
Promising Antigens for the New Frontier of Targeted Immunotherapy in Multiple Myeloma


1
Jerome Lipper Multiple Myeloma Center, Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, 450 Brookline Avenue, Boston, MA 02215, USA
2
Division of Hematology & Oncology, Department of Internal Medicine, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung 807, Taiwan
3
Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung 807, Taiwan
4
Center for Cancer Research, Kaohsiung Medical University, Kaohsiung 807, Taiwan
5
Department of Hematology, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan 250021, China
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(23), 6136; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236136
Submission received: 20 October 2021
/
Revised: 29 November 2021
/
Accepted: 3 December 2021
/
Published: 6 December 2021
(This article belongs to the Special Issue Signaling Pathways in Multiple Myeloma)
Abstract
:Simple Summary
Defining the specificity and biological sequalae induced by receptors differentiated expressed in multiple myeloma cells are critical for the development of effective immunotherapies based on monoclonal antibodies. Ongoing studies continue to discover new antigens with superior tumor selectivity and defined function in regulating the pathophysiology of myeloma cells directly or indirectly in the immunosuppressive bone marrow microenvironment. Meanwhile, it is urgent to identify mechanisms of immune resistance and design more potent immunotherapies, alone and/or with best combination partners to further prolong anti-MM immunity.
Abstract
The incorporation of novel agents in recent treatments in multiple myeloma (MM) has improved the clinical outcome of patients. Specifically, the approval of monoclonal antibody (MoAb) against CD38 (daratumumab) and SLAMF7 (elotuzumab) in relapsed and refractory MM (RRMM) represents an important milestone in the development of targeted immunotherapy in MM. These MoAb-based agents significantly induce cytotoxicity of MM cells via multiple effector-dependent mechanisms and can further induce immunomodulation to repair a dysfunctional tumor immune microenvironment. Recently, targeting B cell maturation antigen (BCMA), an even MM-specific antigen, has shown high therapeutic activities by chimeric antigen receptor T cells (CAR T), antibody-drug conjugate (ADC), bispecific T-cell engager (BiTE), as well as bispecific antibody (BiAb), with some already approved for heavily pretreated RRMM patients. New antigens, such as orphan G protein-coupled receptor class C group 5 member D (GPRC5D) and FcRH5, were identified and rapidly moved to ongoing clinical studies. We here summarized the pathobiological function of key MM antigens and the status of the corresponding immunotherapies. The potential challenges and emerging treatment strategies are also discussed.
Keywords:
multiple myeloma; MM; immunotherapy; tumor target antigen; immunomodulatory drugs; IMiDs; monoclonal antibody; MoAb; CD38; signaling lymphocyte activation molecule family 7; SLAMF7; B cell maturation antigen; BCMA; bone marrow (BM) microenvironment; orphan G protein-coupled receptor class C group 5 member D; GPRC5D; FcRH51. Introduction
The development and introduction of the proteasome inhibitor (PI) bortezomib and immunomodulatory drugs (IMiDs), including thalidomide and lenalidomide, has revolutionized the treatment paradigm for multiple myeloma (MM). Second-generation drugs within the same classes, such as carfilzomib and ixazomib (PIs) and pomalidomide (IMiDs), further improve the response rate, survival, and safety profile [1,2,3]. The incorporation of autologous stem cell transplantation in eligible patients has also prolonged survival with more durable disease control [4,5]. However, disease recurrence remains common for most MM patients. Since drug-resistant clones constantly emerge and evolve, leading to a low 5-year overall survival rate in real-world data [6]. The clinical outcomes of patients with relapsed or refractory MM (RRMM) are dismally poor because of the gradually decreased durability of the response to successive lines of anti-MM therapy [7]. It is thus urgent to further devise novel therapies with different mechanisms of action and optimize treatment efficacy to reduce the risk of disease relapse and deepen response durability.
Accumulating studies for the past decades have defined that the bone marrow (BM) microenvironment is essential in supporting MM cell growth, survival, and drug resistance. MM cells are in close contact with surrounding BM accessory cells through bi-directional interactions, including stromal cells (BMSCs) [8], osteoclasts (OCs) [9,10], regulatory T (Treg) or B (Breg) cells [11,12,13], myeloid-derived suppressor cells (MDSCs) [14], tumor-associated macrophages (TAMs) [15], and plasmacytoid dendritic cells [16]. These non-MM cells, in turn, secrete abnormal levels of a variety of cytokines and growth factors in a paracrine fashion to promote pathogenesis of MM, including interleukin-6 (IL-6), IL-10, MIP-1α/β, transforming growth factor-beta (TGFβ), stromal cell-derived factor-1 (SDF-1), and a proliferation-inducing ligand (APRIL) [9,17,18,19]. Furthermore, changes in BM accessory cells and cytokines, either secreted by accessory cells or MM cells via autocrine or paracrine manners, contribute to myeloma cell immune escape, inhibition of myeloma-specific T effector cells, induction of T-cell anergy, and abnormality in Treg cells, resulting in an immunosuppressive microenvironment that impairs immunotherapy [20].
Monoclonal antibodies (MoAbs) binding to selective molecules on the surface of cancer cells have transformed cancer treatment. In principle, these biologically based molecules/proteins induce tumor cell killing mainly dependent on effector functions, including antibody-dependent cellular cytotoxicity (ADCC) via CD16-expressing effector cells (i.e., NK cells, neutrophils, monocytes), complement-dependent cytotoxicity (CDC), and/or antibody-dependent cellular phagocytosis (ADCP) via macrophages. These primary mechanisms of action are distinct from small molecules used in conventional chemotherapies, which directly induce tumor cell apoptosis and are largely independent of immune effector function. The first two therapeutic MoAbs available for RRMM patients are MoAbs targeting CD38 (daratumumab) and SLAMF7 (also named CS1) (elotuzumab), approved by the U.S. Food and Drug Administration (FDA) in 2015 [21,22]. These represent an important breakthrough for effective targeted immune-based therapies in MM. Importantly, results obtained from preclinical and clinical studies of both MoAbs thus far have shown that these first-generation targeting bio-molecules also affect the immunosuppressive non-MM cell components in addition to MM cells [11,12,13,23,24,25]. These findings have inspired many investigations on identifying the patho-immunological roles of various immune regulatory cell subsets and molecules regulating their function using in vitro, ex vivo, and in vivo models. Data from some of these studies have provided rationales to improve the usage of targeting immunotherapeutic reagents with novel approaches. More detailed results will be highlighted in subsequent sections.
Factors to be considered to enhance the effectiveness of current targeted immunotherapies based on MoAb in MM are as follows. First, these agents should more selectively identify and bind to specific cell surface antigens to trigger MM cell cytotoxicity with a minimal off-target impact on normal tissues. Second, in addition to significantly inducing MM cell lysis, these immunotherapeutic molecules could, at the same time, mitigate an immunocompromised BM microenvironment and revert suppressive immune effector cells. Moreover, a combination of different immunotherapeutic platforms with distinct mechanisms of action against target antigens on MM cells as well as key immune stimulatory or inhibitory cells will enhance the strength and reduce the weakness of individual drugs to further prolong the durability of the responses and reduce non-specific toxicity. Currently, immunotherapeutic modalities based on naked MoAbs, chimeric antigen receptor T cells (CAR-T), bispecific antibody (BiAb), bispecific T-cell engager (BiTE), or antibody-drug conjugates (ADC) are the main research areas under preclinical and clinical development [26,27,28]. These agents are mostly designed to target tumor antigens on MM cells and endowed with the ability to restore and further sustain anti-MM effector function in the suppressive BM milieu.
2. Pathophysiological Function for Validated MM Target Antigens and Their Related Immunotherapies
The anti-MM mechanisms of targeted immunotherapeutic bio-reagents are mainly derived from the MoAbs, which detect and engage with selective proteins on MM cell membrane followed by the induction of NK cell-mediated killing (Figure 1 and Figure 2). These target antigens are chosen based on their differential expression and/or critical roles in growth, survival, and drug resistance in MM cells. The following paragraphs underline the major pathobiological and -immunological characteristics as well as the corresponding targeting immunotherapeutic agents of MM target antigens currently under preclinical and clinical studies.
2.1. CD38
CD38, a type II transmembrane glycoprotein, was first identified as a marker of cell activation and proliferation in lymphocytes. It regulates cell migration [29] and receptor-mediated adhesion via interaction with endothelial CD31 or hyaluronic acid [30]. CD38 receptor also exhibits ecto-enzymatic activity, involved in the metabolism of cytoplasmic nicotinamide adenine dinucleotide phosphate (NADP) and extracellular nicotinamide adenine dinucleotide (NAD+) [31]. In addition, CD38 interacts with its substrate, NAD+, to increase production of Ca2+ mobilizing compounds; i.e., cyclic adenosine diphosphate ribose (CADPR), ADP ribose (ADPR), and nicotinic acid adenine dinucleotide phosphate [32,33,34]. ADPR is further converted to adenosine monophosphate (AMP) by CD203a and then adenosine (ADO), which exhibits immunosuppressive activity via a reduction in immune cell activity and induction of differentiation of osteoclast, one of the most immunosuppressive BM accessory cells [35,36,37,38]. Moreover, the malignant plasma cells further use aerobic glycolysis to promote an acidic BM, which together with CD38 highly expressed on their surface induce the generation of AMP and ADO.
In MM cells, CD38 is downregulated by IL-6, the major MM cell growth and survival factor secreted by BMSCs, via activation of IL-6-induced JAK1/2-STAT1/3 signaling pathways [39,40]. Decreased CD38 expression triggered by IL-6 in MM cells is further associated with reduced sensitivity to anti-CD38 MoAb (i.e., daratumumab), providing a new molecular mechanism to support the immunosuppressive function of IL-6 in the BM microenvironment. Since the JAK1/2 inhibitor ruxolitinib blocks IL-6-induced phosphorylation of STAT1 and STAT3, these data also suggest a potential combination daratumumab with JAK1/2 inhibitor to revert daratumumab resistance.
On the other hand, since CD38 is also expressed at various levels in other normal hematopoietic cells, including NK effector cells, daratumumab-induced MM cell lysis is negatively affected due to daratumumab-induced NK cell depletion, as seen in laboratory studies as well as in patients [41,42,43]. New adoptive immunotherapy using ex vivo expanded human primary NK cells with or without CD38 knockout was recently proposed to boost daratumumab activity in MM [42,43]. In contrast, when compared with immune effector T cells, immune inhibitory Treg and Breg cells express elevated CD38 levels, as high as MM cells, and thereby are preferentially eliminated by CD38 targeting MoAbs and T cell expansion is promoted [11,12,44]. This additional function on targeting key immune suppressive cell subsets further support the therapeutic efficacy of anti-CD38 MoAbs in MM.
