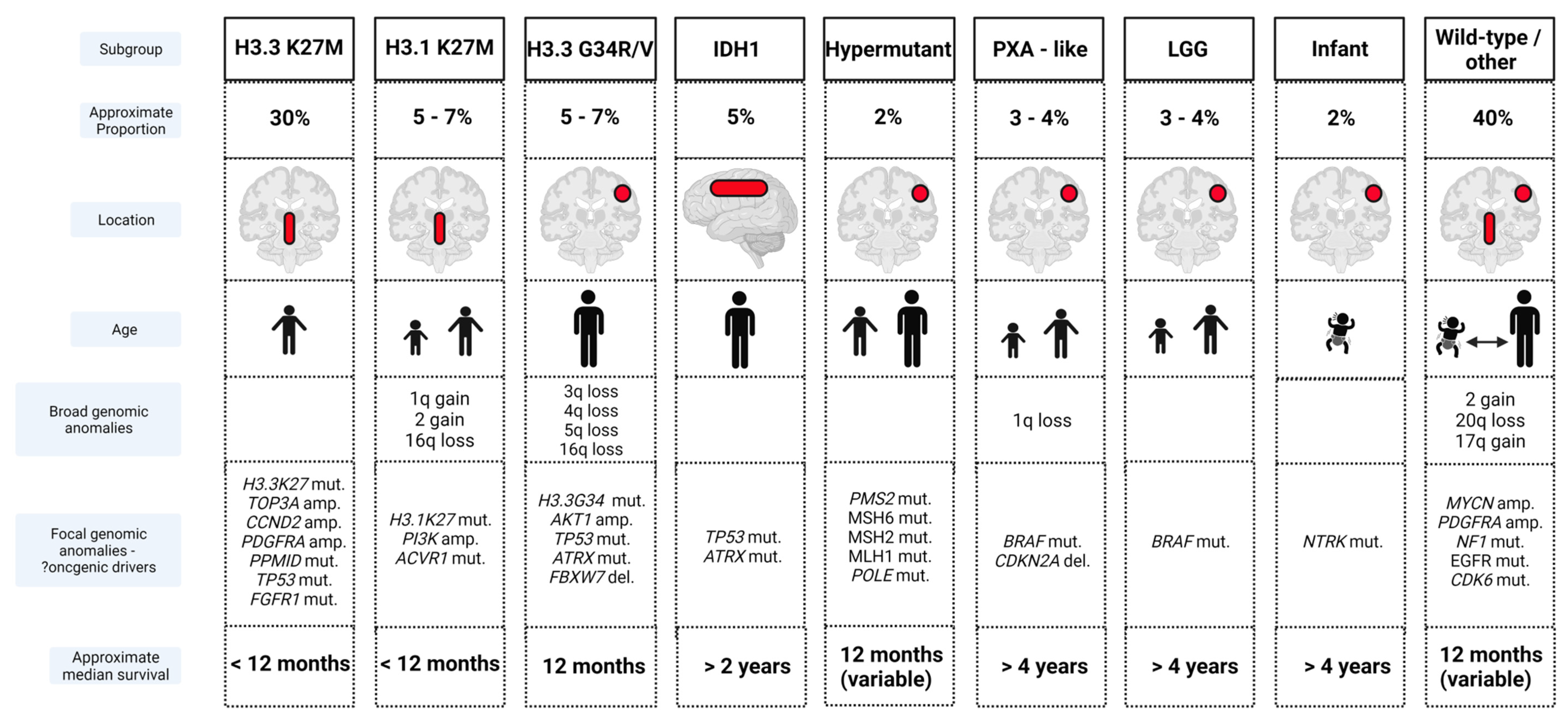{kind=link}
{kind=link}
Table 1.
Multinational collaborative clinical trials in pediatric medulloblastoma, high-grade gliomas and ependymoma, published since 2000.
Table 1.
Multinational collaborative clinical trials in pediatric medulloblastoma, high-grade gliomas and ependymoma, published since 2000.
| Year | Trial | Treatment Strategy | Inclusion Criteria | No. Patients | Results |
|---|---|---|---|---|---|
| Medulloblastoma | |||||
| 1992– 2000 | SIOP PNET III [50] | Randomization Arm 1: RT alone (35 Gy CSI + 20 Gy PF boost) Arm 2: 4 cycles alternating Carbo/VP16 and Cyclo/VP16 followed by RT | Age 3–16 yrs Standard-risk MB | 179 | 5 yr EFS 59.8% vs. 74.2% RT + chemotherapy superior |
| 1996– 2000 | COG A9961 [3] | Radiotherapy: 23.4 Gy CSI + 32.4 Gy PF boost + weekly VCR Continuation chemotherapy randomization: Arm 1: CCNU/Cis/VCR Arm 2: Cis/Cyclo/VCR | Age 3–21 yrs Standard-risk MB | 421 | 10 yr EFS 74% vs. 78% None superior |
| 2001– 2006 | HIT-SIOP PNET-4 [51] | Radiotherapy randomization Arm 1: HFRT (36 Gy CSI, 24 Gy PF boost, 8 Gy TB boost) Arm 2: STRT (23.4 Gy CSI, 30 Gy PF boost) Continuation chemotherapy 8 cycles Cis/CCNU/VCR | Age 4–<22 years Standard-risk MB | 340 | 5 yr EFS 77% vs. 78 None superior |
| 2004– 2016 | COG ACNS0331 [52] | Radiotherapy Children aged 3–7 years randomized: Randomization 1: CSI: Low-dose (LDCSI) 18 Gy vs. Standard dose (SDCSI) 23.4 Gy Randomization 2: Involved field RT boost vs. Standard volume boost Children ≥ 8 yrs receive CSI 23.4 Gy, then randomized: Randomization 3: Involved field RT boost (IFRT) vs. Arm 2: Standard volume boost (PFRT) Continuation chemotherapy 9 cycles (6 × CCNU/Cis/VCR, 3 × Cytoxan/VCR) | Age 3–<21 yrs Standard-risk MB | 513 | 5 yr EFS/OS LDCSI 72.1%/78.1% SDCSI 82.6%/85.9% LDCSI higher event rates and worse survival PFRT 80.8%/85.2% IFRT 82.2%/84.1% None superior |
| 1990– 1996 | POG 9031 [49] | Arm 1: 3 cycles Cis/VP16, followed by RT (CSI 35.2–44.0 Gy, PF dose 53.2–54.4 Gy) then 7 cycles VCR/Cyclo continuation chemotherapy Arm 2: RT (CSI 35.2–44.0 Gy, PF dose 53.2–54.4 Gy) followed by 3 cycles Cis/VP16 and 7 cycles VCR/Cyclo continuation chemotherapy | Age 3–18 yrs High-risk MB | 224 | 5 yr EFS/OS: 66%/73.1% vs. 70%/76.1% None superior |
| 1996– 2007 | SJMB96 [48] | Radiotherapy Risk Stratified: SR: 23.4 Gy, 36 Gy PF dose and 55.8 Gy TB dose; HR: 36–39.6 Gy and 55.8 Gy TB dose (50.4 Gy dose to metastatic sites) Chemotherapy 4 × Cis/Cyclo/VCR with stem cell rescue | Age 3–20 yrs Standard and High-risk MB | 134 | 5 yr EFS/OS: SR 83%/85% HR 70%/70% |
| 2007– 2017 | SJYC07 [38] | Induction chemotherapy LR and IR: MTX/VCR/Cis/Cyclo HR: MTX/VCR/Cis/Cyclo + Vinblastine Consolidation therapy LR: 2 cycles Carbo/Cyclo/VP16 IR ≥ 12 mths old: Focal RT (54 Gy TB dose); IR < 12 months old: 2 × cycles Carbo/Cyclo/VP16 HR < 3 years old: Topo/Cyclo (8 weeks); HR ≥3 years old: could opt for CSI (23.4–39.6 Gy) Continuation chemotherapy All Groups: 6 cycles oral Cyclo/Topo/Erlotinib | Age < 3 yrs newly diagnosed MB OR Age 3–5 yrs -non-metastatic -no high-risk features | 81 | LR: 1 yr EFS 78.3%, (accrual suspended as EFS below stopping rule). 5 yr EFS/OS: LR 55.3%/85.9% IR: 24.6%/52.8% HR: 16.7%/41% |
| 2013– 2016 | ACNS1221 [39] | Induction chemotherapy 3 cycles Cyclo/VCR/MTX/VP16/Carbo Reassessment CR/CCR: No further treatment PRD: Second look surgery + 2 cycles Cyclo/VCR/Carbo/VP16 | Age < 4 yrs Localized ND or MBEN | 25 | 2 yr PFS/OS 52%/92% Failed to achieve 2 yr PFS target of 90%; study closed early |
| 2007– 2018 | ACNS0332 [53] | Randomization Arm 1: Standard treatment (CSI 36 Gy, PF 55.8 Gy + 6 cycles Cis/Cyclo/VCR maintenance) Arm 2: Standard treatment + RT with Carbo Arm 3: Standard treatment + isotretinoin during maintenance Arm 4: Standard treatment + RT with Carbo + isotretinoin during maintenance | 3–21 yrs High-risk MB | 261 | Survival advantage for Grp 3 MB receiving RT with carboplatin. 5 yr EFS/OS: 73.2%/82.3% vs. 53.7%/63.7% Isotretinoin therapy futile |