The incorporation of daratumumab into current anti-MM treatment regimens has significantly prolonged overall survival in RR as well as ND MM patients [22,45,46,47,48]. In 2019, daratumumab is the first MoAb, when used in combination, approved for treatment for NDMM patients who may still exhibit functional immune cells in the less defective immune BM milieu. The second anti-CD38 MoAb, isatuximab, recognizes the non-overlapping CD38 epitope of daratumumab and has shown convincing clinical activity, leading to its approval when combined with pomalidomide and dexamethasone in RRMM in 2020 [49].
Besides the induction of effector-mediated MM cell lysis in a CD38-dependent manner, both daratumumab and isatuximab eliminate high CD38-expressing immune inhibitory cell subsets (i.e., Treg, Breg) in MM patients, a supplementary mechanism to increase immune effector cell number and function [11,12,13]. Compared with NK effector cells, T effector cells express low levels of CD38. A BiTE, AMG 424, with a lower CD38-binding affinity together with CD3 binding to T cells, was recently developed and its preclinical activity was evaluated using in vitro and in vivo models of human MM [50]. This next generation CD38-targeting molecule potently induces a cytotoxic T cell response to kill MM cells via cytolytic cytokine production (i.e., interferon-gamma (IFNγ), granzyme B (GZMB), perforin (PRF1)), with minimal toxicity on T effector cells. Furthermore, anti-CD38 CAR-T cells were reported and induced lysis of CD38+ MM cells with mild effects on other CD3+ T cells [51].
2.2. SLAMF7 (CS1)
SLAMF7 is a member of the immunoglobulin gene superfamily (signaling lymphocyte activation molecule family) and associated with cytotoxic effector, humoral and auto-immunity, and cell survival/adhesion, as well as lymphocyte development [52,53]. SLAMF7 is also expressed on the surface of certain subsets of immune cells, including NK cells, cytotoxic T lymphocytes (CD8+ cells), B lymphocytes, and mature dendritic cells. SLAMF7 itself can serve as a receptor of NK cell and it is a self-ligand that exhibits a homophilic interaction to augment NK cell-mediated cytotoxicity [54]. Moreover, SLAMF7 on the surface of B cells is upregulated during B cell activation to promote proliferation of naive and memory B cells and cytokine production [55].
SLAMF7 is highly expressed on patient MM cells from ND to RRMM patients, independent of the cytogenetic risk classification [24,56]. Soluble SLAMF7 (sSLAMF7) is detected in MM patients rather than normal individuals [24] and could promote MM cell growth via activating the SHP-2 and ERK signaling pathways via homophilic interaction [57]. Targeting IKZF1 (Ikaros 1), a critical transcriptional activator of SLAMF7, by IMiDs downregulates SLAMF7 expression and ameliorated the response of MM cells to sSLAMF7. MM cells with t(4;14) translocations (15% of all MM cases) have higher SLAMF7 expression, associated with MMSET overexpression [58]. SLAMF7 knockdown by its shRNA inhibits colony formation and induces cell cycle arrest followed by apoptosis of t(4;14) plasma cells, indicating elevated SLAMF7 expression in promoting the growth of MM cells. Furthermore, SLAMF7 promotes the adhesion of plasma cells to BM stromal cells to support survival and proliferation of MM cells in the BM; conversely, an antagonistic anti-SLAMF7 MoAb prevents MM cells from adhesion to BMSCs and induces ADCC to lyse MM cells [24,59]. Most recently, SLAMF7 was found highly expressed on immunosuppressive CD8+CD28-CD57+ Tregs in MM patients and these cells could be eliminated using anti-SLAMF7 MoAb elotuzumab [25].
Elotuzumab, the first-in-class humanized immunoglobulin G1 anti-SLAMF7 MoAb, exhibits immunomodulatory effects on NK cells via activation of SLAMF7/EAT2 signaling without negative impacts on NK cell number and survival [24,56,60]. Combination of elotuzumab with IMiDs (lenalidomide, pomalidomide) with low-dose dexamethasone significantly improved the clinical outcome of RRMM patients in the ELOQUENT-2 trial [21,61], leading to its approval for the treatment in RRMM. SLAMF7-targeting agents using ADC, CAR-T, or BiAb are under preclinical and/or clinical investigations.
2.3. CD138 (Syndecan-1)
CD138 (syndecan 1), a member of the syndecan family of type I transmembrane proteoglycan, has been commonly used as a prognostic marker in MM, since its expression level is elevated in malignant versus normal plasma cells [62]. CD138 modulates various biological processes, including proliferation [63], adhesion [64], migration [65], endocytosis [66], macropinocytosis [67], immunomodulation [68], and regulation of heparan sulfate proteoglycans [69]. Increased CD38 expression promotes proliferation and survival of MM cells, as well as angiogenesis and IL-6 receptor sensitivity in MM cells [70,71]. IL-6-induced growth and survival signaling cascades upon binding to IL-6R is further augmented in MM cells overexpressing CD138, indicating cross-talks between CD138 and IL6R in the progression of MM. Importantly, high CD138 expression is linked to enhanced malignant plasma cell growth and disease burden in patients. Since CD138 is cleaved by metalloproteinases and heparanase, soluble CD138 (sCD138) is detected in patient serum samples and its levels are associated with the prognosis of MM, with shorter survival in patients with higher levels [72]. Significantly, shedding of CD138 (sCD138) from MM cells stimulates myeloma cell growth by positive regulation and interaction with other MM-promoting factors (i.e., IL-6, vascular endothelial growth factor (VEGF), APRIL) in the BM microenvironment [62,70,73].
Besides plasma cells, CD138 is expressed in other normal and malignant human tissues at various levels, including normal squamous epithelial, goblet, and columnar cells in the gastrointestinal tract, as well as tumor cells, including squamous cell carcinoma and adenocarcinoma [67,74,75]. As reported in previous clinical studies testing the first CD138-targeting ADC, BT062, described below, high expression of CD138 on epithelial cells was associated with increased risk of treatment toxicity (such as mucositis or diarrhea) [76].
BT062/Indatuximab ravtansine, an ADC with cytotoxic maytansinoid DM4, was the first CD138-targeting drug tested in an MM clinical trial [76,77]. Most of the adverse events were mild (grade 1 or 2), with diarrhea and fatigue the most common. The response rate of monotherapy or combination therapy (with lenalidomide or pomalidomide and dexamethasone) in RRMM patients were 14.7% and 77%, respectively [76,77]. Besides ADC, CD138-targeting therapy under clinical and preclinical investigation include CAR-NK and CAR-T cell therapy [78,79,80]. A case report demonstrated a patient with extensive extramedullary MM involvement receiving anti-CD138 CAR-T cell infusion (total: 1.5 × 108) after a conditioning regimen of cyclophosphamide and fludarabine. The patient experienced grade 2 cytokine release syndrome (CRS) and received anti-IL-6R MoAb tocilizumab treatment with clinical partial response (PR) [81].
Recently, a bispecific antibody and CAR-T cell based on a new anti-CD138 MoAb showed significant anti-MM activity and an immunomodulatory effect in preclinical studies [78]. A novel naked MoAb VIS832 was recently made, showing enhanced membrane CD138-binding affinity compared to the MoAb portion of BT062 [82]. VIS832, as a naked IgG1 MoAb, potently induced ADCC and ADCP against MM cells, including resistant cell lines or patient MM cells, without directly impacting MM cells. No toxicity was seen in NK cells treated with VIS832, confirming the absence of CD138 expression on immune effector cells. Anti-MM activity of VIS832 in the in vitro and in vivo models was further augmented when combining with lenalidomide or bortezomib in the preclinical study. These data provide a clinical rationale to test this new anti-CD138 MoAb, alone or in combination with current standard-of-care anti-MM drugs in MM.
2.4. B-Cell Maturation Antigen (BCMA)
BCMA, also called tumor necrosis factor receptor superfamily member 17 (TNFRS17) or CD269, is a type III transmembrane protein with extracellular domains rich in cysteine without a signal peptide. BCMA, closely related to B-cell activation factor receptor (BAFF-R), and transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI), regulates B cell proliferation and survival, as well as maturation and differentiation into plasma cells [83,84,85]. These three functionally related receptors bind to their cognate ligands, BAFF and/or APRIL, with different affinities, to support long-term survival of B cells at different stages of development. Specifically, BCMA, but not BAFF-R or TACI, is crucial for the long-term survival of plasma cells, but not overall B cell homeostasis [85]. During the differentiation of B cells into plasma cells, the expression of BCMA is induced from late memory cell, while BAFF-R is concomitantly extinguished. BCMA expression is regulated by B-lymphocyte-induced maturation protein 1 (BLIMP1), an important plasma cell transcriptional factor [86]. Under normal physiological conditions, the membrane BCMA is cleaved by gamma-secretase to form the soluble BCMA (sBCMA) [87]. Serum levels of sBCMA are significantly higher in MM patients than healthy individuals and associated with immune deficiency in the tumor microenvironment [88,89]. Elevated sBCMA levels are positively linked to increased tumor burden and poorer overall or progression-free survival [89]. Furthermore, the post-treatment levels of sBCMA could be used as a predictive marker for treatment response [90,91,92].
As in the case for CD38, SLAMF7, and CD138, expression of BCMA is increased in patient MM cells. The evaluation of patient samples from various cohorts further confirmed that expression of BCMA at the transcript and protein level is more restrictively expressed on plasma cells but no other normal tissues, when compared with the above MM antigens included in the previous paragraphs [93,94,95]. Among other normal tissues, BCMA transcript and protein are only weakly detected on plasmacytoid dendritic cells, a minute BM cell subset that promote MM cell growth, survival, and resistance to anti-MM drugs [95]. Patients with higher BCMA expression from their CD138+ plasma cells also have elevated BCMA levels in autologous plasmacytoid dendritic cells, further supporting the patho-biological role of BCMA in MM. In addition, BCMA is co-immunoprecipitated with interferon regulatory factor 4, a major transcription factor mediating survival of MM cells [96]. Overexpression of BCMA directly augments MM cell growth and survival via induction of protein kinase B (AKT), MAPK, and nuclear factor (NF)-κB signaling cascades, followed by upregulation of the gene expression of molecules critical in growth and anti-apoptosis [18,19,84]. It further enhances expression of genes related to activation of OCs, adhesion, angiogenesis, and metastasis, as well as development of immunosuppressive characters, including programmed death ligand 1 (PD-L1), TGFβ, and IL-10. Moreover, overexpression of BCMA in the MM cell line expressing low BCMA levels induces early onset and increased volume of xenografted tumors with increased CD31/microvessel density and VEGF in a murine model of RPMI8226 MM cells, confirming its tumor-promoting effects in vivo.
As a critical plasma cell receptor, BCMA binds to its cognate ligands BAFF or APRIL to activate AKT, ERK1/2, and NFκB pathways in MM cells [18,97]. These two ligands are mainly secreted by non-MM cells in the BM and with differential influences on the biology of MM cells. First, BAFF binds to BAFF-R, BCMA, and TACI, to promote adhesion of MM cell to BM stromal cells, but with a significantly higher selectivity (~100-fold) to BAFF-R, which is hardly detectable in MM cells [97,98]. In contrast, APRIL cannot bind to BAFF-R and preferentially binds to BCMA or TACI, the latter of which is less frequently expressed when compared with the former in MM cells. Specifically, APRIL preferably binds to BCMA with much higher affinity than BAFF [83,99] and is predominantly produced by myeloid cells, macrophages, OC precursor cells, and OCs which play central pathophysiological roles in MM-induced bone lesions. All these data indicate that APRIL may be a more significant factor than BAFF in the development and progression of MM. Importantly, APRIL binding to BCMA triggers multiple signaling pathways to further promote drug resistance of MM cells and the progression of immunosuppressive BM milieu via induction of the key downstream anti-apoptotic genes (Mcl-1, Bcl-2/Bcl-xL) and immune regulatory genes (IL-10, PD-L1, VEGF, TGF-β) in MM cells [18,19]. Moreover, APRIL directly impacts Treg cells that express no BCMA, to promote an immunosuppressive BM microenvironment via binding to TACI whose expression levels are significantly correlated with upregulated Treg markers, including Foxp3 and CTLA-4 [44]. Besides TACI-dependent induction of cell cycle progression and anti-apoptosis genes, APRIL specifically augments expression of Foxp3, IL-10, TGFβ1, and PD-L1 in Tregs to further augment Treg-inhibited conventional T cells proliferation and cytolytic function. APRIL also enhances IL-10-producing Breg cells via TACI in the BM of MM patients. All these results strongly support targeting APRIL/BCMA and APRIL/TACI systems for novel MM immunotherapies.
The first therapeutic anti-BCMA MoAb J6M0 was selected based on its significant blocking activity induced by BCMA binding to BAFF or APRIL in the single to sub-digit nanomolar range [95]. J6M0 was subsequently conjugated via non-cleavable linker with a novel anti-tubulin drug monomethyl auristatin F (MMAF) (GSK2857916, now Belantamab mafodotin). GSK2857916 showed more selective and potent anti-MM killing than its monomethyl auristatin E (MMAE) ADC homolog in the preclinical study, thereby moving forward to the first clinical trial of BCMA-targeting ADC in MM [83,95]. GSK2857916 directly induces MM cell apoptosis and simultaneously stimulates ADCC and ADCP via NK and macrophages, respectively. Since 2015 [83], there have been overwhelming advances of new BCMA-based immunotherapeutic agents, including ADC delivering novel potent drugs with different mechanisms to induce MM cell apoptosis, CAR-T, or NK cells, as well as BiTEs and BiAbs engaging T or NK effector cells. Impressively, preclinical studies of these innovative agents utilizing various in vitro and in vivo models consistently demonstrate robust anti-MM cytotoxicity with some having been further translated into significant clinical activities [93,94,95,100,101,102]. All early phase clinical studies in small patient cohorts showed a promising high response rate and durable disease control in heavily pretreated RRMM patients [27,103,104]. Several anti-BCMA agents have completed or entered phase 2 or 3 studies [27]. In 2020, belantamab mafodotin and idecabtagene vicleucel (ide-cel; formerly bb2121) were the first anti-BCMA ADC and CAR-T therapy, respectively, approved by FDA as a single agent for heavily pretreated RRMM patients. Most recently, teclistamab, a BiAb targeting BCMA and CD3 (a Humanized BCMA CD3 DuoBody® Antibody), received FDA breakthrough therapy designations for RRMM [105]. Today, BCMA targeting immunotherapy is the first targeted therapy inducing an impressive clinical response as monotherapy in heavily pretreated MM patients who have no more treatment options left.
3. Other MM Tumor Antigens for Emerging Targeted Immunotherapy
The success of recent CAR-T, BiTE, and ADC, based on BCMA-targeting therapies, has quickly stimulated further development of immunotherapy targeting other novel antigens. Data of clinical studies further confirm that exclusive and high expression of tumor antigens on cancer cell is a key factor for new target selection to maximize the potency while minimizing the risk of off-target toxicity. Orphan G protein-coupled receptor, class C group 5 member D (GPRC5D), is a newly identified MM antigen that is highly expressed on MM cells in the BM but not normal tissue, although weakly expressed in hair follicles [106]. GPRC5D CAR-T exhibiting potent anti-MM activity in a preclinical study has led to ongoing clinical studies. BiAbs targeting GPRC5D and CD3 (talquetamab/JNJ-64407564 and GPRC5D TRAB) have also shown potent T-cell-mediated killing of GPRC5D+ MM cells and proliferation/activation of T cells in the preclinical and ongoing clinical studies [107,108]. Furthermore, the expression level of GPRC5D on MM cells and the BM microenvironment-related factors contribute to a different degree of responses to JNJ-7564 [109]. The early phase clinical trials of talquetamab (JNJ-64407564), as a monotherapy (NCT03399799) or combined with other anti-MM agents (NCT04108195), are ongoing, with already significant clinical activity.
Another potential antigen, integrin β7 (ITGB7), is associated with adhesion of MM cells to extra-cellular matrix elements, migration, invasion, and drug resistance [110]. In the in vitro study, novel ITGB7 targeting MMG49-derived CAR T cells showed specific MM cell lysis without damaging normal hematopoietic cells [111].
Natural Killer Group 2D (NKG2D) ligand, expressed on about 80% of MM cells, can bind to NKG2D on natural killer cells, leading to immune escape and tumor growth [112,113]. A BiAb targeting NKG2D and CS1 showed significant immune synapse between CS1+ MM cells and NKG2D+ immune cells, leading to effective MM lysis [114]. NKG2D-CAR T cells was also evaluated in a clinical trial, which showed good safety, but no objective response was observed (NCT02203825) [115].
CD229, a member of the SLAM family, is highly and homogenously expressed on MM cells and myeloma precursors, but not on other normal tissues [116,117]. A preclinical study showed anti-CD229 CAR-T cells exhibited potent in vitro and in vivo activity to against MM and MM-propagating cells, with minimal damage to normal T cells [118].
FcRH5 (or FcRL5, CD307), a membrane protein highly expressed on mature B cells and plasma cells, was also evaluated in MM treatment [119]. A BiAb, cevostamab (BFCR4350A), targeting FcRH5 and CD3, was constructed and showed significant in vitro and in vivo anti-MM activity, as well as T cell activation/proliferation in a preclinical study [120]. The phase 1 trial evaluating the safety and efficacy of cevostamab in RRMM is ongoing (NCT03275103).
4. Potential Challenges and Strategies
Multiple factors are associated with lower treatment efficacy and immune resistance, frequently seen in heavily pretreated patients. First, downregulation of tumor antigen reduces the binding affinity of target agents, resulting in lower tumor killing. Low expression of CD38, together with increased membrane expression of CD55 and CD59, two important immune inhibitory molecules in complement-mediated tumor cell lysis, were associated with treatment resistance to daratumumab [41]. Second, increased shedding of tumor antigens into a soluble form in the serum could act as a decoy, which naturalizes the immune-targeted agents and negatively affects the pharmacokinetic profile and treatment response. High serum levels of soluble SLAMF7 have been associated with a poorer response to elotuzumab and shorter survival [125]. Likewise, sBCMA and sCD38 could reduce the anti-MM activity of anti-BCMA BiTE [102] or daratumumab [126], respectively. Another mechanism contributing to resistance is antigen escape (or loss), which was recently reported in plasma cells in relapsed MM patients with a very low level or loss of BCMA expression, mostly in anti-BCMA CAR-T cell trials [90,91,127]. These low or no antigen-expressing MM cells may be selected out and proliferate after normal or high tumor antigen-expressing cells were eradicated by potent immunotherapy, leading to disease progression. A study using a mouse model revealed that both CD28- and 4-1BB-based CARs were able to induce reversible antigen loss by trogocytosis, which promote the transfer of target antigens to T cells [128]. Most recently, two correlative studies using a sequencing technique to analyze patient samples sequentially collected revealed that biallelic loss or homozygous gene deletion of BCMA play a critical role in antigen escape [129,130]. In MM patients who have not been previously treated with BCMA-targeting therapies, BCMA loss or monosomy 16 was observed in 22% (37/168) of them. Moreover, a significantly higher percentage was noted in patients with hyperdiploid cytogenetics (84.8%, 28/33) [130]. Furthermore, the immunocompromised BM microenvironment is aggravated by MM cell-induced abnormal increased Treg cells, MDSCs, OCs, and/or Breg cells, as well as upregulation of their secretory immunosuppressive cytokines, i.e., IL-6, IL-10, and TGFβ. Enhanced PD-L1 expression on MM cells and other BM accessory cells engages PD-1 on activated effector T cells to suppress their proliferation and production of cytolytic cytokines, leading to a functionally exhausted state and further apoptosis [9,11,19,131,132]. In addition to PD-1, aberrant enhanced or prolonged expression of other immune checkpoint molecules, i.e., TIM-3, CTLA-4, and LAG-3, in NK and T effector cells further inhibit their number and effector function upon binding to their cognate ligands upregulated in MM cells [133].
Several strategies are and/or have been under preclinical or clinical investigations. For decreased MM antigen expression, selective therapeutic agents are shown to enhance target expression on the MM cell surface. In preclinical studies, all-trans retinoic acid, histone deacetylase (HDAC) inhibitors, and JAK/STAT3 inhibitors were reported to induce CD38 expression, which was associated with enhanced CD38 targeting by daratumumab [134,135,136,137]. Since CD38 is a NAD+-degrading enzyme producing ADO, which is immunosuppressive, as mentioned in the above section, the depletion of NAD+ may decrease generation of ADO and allow more CD38 targeting by anti-CD38-based immunotherapies [35]. Most recently, a new DNA-damaging drug specifically delivered to MM cells through BCMA targeting was reported to increase the CD38 levels and further overcome daratumumab insensitivity in MM cell lines and patient MM cells [40]. For BCMA-based immunotherapies, gamma-secretase inhibitors could reduce the shedding of BCMA from MM cells, associated with increased levels of cell membrane BCMA and MM cell killing [87,138]. The preliminary result is promising in an early phase trial evaluating the combination effect of anti-BCMA CAR-T cell therapy and gamma-secretase inhibitor in RRMM patients (NCT03502577) [139].