| High-Grade Gliomas | |||||
| 2004– 2005 | ACNS0126 [54] | RT (HGG 54 Gy, DIPG 59.4 Gy) + concomitant low-dose TMZ, followed by 10 cycles of higher dose TMZ continuation therapy | Age 3–≤22 yrs | HGG = 107 DIPG = 63 | 1 yr EFS/OS 14%/40% No improvement vs. historical controls |
| 2005– 2007 | ACNS0423 [55] | RT (GTR 54 Gy, STR 59.4 Gy, spinal cord lesions 50.4–54 Gy) + concomitant low-dose TMZ, followed by up to 6 cycles of higher dose TMZ + CCNU continuation | Age 3–≤22 yrs | 108 | 3 yr EFS/OS 22%/19% Improved vs. ACNS0126 |
| 2007– 2008 | ACNS0222 [56] | RT (54 Gy) with motexafin-gadolinium as a potent radiosensitizer | Age ≤ 21 yrs Unifocal DIPG | 60 | 1 yr EFS/OS 18%/53% No Improvement |
| 2011– 2015 | HERBY [57] | Randomization Arm 1: RT (54 Gy) + low-dose TMZ, continuation high-dose TMZ 12 months Arm 2: RT (54 Gy) + low-dose TMZ + Bev, continuation high-dose TMZ + Bev 12 mnths | Age ≥ 3–≤18 yrs Non–brainstem | 116 | 1 yr median EFS 11.8 vs. 8.2 mnths No improvement |
| 2014– 2020 | BIOMEDE 1 [58] | Randomization Arm 1: RT + Everolimus Arm 2: RT + Dasatinib Arm 3: RT + Erlotinib | Age 6 mths–25 yrs DIPG | 193 | Median OS Arms 1, 2, 3 10.9, 9.5 and 9 mnths No improvement |
| Ependymoma | |||||
| 2003– 2007 | ACNS0121 [59] | Stratum 1: Completely resected differentiated, ST ependymoma undergo observation Stratum 2: Incompletely resected ependymoma undergo chemotherapy, second surgery and RT Stratum 3: Near-total or macroscopic GTR undergo conformal RT Stratum 4: Microscopic GTR undergo conformal RT, excluding differentiated, ST lesions | Age 1–21 yrs | 356 | 5 yr EFS/OS Strata 1: 61%/100% Strata 2: 37.2%/70.2% Strata 3: 67%/83.3% Strata 4: 70%/88.3% |
| 2010– 2017 | ACNS0831 [60] | PF tumours gross/near total resection: randomization Arm 1: RT alone Arm 2: RT + 4 cycles VCR/Cis/Cyclo/VP16 | Age 1–21 yrs | 451 | 3 yr EFS 71% vs. 80% ? chemotherapy superior |
RT: radiotherapy; CSI: craniospinal irradiation; PF: posterior fossa; Carbo: carboplatin; VP16: etoposide; Cyclo: cyclophosphamide; MB; medulloblastoma; EFS: event-free survival; VCR; vincristine; CCNU: lomustine; Cis: cisplatin; HFRT: hyper-fractionated radiotherapy; STRT: standard radiotherapy; TB: tumor bed; OS: overall survival; SR: standard risk; HR: high risk; MTX: methotrexate; LR: low risk; IR: intermediate risk; Topo: topotecan; CR: complete response; CCR: continuous complete response; PRD: persistent residual disease; Ifos: ifosfamide; GTR: gross total resection; DIPG: diffuse intrinsic pontine glioma; yrs: years; mnths: months.
Table 2.
Future clinical challenges for pediatric malignant brain tumors.
| Tumor Group | Future Clinical Challenge |
|---|---|
| ALL |
|
| Medulloblastoma | |
| WNT |
|
| SHH |
|
| Group 3 |
|
| Group 4 |
|
| High-grade gliomas |
|
| Ependymoma | |
| PF-A |
|
| PF-B |
|
| ST-ZFTA |
|
| ST-YAP1 |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Thorbinson, C.; Kilday, J.-P. Childhood Malignant Brain Tumors: Balancing the Bench and Bedside. Cancers 2021, 13, 6099. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236099
AMA Style
Thorbinson C, Kilday J-P. Childhood Malignant Brain Tumors: Balancing the Bench and Bedside. Cancers. 2021; 13(23):6099. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236099
Chicago/Turabian StyleThorbinson, Colin, and John-Paul Kilday. 2021. "Childhood Malignant Brain Tumors: Balancing the Bench and Bedside" Cancers 13, no. 23: 6099. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236099
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.