Modification of the MoAb structure via protein engineering to increase the binding affinity to the membranous form of the target antigens could significantly augment MM cell targeting, as reported for a novel anti-BCMA ADC [140] and anti-CD138 VIS832 [82]. For antigen loss identified in patients refractory to BCMA CAR T treatment, preclinical studies have demonstrated that targeting the new antigen GPRC5D using CAR-T or BiTE could still effectively induce lysis of the BCMA knockout MM cell lines [106,108]. A preclinical study evaluating bispecific CAR-T cells simultaneously targeting both BCMA and SLAMF7 on MM cells showed promising results to prevent antigen escape and better control MM cells heterogeneously expressing these antigens [141]. Significantly, BCMA/CS1 bispecific CAR-T cells induce superior CAR expression and function compared to T cells co-expressing individual BCMA and CS1 CARs. Furthermore, combination of an antagonistic anti-PD-1 MoAb with BCMA/CS1 bispecific CAR-T cells accelerates the rate of initial tumor clearance in a murine model, while CAR-T cell treatment alone achieves durable tumor-free host survival even upon tumor re-challenge.
A novel anti-BCMA pyrrolobenzodiazepine (PBD) ADC, MEDI2228, preferentially binds to membrane BCMA [142] and further induces DNA damage-induced ATM/ATR-CHK1/2, cGAS-STING-TBK1-IRF3, and STAT1-IRF1-signaling cascades to activate IFN-related molecules [40,143] (Figure 3). Significantly, MEDI2228 upregulates expression of CD38 and multiple NKG2D ligands on the MM cell membrane in vitro and in a xenograft murine model of human MM. It overcomes CD38 downregulation triggered by IL6 via activation of STAT1/IRF1 and further restores daratumumab-induced ADCC against resistant MM cell lines and patient MM cells. Unlike daratumumab, which depletes NK cells due to CD38 expression, MEDI2228 has no impact on NK cells. Upregulation of NKG2D ligands as “eat me” signals, including MICA/B and ULBP2/3/5, further augment binding of MEDI2228-treated MM cells to the NKG2D receptor on NK cells, resulting in increased NK immune surveillance as well as enhanced daratumumab-induced MM cell killing in in vitro and in vivo preclinical study models. Importantly, combination daratumumab and MEDI2228 treatment led to all mice bearing MM1S tumors becoming tumor-free with 100% survival. These results further support clinical rationales to test the combination CD38- and BCMA-targeting immunotherapies to achieve effective and durable anti-MM activity. Such combination clinical studies are ongoing based on belantamab mafodotin, the first approved BCMA ADC, in MM, including NCT04246047 with daratumumab and NCT04643002 with isatuximab.
Many ongoing investigations (mainly CAR-T cell therapy) are to evaluate dual-targeting strategies with various combinations based on BCMA, CD19, CD138, and CS1 (SLAMF7), together or in sequence (Table 1), and some already showed improved clinical activity in early phase clinical studies [27,124]. Regarding treatment approaches to augment anti-MM activity of a single targeted immunotherapeutic agent or to overcome an immunosuppressive BM microenvironment, a combination of different anti-MM agents with distinct mechanisms of action remains very attractive strategies. For example, IMiDs, which enhance immune effector cell function and block immunosuppressive BM accessory cells, have been commonly combined with many current anti-MM treatments [144,145]. A preclinical study has shown that lenalidomide enhances cytotoxic effect anti-CS1 CAR-T cells in in vitro and in vivo MM models [146]. Recent studies also demonstrated that lenalidomide or pomalidomide, as well as an PD-L1 inhibitor, further augment T-cell mediated MM cell lysis and immune modulatory function of half-life-extended anti-BCMA BiTE AMG 701 [100,101]. IMiDs further upregulated AMG 701-induced patient T-cell differentiation toward memory phenotypes, associated with increased CD8/CD4 ratios and stem-like T cells (Tscm), as well as decreased IL-10+ T and Treg cells that downregulate T effector cells. Importantly, the combination of AMG 701 with lenalidomide further prolonged host survival following sustained inhibition of MM cell growth in SCID mice reconstituted with human T cells. As previously mentioned, bortezomib or daratumumab also enhance the anti-MM effect of an anti-BCMA ADC MEDI2228 in preclinical studies [40,143]. Furthermore, VIS832 with superior CD138-binding affinity, significantly augments MM cell killing in vitro and in vivo when combined with lenalidomide or bortezomib [82].
5. Perspectives
Since the approval of daratumumab and elotuzumab in 2015, more targeted approaches based on the highlighted antigens above have been generated and investigated in different phases of clinical development, as monotherapy and/or in combination (Figure 4, Table 1). To date, BCMA-based immunotherapy represents the most promising targeted approach, as many clinical studies of different immunotherapeutic formats have showed impressive overall response rates (>70–90%) with prolonged disease control duration, achieving minimal residual disease (MRD) negativity in RRMM patients [27,104,147]. However, a significant portion of patients still suffered from progression or relapse of disease; thus, further optimization of the current treatment approaches, using various immunotherapeutic forms targeting single, dual, or multiple antigens and/or in combination, are urgently needed to prevent disease recurrence and deepen treatment responses.
For the anti-CD38 treatment, the MoAbs recognizing the distinct epitope of CD38, such as isatuximab and MOR202, have been under several clinical investigations, with isatuximab approved in August 2021. Like daratumumab, isatuximab is characterized by multiple effector-dependent anti-MM mechanisms (ADCC, ADCP, and CDC). In contrast, isatuximab, in the absence of effector cells, can further induce apoptosis of MM, Treg, and Breg cells, expressing even higher CD38 levels, while daratumumab cannot [148,149]. A new humanized IgG1 anti-CD38 MoAb, mezagitamab (TAK 079), also showed robust anti-MM activity in RRMM samples and immunomodulatory effects, including activation of NK and T cells, as well as suppression of immune inhibitory cells [150]. The early result of a phase 1 study (NCT03439280) is encouraging, with overall response rate of 31% and a good safety profile [151]. Another novel anti-CD38 ADC, TAK-169, using deimmunized Shiga-like toxin A subunit as the payload, also demonstrated a remarkable in vitro anti-MM effect [152]. TAK-573, a humanized, anti-CD38, IgG4 MoAb genetically fused to two attenuated IFN alpha-2b molecules, showed further increased cytotoxic potential of CD8 T cells via modulation of the IFN-α receptor pathway after treatment in an early phase clinical study [153]. Strategies to modulate the glycosylation of the Fc portion (glyco-engineering) may enhance the affinity of the antibody for FcγRs, thereby resulting in more potent direct and immune effector cell-mediated cytotoxicity. A novel, hexamerization-enhanced human IgG1 anti-CD38 antibody (GEN3014) with an E430G mutation to enhance intermolecular Fc–Fc interactions showed potent CDC activity and ADCC in a preclinical study [154]; an early phase clinical study was recently initiated (NCT04824794). Furthermore, a preclinical study evaluated a BiAb targeting CD38 and CD59 showed a significant increased CDC activity, which was mediated by simultaneously binding to CD38 and neutralization of CD59, which is associated with daratumumab resistance [155].
Next-generation modalities targeting SLAMF7 are also under development. ABBV-838, the first anti-SLAMF7/CS1 ADC, is characterized by cytotoxic MMAE as the payload and an enzymatic cleavable valine-citrulline linker, despite the first-in-human clinical trial of this ADC showing a low response rate in RRMM patients [156]. Other anti-CS1 agents, such as CAR-T cell therapy or anti-CS1/NKG2D BiAb, also showed significant in vitro and in vivo anti-MM activity in preclinical studies [114,146]. Anti-CS1 CAR-T cell therapy, including allogeneic CAR-T cells (NCT04142619), is currently under clinical investigation.
For further improvement of anti-BCMA immunotherapy, the novel ADC MEDI2228 preferentially binds to membrane BCMA showed significant clinical activity in RRMM patients, with an overall response rate of 61% and no keratopathy reported in an early phase clinical study [157]. MEDI2228 can be combined with bortezomib and further upregulates CD38 in MM cells and increased immune surveillance via NK cells to overcome daratumumab resistance [40]. Regarding BiTE therapy, a novel half-life-extended anti-BCMA (AMG 701), with a better pharmacodynamic profile, has demonstrated remarkable anti-MM activity in vitro and in vivo [100,101]. The early phase clinical trial investigating weekly dosing of AMG 701 showed promising results and a good safety profile in heavily pretreated RRMM patients (NCT03287908) [147]. For better outcome of anti-BCMA CAR-T cell therapy, structural or protocol modification are also under evaluation. First, a fully humanized and smaller size scFv may reduce the immune response against murine scFv, to improve post-infusion persistence and overcome treatment failure [158]. For example, CART-ddBCMA, a CAR-T cell characterized by utilization of 73 amino acids as the binding domain rather than conventional scFv to reduce immunogenicity, also showed a high response and good safety profile in RRMM patients with high tumor burden (NCT04155749) [159]. Second, a recent study evaluating the pre- and post-infusion sample in the bb21217 trial revealed that the presence of early memory-like T cells in peripheral blood mononuclear cells may be linked to high peak expansion and better disease control, but highly differentiated or senescent T cells exhibited a negative effect [160]. For better treatment safety, novel mRNA-generated CAR-T cells (Descartes-08), to limit excessive proliferation to reduce the risk of cytokine-releasing syndrome (CRS), has shown robust in vitro and in vivo anti-MM activity [161]. The early phase clinical study is ongoing (NCT03448978). In addition to autologous CAR-T cells, the clinical investigation of allogeneic CAR-T cell ALLO-715 is ongoing to test whether off-shelf CAR-T products are feasible to reduce the cost, time, and success of CAR-T generation (NCT04093596). Moreover, NK-based cellular therapy may also exhibit potent anti-MM killing but with a lower risk of fetal CRS than CAR-T cell treatment, since NK cells tend to survive shorter than T cells after infusion (NCT03940833, NCT05008536).
Several strategies to further optimize anti-MM immune targeted therapy are also under exploration. In the BiAb approach, novel treatment approaches, such as BiAb-armed T cells, are emerging in MM treatment. To avoid antigen-loss-related treatment failure, bicistronic CAR, bivalent “tandem CARs”, or CARs with three specificities are under development [162]. Clinical studies of the above novel concept using these validated MM antigens are expected.
Moreover, the first off-the-shelf multiplexed engineered NK cell therapy (FT538) generated from a clonal master engineered-induced pluripotent stem cell is characterized with CD38 knock-out to avoid damage from anti-CD38 MoAb. An early phase of FT538 combined with daratumumab or elotuzumab in RRMM patients is ongoing (NCT04614636) [163].
Besides the therapeutic role, the exclusive expression of MM-specific antigens on MM cells provides a rationale to develop diagnostic tools. ImmunoPET imaging, which utilized the conjugation of deferoxamine-p-benzyl-isothiocyanate to elotuzumab or daratumumab to enable radiolabeling of zirconium-89, showed the optimal detection ability of MM cells in preclinical studies [164,165]. The clinical study to explore the diagnostic role of this novel technique is ongoing (NCT04814615). The combination of these state-of-the-art imaging tools with minimal residual disease assessment may provide more valuable clinical information.
6. Conclusions
Landscapes of targeted treatments using immunotherapeutic approaches is continuously evolving, with increasing numbers of novel effective agents granted for approval. Research areas continue to be focused on identification of more specific new target antigens, inhibition of shedding and/or escape/loss of target antigens, modulation of binding affinity to validated and/or new target antigens on MM cells and immune molecules on immune effector or suppressive cells using various immunotherapeutic platforms, and blockage of immune inhibitory cytokines, as well as reprogramming the tumor immune microenvironment in favor of persistent anti-MM immunity. Combinations within individual targeted reagents and/or with broad immunoregulators (IMiDs), or chemotherapies with distinct mechanisms of action, remain the most promising strategies due to complex bi- or multi-directional interactions between MM cells with heterogenous genetic backgrounds and different accessory cells via multiple regulatory levels of ligand–receptor engagements in the BM microenvironment. Moreover, using more comprehensive next-generation proteomic and transcriptomic analysis from data longitudinally collected from patients in ongoing trials, novel druggable molecules will be discovered and new subsets of immune cells with more potent activity in regulating intrinsic and/or acquired immune resistance will be further identified.
Author Contributions
S.-F.C. and Y.-T.T. drafted, wrote, and edited the manuscript; S.-F.C., L.X. and Y.-T.T. prepared the tables and figures; K.C.A. reviewed and edited the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by in part by grants from the National Institutes of Health Specialized Programs of Research Excellence (SPORE) P50 CA100707, P01CA155258, and RO1 CA 207237. S.-F.C. was supported by a grant from the Kaohsiung Medical University Hospital (KMUH109-9M22 and SA10902).
Conflicts of Interest
K.C. Anderson reports personal fees from AstraZeneca, Amgen, Pfizer, C4 Therapeutics, Oncopep, Mana Therapeutics, Precision Biosciences, Raqia and Janssen. The other authors declare no competing interest.
References
- Stewart, A.K.; Rajkumar, S.V.; Dimopoulos, M.A.; Masszi, T.; Spicka, I.; Oriol, A.; Hajek, R.; Rosinol, L.; Siegel, D.S.; Mihaylov, G.G.; et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2015, 372, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Oriol, A.; Beksac, M.; Liberati, A.M.; Galli, M.; Schjesvold, F.; Lindsay, J.; Weisel, K.; White, D.; Facon, T.; et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with lenalidomide (OPTIMISMM): A randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 781–794. [Google Scholar] [CrossRef]
- Al Hamed, R.; Bazarbachi, A.H.; Malard, F.; Harousseau, J.L.; Mohty, M. Current status of autologous stem cell transplantation for multiple myeloma. Blood Cancer J. 2019, 9, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, K.K.; Barlogie, B.; van Rhee, F.; Zangari, M.; Walker, B.A.; Rosenthal, A.; Schinke, C.; Thanendrarajan, S.; Davies, F.E.; Hoering, A.; et al. Long-term outcomes after autologous stem cell transplantation for multiple myeloma. Blood Adv. 2020, 4, 422–431. [Google Scholar] [CrossRef]
- Fonseca, R.; Abouzaid, S.; Bonafede, M.; Cai, Q.; Parikh, K.; Cosler, L.; Richardson, P. Trends in overall survival and costs of multiple myeloma, 2000–2014. Leukemia 2017, 31, 1915–1921. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.K.; Dimopoulos, M.A.; Kastritis, E.; Terpos, E.; Nahi, H.; Goldschmidt, H.; Hillengass, J.; Leleu, X.; Beksac, M.; Alsina, M.; et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: A multicenter IMWG study. Leukemia 2017, 31, 2443–2448. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Bergsagel, P.L.; Kuehl, W.M.; Anderson, K.C. Advances in biology of multiple myeloma: Clinical applications. Blood 2004, 104, 607–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.T.; Cho, S.F.; Anderson, K.C. Osteoclast Immunosuppressive Effects in Multiple Myeloma: Role of Programmed Cell Death Ligand 1. Front. Immunol. 2018, 9, 1822. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef] [Green Version]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Tai, Y.T.; Ho, M.; Xing, L.; Chauhan, D.; Gang, A.; Qiu, L.; Anderson, K.C. Regulatory B cell-myeloma cell interaction confers immunosuppression and promotes their survival in the bone marrow milieu. Blood Cancer J. 2017, 7, e547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorgun, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [Green Version]
- De Beule, N.; de Veirman, K.; Maes, K.; de Bruyne, E.; Menu, E.; Breckpot, K.; de Raeve, H.; van Rampelbergh, R.; van Ginderachter, J.A.; Schots, R.; et al. Tumour-associated macrophage-mediated survival of myeloma cells through STAT3 activation. J. Pathol. 2017, 241, 534–546. [Google Scholar] [CrossRef]
- Chauhan, D.; Singh, A.V.; Brahmandam, M.; Carrasco, R.; Bandi, M.; Hideshima, T.; Bianchi, G.; Podar, K.; Tai, Y.T.; Mitsiades, C.; et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: A therapeutic target. Cancer Cell 2009, 16, 309–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hideshima, T.; Mitsiades, C.; Tonon, G.; Richardson, P.G.; Anderson, K.C. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat. Rev. Cancer 2007, 7, 585–598. [Google Scholar] [CrossRef]
- Tai, Y.T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.T.; Anderson, K.C. B cell maturation antigen (BCMA)-based immunotherapy for multiple myeloma. Expert Opin. Biol. Ther. 2019, 19, 1143–1156. [Google Scholar] [CrossRef]
- Neri, P.; Bahlis, N.J.; Lonial, S. New Strategies in Multiple Myeloma: Immunotherapy as a Novel Approach to Treat Patients with Multiple Myeloma. Clin. Cancer Res. 2016, 22, 5959–5965. [Google Scholar] [CrossRef] [Green Version]
- Lonial, S.; Dimopoulos, M.; Palumbo, A.; White, D.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; Moreau, P.; Mateos, M.V.; Magen, H.; et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2015, 373, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Pazina, T.; James, A.M.; MacFarlane, A.W., IV; Bezman, N.A.; Henning, K.A.; Bee, C.; Graziano, R.F.; Robbins, M.D.; Cohen, A.D.; Campbell, K.S. The anti-SLAMF7 antibody elotuzumab mediates NK cell activation through both CD16-dependent and -independent mechanisms. Oncoimmunology 2017, 6, e1339853. [Google Scholar] [CrossRef]
- Tai, Y.T.; Dillon, M.; Song, W.; Leiba, M.; Li, X.F.; Burger, P.; Lee, A.I.; Podar, K.; Hideshima, T.; Rice, A.G.; et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 2008, 112, 1329–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awwad, M.H.S.; Mahmoud, A.; Bruns, H.; Echchannaoui, H.; Kriegsmann, K.; Lutz, R.; Raab, M.S.; Bertsch, U.; Munder, M.; Jauch, A.; et al. Selective elimination of immunosuppressive T cells in patients with multiple myeloma. Leukemia 2021, 35, 2602–2615. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.F.; Lin, L.; Xing, L.; Yu, T.; Wen, K.; Anderson, K.C.; Tai, Y.T. Monoclonal Antibody: A New Treatment Strategy against Multiple Myeloma. Antibodies 2017, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.F.; Lin, L.; Xing, L.; Li, Y.; Yu, T.; Anderson, K.C.; Tai, Y.T. BCMA-Targeting Therapy: Driving a New Era of Immunotherapy in Multiple Myeloma. Cancers 2020, 12, 1473. [Google Scholar] [CrossRef]
- Gagelmann, N.; Riecken, K.; Wolschke, C.; Berger, C.; Ayuk, F.A.; Fehse, B.; Kroger, N. Development of CAR-T cell therapies for multiple myeloma. Leukemia 2020, 34, 2317–2332. [Google Scholar] [CrossRef] [PubMed]
- Frasca, L.; Fedele, G.; Deaglio, S.; Capuano, C.; Palazzo, R.; Vaisitti, T.; Malavasi, F.; Ausiello, C.M. CD38 orchestrates migration, survival, and Th1 immune response of human mature dendritic cells. Blood 2006, 107, 2392–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deaglio, S.; Morra, M.; Mallone, R.; Ausiello, C.M.; Prager, E.; Garbarino, G.; Dianzani, U.; Stockinger, H.; Malavasi, F. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J. Immunol. 1998, 160, 395–402. [Google Scholar]
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Aarhus, R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. J. Biol. Chem. 1995, 270, 2152–2157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.C.; Aarhus, R.; Graeff, R.M. Sensitization of calcium-induced calcium release by cyclic ADP-ribose and calmodulin. J. Biol. Chem. 1995, 270, 9060–9066. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.Y.; Cho, B.H.; Kim, U.H. CD38-mediated Ca2+ signaling contributes to angiotensin II-induced activation of hepatic stellate cells: Attenuation of hepatic fibrosis by CD38 ablation. J. Biol. Chem. 2010, 285, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, B.E.; Sadek, M.; Elnenaei, M.O.; Reiman, A.; Gujar, S.A. Targeting NAD(+) Synthesis to Potentiate CD38-Based Immunotherapy of Multiple Myeloma. Trends Cancer 2020, 6, 9–12. [Google Scholar] [CrossRef]
- Mediero, A.; Cronstein, B.N. Adenosine and bone metabolism. Trends Endocrinol. Metab. 2013, 24, 290–300. [Google Scholar] [CrossRef] [Green Version]
- Buckley, K.A.; Hipskind, R.A.; Gartland, A.; Bowler, W.B.; Gallagher, J.A. Adenosine triphosphate stimulates human osteoclast activity via upregulation of osteoblast-expressed receptor activator of nuclear factor-kappa B ligand. Bone 2002, 31, 582–590. [Google Scholar] [CrossRef]
- Mastelic-Gavillet, B.; Navarro Rodrigo, B.; Decombaz, L.; Wang, H.; Ercolano, G.; Ahmed, R.; Lozano, L.E.; Ianaro, A.; Derre, L.; Valerio, M.; et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor-infiltrating CD8(+) T cells. J. Immunother. Cancer 2019, 7, 257. [Google Scholar] [CrossRef]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: Therapeutic implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef]
- Xing, L.; Wang, S.; Liu, J.; Yu, T.; Chen, H.; Wen, K.; Li, Y.; Lin, L.; Hsieh, P.A.; Cho, S.F.; et al. BCMA-Specific ADC MEDI2228 and Daratumumab Induce Synergistic Myeloma Cytotoxicity via IFN-Driven Immune Responses and Enhanced CD38 Expression. Clin. Cancer Res. 2021, 27, 5376–5388. [Google Scholar] [CrossRef] [PubMed]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef] [Green Version]
- Naeimi Kararoudi, M.; Nagai, Y.; Elmas, E.; de Souza Fernandes Pereira, M.; Ali, S.A.; Imus, P.H.; Wethington, D.; Borrello, I.M.; Lee, D.A.; Ghiaur, G. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood 2020, 136, 2416–2427. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Hughes, T.; Zhang, J.; Caligiuri, M.A.; Benson, D.M.; Yu, J. Fratricide of NK Cells in Daratumumab Therapy for Multiple Myeloma Overcome by Ex Vivo-Expanded Autologous NK Cells. Clin. Cancer Res. 2018, 24, 4006–4017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, Y.T.; Lin, L.; Xing, L.; Cho, S.F.; Yu, T.; Acharya, C.; Wen, K.; Hsieh, P.A.; Dulos, J.; van Elsas, A.; et al. APRIL signaling via TACI mediates immunosuppression by T regulatory cells in multiple myeloma: Therapeutic implications. Leukemia 2019, 33, 426–438. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Voorhees, P.M.; Kaufman, J.L.; Laubach, J.; Sborov, D.W.; Reeves, B.; Rodriguez, C.; Chari, A.; Silbermann, R.; Costa, L.J.; Anderson, L.D., Jr.; et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: The GRIFFIN trial. Blood 2020, 136, 936–945. [Google Scholar] [CrossRef]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): A randomised, multicentre, open-label, phase 3 study. Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- Zuch de Zafra, C.L.; Fajardo, F.; Zhong, W.; Bernett, M.J.; Muchhal, U.S.; Moore, G.L.; Stevens, J.; Case, R.; Pearson, J.T.; Liu, S.; et al. Targeting Multiple Myeloma with AMG 424, a Novel Anti-CD38/CD3 Bispecific T-cell-recruiting Antibody Optimized for Cytotoxicity and Cytokine Release. Clin. Cancer Res. 2019, 25, 3921–3933. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Groen, R.W.; Noort, W.A.; Themeli, M.; Lammerts van Bueren, J.J.; Parren, P.W.; Kuball, J.; Sebestyen, Z.; Yuan, H.; de Bruijn, J.; et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica 2016, 101, 616–625. [Google Scholar] [CrossRef] [Green Version]
- Boles, K.S.; Mathew, P.A. Molecular cloning of CS1, a novel human natural killer cell receptor belonging to the CD2 subset of the immunoglobulin superfamily. Immunogenetics 2001, 52, 302–307. [Google Scholar] [CrossRef]
- Bouchon, A.; Cella, M.; Grierson, H.L.; Cohen, J.I.; Colonna, M. Activation of NK cell-mediated cytotoxicity by a SAP-independent receptor of the CD2 family. J. Immunol. 2001, 167, 5517–5521. [Google Scholar] [CrossRef] [Green Version]
- Kumaresan, P.R.; Lai, W.C.; Chuang, S.S.; Bennett, M.; Mathew, P.A. CS1, a novel member of the CD2 family, is homophilic and regulates NK cell function. Mol. Immunol. 2002, 39, 1–8. [Google Scholar] [CrossRef]
- Lee, J.K.; Mathew, S.O.; Vaidya, S.V.; Kumaresan, P.R.; Mathew, P.A. CS1 (CRACC, CD319) induces proliferation and autocrine cytokine expression on human B lymphocytes. J. Immunol. 2007, 179, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Hsi, E.D.; Steinle, R.; Balasa, B.; Szmania, S.; Draksharapu, A.; Shum, B.P.; Huseni, M.; Powers, D.; Nanisetti, A.; Zhang, Y.; et al. CS1, a potential new therapeutic antibody target for the treatment of multiple myeloma. Clin. Cancer Res. 2008, 14, 2775–2784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, J.; Hori, M.; Iha, H.; Toyama-Sorimachi, N.; Hagiwara, S.; Kuroda, Y.; Koyama, D.; Izumi, T.; Yasui, H.; Suzuki, A.; et al. Soluble SLAMF7 promotes the growth of myeloma cells via homophilic interaction with surface SLAMF7. Leukemia 2020, 34, 180–195. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Gunaratne, J.; Cheong, L.L.; Liu, S.C.; Koh, T.L.; Huang, G.; Blackstock, W.P.; Chng, W.J. Plasma membrane proteomics identifies biomarkers associated with MMSET overexpression in T(4;14) multiple myeloma. Oncotarget 2013, 4, 1008–1018. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.T.; Soydan, E.; Song, W.; Fulciniti, M.; Kim, K.; Hong, F.; Li, X.F.; Burger, P.; Rumizen, M.J.; Nahar, S.; et al. CS1 promotes multiple myeloma cell adhesion, clonogenic growth, and tumorigenicity via c-maf-mediated interactions with bone marrow stromal cells. Blood 2009, 113, 4309–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, S.M.; Bakan, C.E.; Swartzel, G.D.; Hofmeister, C.C.; Efebera, Y.A.; Kwon, H.; Starling, G.C.; Ciarlariello, D.; Bhaskar, S.; Briercheck, E.L.; et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: Evidence for augmented NK cell function complementing ADCC. Cancer Immunol. Immunother. 2013, 62, 1841–1849. [Google Scholar] [CrossRef] [Green Version]
- Dimopoulos, M.A.; Lonial, S.; White, D.; Moreau, P.; Weisel, K.; San-Miguel, J.; Shpilberg, O.; Grosicki, S.; Spicka, I.; Walter-Croneck, A.; et al. Elotuzumab, lenalidomide, and dexamethasone in RRMM: Final overall survival results from the phase 3 randomized ELOQUENT-2 study. Blood Cancer J. 2020, 10, 91. [Google Scholar] [CrossRef]
- Wijdenes, J.; Vooijs, W.C.; Clement, C.; Post, J.; Morard, F.; Vita, N.; Laurent, P.; Sun, R.X.; Klein, B.; Dore, J.M. A plasmocyte selective monoclonal antibody (B-B4) recognizes syndecan-1. Br. J. Haematol. 1996, 94, 318–323. [Google Scholar] [CrossRef]
- Shi, S.; Zhong, D.; Xiao, Y.; Wang, B.; Wang, W.; Zhang, F.; Huang, H. Syndecan-1 knockdown inhibits glioma cell proliferation and invasion by deregulating a c-src/FAK-associated signaling pathway. Oncotarget 2017, 8, 40922–40934. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.J.; Jang, B.; Yi, J.Y.; Han, I.O.; Oh, E.S. Syndecans play dual roles as cell adhesion receptors and docking receptors. FEBS Lett. 2012, 586, 2207–2211. [Google Scholar] [CrossRef] [Green Version]
- Altemeier, W.A.; Schlesinger, S.Y.; Buell, C.A.; Parks, W.C.; Chen, P. Syndecan-1 controls cell migration by activating Rap1 to regulate focal adhesion disassembly. J. Cell Sci. 2012, 125, 5188–5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Williams, K.J. Molecular mediators for raft-dependent endocytosis of syndecan-1, a highly conserved, multifunctional receptor. J. Biol. Chem. 2013, 288, 13988–13999. [Google Scholar] [CrossRef] [Green Version]
- Yao, W.; Rose, J.L.; Wang, W.; Seth, S.; Jiang, H.; Taguchi, A.; Liu, J.; Yan, L.; Kapoor, A.; Hou, P.; et al. Syndecan 1 is a critical mediator of macropinocytosis in pancreatic cancer. Nature 2019, 568, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.E.; Gadalla, R.; Hassan, H.; Afifi, A.; Gotte, M.; El-Shinawi, M.; Mohamed, M.M.; Ibrahim, S.A. The immunomodulatory role of tumor Syndecan-1 (CD138) on ex vivo tumor microenvironmental CD4+ T cell polarization in inflammatory and non-inflammatory breast cancer patients. PLoS ONE 2019, 14, e0217550. [Google Scholar] [CrossRef] [Green Version]
- Szatmari, T.; Dobra, K. The role of syndecan-1 in cellular signaling and its effects on heparan sulfate biosynthesis in mesenchymal tumors. Front. Oncol. 2013, 3, 310. [Google Scholar] [CrossRef] [Green Version]
- Akhmetzyanova, I.; McCarron, M.J.; Parekh, S.; Chesi, M.; Bergsagel, P.L.; Fooksman, D.R. Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia 2020, 34, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Andersen, N.F.; Standal, T.; Nielsen, J.L.; Heickendorff, L.; Borset, M.; Sorensen, F.B.; Abildgaard, N. Syndecan-1 and angiogenic cytokines in multiple myeloma: Correlation with bone marrow angiogenesis and survival. Br. J. Haematol. 2005, 128, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Seidel, C.; Sundan, A.; Hjorth, M.; Turesson, I.; Dahl, I.M.; Abildgaard, N.; Waage, A.; Borset, M. Serum syndecan-1: A new independent prognostic marker in multiple myeloma. Blood 2000, 95, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Dhodapkar, M.V.; Kelly, T.; Theus, A.; Athota, A.B.; Barlogie, B.; Sanderson, R.D. Elevated levels of shed syndecan-1 correlate with tumour mass and decreased matrix metalloproteinase-9 activity in the serum of patients with multiple myeloma. Br. J. Haematol. 1997, 99, 368–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinkes, M.T.; Goldberger, O.A.; Neumann, P.E.; Kokenyesi, R.; Bernfield, M. Organization and promoter activity of the mouse syndecan-1 gene. J. Biol. Chem. 1993, 268, 11440–11448. [Google Scholar] [CrossRef]
- Kind, S.; Merenkow, C.; Buscheck, F.; Moller, K.; Dum, D.; Chirico, V.; Luebke, A.M.; Hoflmayer, D.; Hinsch, A.; Jacobsen, F.; et al. Prevalence of Syndecan-1 (CD138) Expression in Different Kinds of Human Tumors and Normal Tissues. Dis. Markers 2019, 2019, 4928315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannath, S.; Heffner, L.T., Jr.; Ailawadhi, S.; Munshi, N.C.; Zimmerman, T.M.; Rosenblatt, J.; Lonial, S.; Chanan-Khan, A.; Ruehle, M.; Rharbaoui, F.; et al. Indatuximab Ravtansine (BT062) Monotherapy in Patients With Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 372–380. [Google Scholar] [CrossRef]
- Kelly, K.R.; Siegel, D.S.; Chanan-Khan, A.A.; Somlo, G.; Heffner, L.T.; Jagannath, S.; Zimmerman, T.; Munshi, N.C.; Madan, S.; Mohrbacher, A.; et al. Indatuximab Ravtansine (BT062) in Combination with Low-Dose Dexamethasone and Lenalidomide or Pomalidomide: Clinical Activity in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2016, 128, 4486. [Google Scholar] [CrossRef]
- Sun, C.; Mahendravada, A.; Ballard, B.; Kale, B.; Ramos, C.; West, J.; Maguire, T.; McKay, K.; Lichtman, E.; Tuchman, S.; et al. Safety and efficacy of targeting CD138 with a chimeric antigen receptor for the treatment of multiple myeloma. Oncotarget 2019, 10, 2369–2383. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Zhang, W.; Shang, P.; Zhang, H.; Fu, W.; Ye, F.; Zeng, T.; Huang, H.; Zhang, X.; Sun, W.; et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014, 8, 297–310. [Google Scholar] [CrossRef]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Lutz, R.J.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Vallet, S.; Pozzi, S.; Santo, L.; et al. The monoclonal antibody nBT062 conjugated to cytotoxic Maytansinoids has selective cytotoxicity against CD138-positive multiple myeloma cells in vitro and in vivo. Clin. Cancer Res. 2009, 15, 4028–4037. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Yang, H.; Zhu, L.; Zhang, Q.; Cao, Z.; Zhang, Y. Anti-CD138 chimeric antigen receptor-modified T cell therapy for multiple myeloma with extensive extramedullary involvement. Ann. Hematol. 2017, 96, 1407–1410. [Google Scholar] [CrossRef]
- Yu, T.; Chaganty, B.; Lin, L.; Xing, L.; Ramakrishnan, B.; Wen, K.; Hsieh, P.A.; Wollacott, A.; Viswanathan, K.; Adari, H.; et al. VIS832, a novel CD138-targeting monoclonal antibody, potently induces killing of human multiple myeloma and further synergizes with IMiDs or bortezomib in vitro and in vivo. Blood Cancer J. 2020, 10, 110. [Google Scholar] [CrossRef]
- Tai, Y.T.; Anderson, K.C. Targeting B-cell maturation antigen in multiple myeloma. Immunotherapy 2015, 7, 1187–1199. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef]
- O’Connor, B.P.; Raman, V.S.; Erickson, L.D.; Cook, W.J.; Weaver, L.K.; Ahonen, C.; Lin, L.L.; Mantchev, G.T.; Bram, R.J.; Noelle, R.J. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 2004, 199, 91–98. [Google Scholar] [CrossRef]
- Deng, S.; Yuan, T.; Cheng, X.; Jian, R.; Jiang, J. B-lymphocyte-induced maturation protein1 up-regulates the expression of B-cell maturation antigen in mouse plasma cells. Mol. Biol. Rep. 2010, 37, 3747–3755. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.A.; Hoffmann, F.S.; Kuhn, P.H.; Cheng, Q.; Chu, Y.; Schmidt-Supprian, M.; Hauck, S.M.; Schuh, E.; Krumbholz, M.; Rubsamen, H.; et al. gamma-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat. Commun. 2015, 6, 7333. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Gillespie, A.; Tang, G.; Ferros, M.; Harutyunyan, N.M.; Vardanyan, S.; Gottlieb, J.; Li, M.; Wang, C.S.; Chen, H.; et al. Soluble B-Cell Maturation Antigen Mediates Tumor-Induced Immune Deficiency in Multiple Myeloma. Clin. Cancer Res. 2016, 22, 3383–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghermezi, M.; Li, M.; Vardanyan, S.; Harutyunyan, N.M.; Gottlieb, J.; Berenson, A.; Spektor, T.M.; Andreu-Vieyra, C.; Petraki, S.; Sanchez, E.; et al. Serum B-cell maturation antigen: A novel biomarker to predict outcomes for multiple myeloma patients. Haematologica 2017, 102, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Invest. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti-B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Kronke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti-B-Cell Maturation Antigen BiTE Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-cell maturation antigen is a promising target for adoptive T-cell therapy of multiple myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti-B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Emre, N.C.; Lamy, L.; Ngo, V.N.; Wright, G.; Xiao, W.; Powell, J.; Dave, S.; Yu, X.; Zhao, H.; et al. IRF4 addiction in multiple myeloma. Nature 2008, 454, 226–231. [Google Scholar] [CrossRef]
- Tai, Y.T.; Li, X.F.; Breitkreutz, I.; Song, W.; Neri, P.; Catley, L.; Podar, K.; Hideshima, T.; Chauhan, D.; Raje, N.; et al. Role of B-cell-activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006, 66, 6675–6682. [Google Scholar] [CrossRef] [Green Version]
- Day, E.S.; Cachero, T.G.; Qian, F.; Sun, Y.; Wen, D.; Pelletier, M.; Hsu, Y.M.; Whitty, A. Selectivity of BAFF/BLyS and APRIL for binding to the TNF family receptors BAFFR/BR3 and BCMA. Biochemistry 2005, 44, 1919–1931. [Google Scholar] [CrossRef]
- Wallweber, H.J.; Compaan, D.M.; Starovasnik, M.A.; Hymowitz, S.G. The crystal structure of a proliferation-inducing ligand, APRIL. J. Mol. Biol. 2004, 343, 283–290. [Google Scholar] [CrossRef]
- Cho, S.F.; Lin, L.; Xing, L.; Li, Y.; Wen, K.; Yu, T.; Hsieh, P.A.; Munshi, N.; Wahl, J.; Matthes, K.; et al. The immunomodulatory drugs lenalidomide and pomalidomide enhance the potency of AMG 701 in multiple myeloma preclinical models. Blood Adv. 2020, 4, 4195–4207. [Google Scholar] [CrossRef]
- Goldstein, R.L.; Goyos, A.; Li, C.M.; Deegen, P.; Bogner, P.; Sternjak, A.; Thomas, O.; Klinger, M.; Wahl, J.; Friedrich, M.; et al. AMG 701 induces cytotoxicity of multiple myeloma cells and depletes plasma cells in cynomolgus monkeys. Blood Adv. 2020, 4, 4180–4194. [Google Scholar] [CrossRef] [PubMed]
- Hipp, S.; Tai, Y.T.; Blanset, D.; Deegen, P.; Wahl, J.; Thomas, O.; Rattel, B.; Adam, P.J.; Anderson, K.C.; Friedrich, M. A novel BCMA/CD3 bispecific T-cell engager for the treatment of multiple myeloma induces selective lysis in vitro and in vivo. Leukemia 2017, 31, 1743–1751. [Google Scholar] [CrossRef]
- Yu, B.; Jiang, T.; Liu, D. BCMA-targeted immunotherapy for multiple myeloma. J. Hematol. Oncol. 2020, 13, 125. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Chari, A.; Scott, E.; Mezzi, K.; Usmani, S.Z. B-cell maturation antigen (BCMA) in multiple myeloma: Rationale for targeting and current therapeutic approaches. Leukemia 2020, 34, 985–1005. [Google Scholar] [CrossRef] [PubMed]
- Killock, D. MajesTEC results with teclistamab in RRMM. Nat. Rev. Clin. Oncol. 2021, 18, 604. [Google Scholar] [CrossRef]
- Smith, E.L.; Harrington, K.; Staehr, M.; Masakayan, R.; Jones, J.; Long, T.J.; Ng, K.Y.; Ghoddusi, M.; Purdon, T.J.; Wang, X.; et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Pillarisetti, K.; Edavettal, S.; Mendonca, M.; Li, Y.; Tornetta, M.; Babich, A.; Majewski, N.; Husovsky, M.; Reeves, D.; Walsh, E.; et al. A T-cell-redirecting bispecific G-protein-coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood 2020, 135, 1232–1243. [Google Scholar] [CrossRef]
- Kodama, T.; Kochi, Y.; Nakai, W.; Mizuno, H.; Baba, T.; Habu, K.; Sawada, N.; Tsunoda, H.; Shima, T.; Miyawaki, K.; et al. Anti-GPRC5D/CD3 Bispecific T-Cell-Redirecting Antibody for the Treatment of Multiple Myeloma. Mol. Cancer Ther. 2019, 18, 1555–1564. [Google Scholar] [CrossRef]
- Verkleij, C.P.M.; Broekmans, M.E.C.; van Duin, M.; Frerichs, K.A.; Kuiper, R.; de Jonge, A.V.; Kaiser, M.; Morgan, G.; Axel, A.; Boominathan, R.; et al. Preclinical activity and determinants of response of the GPRC5DxCD3 bispecific antibody talquetamab in multiple myeloma. Blood Adv. 2021, 5, 2196–2215. [Google Scholar] [CrossRef]
- Neri, P.; Ren, L.; Azab, A.K.; Brentnall, M.; Gratton, K.; Klimowicz, A.C.; Lin, C.; Duggan, P.; Tassone, P.; Mansoor, A.; et al. Integrin beta7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood 2011, 117, 6202–6213. [Google Scholar] [CrossRef] [Green Version]
- Hosen, N.; Matsunaga, Y.; Hasegawa, K.; Matsuno, H.; Nakamura, Y.; Makita, M.; Watanabe, K.; Yoshida, M.; Satoh, K.; Morimoto, S.; et al. The activated conformation of integrin beta7 is a novel multiple myeloma-specific target for CAR T cell therapy. Nat. Med. 2017, 23, 1436–1443. [Google Scholar] [CrossRef] [PubMed]
- Carbone, E.; Neri, P.; Mesuraca, M.; Fulciniti, M.T.; Otsuki, T.; Pende, D.; Groh, V.; Spies, T.; Pollio, G.; Cosman, D.; et al. HLA class I, NKG2D, and natural cytotoxicity receptors regulate multiple myeloma cell recognition by natural killer cells. Blood 2005, 105, 251–258. [Google Scholar] [CrossRef] [Green Version]
- El-Sherbiny, Y.M.; Meade, J.L.; Holmes, T.D.; McGonagle, D.; Mackie, S.L.; Morgan, A.W.; Cook, G.; Feyler, S.; Richards, S.J.; Davies, F.E.; et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res. 2007, 67, 8444–8449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, W.K.; Kang, S.; Youssef, Y.; Glankler, E.N.; Barrett, E.R.; Carter, A.M.; Ahmed, E.H.; Prasad, A.; Chen, L.; Zhang, J.; et al. A CS1-NKG2D Bispecific Antibody Collectively Activates Cytolytic Immune Cells against Multiple Myeloma. Cancer Immunol. Res. 2018, 6, 776–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumeister, S.H.; Murad, J.; Werner, L.; Daley, H.; Trebeden-Negre, H.; Gicobi, J.K.; Schmucker, A.; Reder, J.; Sentman, C.L.; Gilham, D.E.; et al. Phase I Trial of Autologous CAR T Cells Targeting NKG2D Ligands in Patients with AML/MDS and Multiple Myeloma. Cancer Immunol. Res. 2019, 7, 100–112. [Google Scholar] [CrossRef]
- Carulli, G.; Buda, G.; Azzara, A.; Ciancia, E.M.; Sammuri, P.; Domenichini, C.; Guerri, V.; Petrini, M. CD229 Expression on Bone Marrow Plasma Cells from Patients with Multiple Myeloma and Monoclonal Gammopathies of Uncertain Significance. Acta Haematol. 2016, 135, 11–14. [Google Scholar] [CrossRef]
- Atanackovic, D.; Panse, J.; Hildebrandt, Y.; Jadczak, A.; Kobold, S.; Cao, Y.; Templin, J.; Meyer, S.; Reinhard, H.; Bartels, K.; et al. Surface molecule CD229 as a novel target for the diagnosis and treatment of multiple myeloma. Haematologica 2011, 96, 1512–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radhakrishnan, S.V.; Luetkens, T.; Scherer, S.D.; Davis, P.; Vander Mause, E.R.; Olson, M.L.; Yousef, S.; Panse, J.; Abdiche, Y.; Li, K.D.; et al. CD229 CAR T cells eliminate multiple myeloma and tumor propagating cells without fratricide. Nat. Commun. 2020, 11, 798. [Google Scholar] [CrossRef]
- Franco, A.; Damdinsuren, B.; Ise, T.; Dement-Brown, J.; Li, H.; Nagata, S.; Tolnay, M. Human Fc receptor-like 5 binds intact IgG via mechanisms distinct from those of Fc receptors. J. Immunol. 2013, 190, 5739–5746. [Google Scholar] [CrossRef]
- Li, J.; Stagg, N.J.; Johnston, J.; Harris, M.J.; Menzies, S.A.; DiCara, D.; Clark, V.; Hristopoulos, M.; Cook, R.; Slaga, D.; et al. Membrane-Proximal Epitope Facilitates Efficient T Cell Synapse Formation by Anti-FcRH5/CD3 and Is a Requirement for Myeloma Cell Killing. Cancer Cell 2017, 31, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Casucci, M.; Falcone, L.; Camisa, B.; Norelli, M.; Porcellini, S.; Stornaiuolo, A.; Ciceri, F.; Traversari, C.; Bordignon, C.; Bonini, C.; et al. Extracellular NGFR Spacers Allow Efficient Tracking and Enrichment of Fully Functional CAR-T Cells Co-Expressing a Suicide Gene. Front. Immunol. 2018, 9, 507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, A.T.; Jones, D.R.; Raison, R.L. Preclinical and clinical development of an anti-kappa free light chain mAb for multiple myeloma. Mol. Immunol. 2015, 67, 89–94. [Google Scholar] [CrossRef]
- Garfall, A.L.; Stadtmauer, E.A.; Hwang, W.T.; Lacey, S.F.; Melenhorst, J.J.; Krevvata, M.; Carroll, M.P.; Matsui, W.H.; Wang, Q.; Dhodapkar, M.V.; et al. Anti-CD19 CAR T cells with high-dose melphalan and autologous stem cell transplantation for refractory multiple myeloma. JCI Insight 2019, 4, e120505. [Google Scholar] [CrossRef]
- Yan, Z.; Cao, J.; Cheng, H.; Qiao, J.; Zhang, H.; Wang, Y.; Shi, M.; Lan, J.; Fei, X.; Jin, L.; et al. A combination of humanised anti-CD19 and anti-BCMA CAR T cells in patients with relapsed or refractory multiple myeloma: A single-arm, phase 2 trial. Lancet Haematol. 2019, 6, e521–e529. [Google Scholar] [CrossRef]
- Ishibashi, M.; Soeda, S.; Sasaki, M.; Handa, H.; Imai, Y.; Tanaka, N.; Tanosaki, S.; Ito, S.; Odajima, T.; Sugimori, H.; et al. Clinical impact of serum soluble SLAMF7 in multiple myeloma. Oncotarget 2018, 9, 34784–34793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nooka, A.K.; Kaufman, J.L.; Hofmeister, C.C.; Joseph, N.S.; Heffner, T.L.; Gupta, V.A.; Sullivan, H.C.; Neish, A.S.; Dhodapkar, M.V.; Lonial, S. Daratumumab in multiple myeloma. Cancer 2019, 125, 2364–2382. [Google Scholar] [CrossRef]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Hamieh, M.; Dobrin, A.; Cabriolu, A.; van der Stegen, S.J.C.; Giavridis, T.; Mansilla-Soto, J.; Eyquem, J.; Zhao, Z.; Whitlock, B.M.; Miele, M.M.; et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 2019, 568, 112–116. [Google Scholar] [CrossRef]
- Samur, M.K.; Fulciniti, M.; Aktas Samur, A.; Bazarbachi, A.H.; Tai, Y.T.; Prabhala, R.; Alonso, A.; Sperling, A.S.; Campbell, T.; Petrocca, F.; et al. Biallelic loss of BCMA as a resistance mechanism to CAR T cell therapy in a patient with multiple myeloma. Nat. Commun. 2021, 12, 868. [Google Scholar] [CrossRef]
- Da Via, M.C.; Dietrich, O.; Truger, M.; Arampatzi, P.; Duell, J.; Heidemeier, A.; Zhou, X.; Danhof, S.; Kraus, S.; Chatterjee, M.; et al. Homozygous BCMA gene deletion in response to anti-BCMA CAR T cells in a patient with multiple myeloma. Nat. Med. 2021, 27, 616–619. [Google Scholar] [CrossRef]
- Chen, D.; Tang, P.; Liu, L.; Wang, F.; Xing, H.; Sun, L.; Jiang, Z. Bone marrow-derived mesenchymal stem cells promote cell proliferation of multiple myeloma through inhibiting T cell immune responses via PD-1/PD-L1 pathway. Cell Cycle 2018, 17, 858–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connell, P.; Hyslop, S.; Blake, M.K.; Godbehere, S.; Amalfitano, A.; Aldhamen, Y.A. SLAMF7 Signaling Reprograms T Cells toward Exhaustion in the Tumor Microenvironment. J. Immunol. 2021, 206, 193–205. [Google Scholar] [CrossRef]
- Lee, L.; Alrasheed, N.; Khandelwal, G.; Fitzsimons, E.; Richards, H.; Wilson, W.; Chavda, S.J.; Henry, J.; Conde, L.; de Massy, M.R.; et al. Increased Immune-Regulatory Receptor Expression on Effector T Cells as Early Indicators of Relapse Following Autologous Stem Cell Transplantation for Multiple Myeloma. Front. Immunol. 2021, 12, 618610. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, P.; Mariano, M.C.; Geng, H.; Martin, T.G., 3rd; Wolf, J.L.; Wong, S.W.; Shah, N.; Wiita, A.P. DNA methyltransferase inhibitors upregulate CD38 protein expression and enhance daratumumab efficacy in multiple myeloma. Leukemia 2020, 34, 938–941. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Groen, R.W.; Lokhorst, H.M.; van Kessel, B.; Bloem, A.C.; van Velzen, J.; de Jong-Korlaar, R.; Yuan, H.; Noort, W.A.; Klein, S.K.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Bat-Erdene, A.; Nakamura, S.; Oda, A.; Iwasa, M.; Teramachi, J.; Ashtar, M.; Harada, T.; Miki, H.; Tenshin, H.; Hiasa, M.; et al. Class 1 HDAC and HDAC6 inhibition inversely regulates CD38 induction in myeloma cells via interferon-alpha and ATRA. Br. J. Haematol. 2019, 185, 969–974. [Google Scholar] [CrossRef]
- Garcia-Guerrero, E.; Gogishvili, T.; Danhof, S.; Schreder, M.; Pallaud, C.; Perez-Simon, J.A.; Einsele, H.; Hudecek, M. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood 2017, 129, 3386–3388. [Google Scholar] [CrossRef]
- Pont, M.J.; Hill, T.; Cole, G.O.; Abbott, J.J.; Kelliher, J.; Salter, A.I.; Hudecek, M.; Comstock, M.L.; Rajan, A.; Patel, B.K.R.; et al. gamma-Secretase inhibition increases efficacy of BCMA-specific chimeric antigen receptor T cells in multiple myeloma. Blood 2019, 134, 1585–1597. [Google Scholar] [CrossRef]
- Cowan, A.J.; Pont, M.; Sather, B.D.; Turtle, C.J.; Till, B.G.; Nagengast, A.M.; Libby, E.N., III; Becker, P.S.; Coffey, D.G.; Tuazon, S.A.; et al. Efficacy and Safety of Fully Human Bcma CAR T Cells in Combination with a Gamma Secretase Inhibitor to Increase Bcma Surface Expression in Patients with Relapsed or Refractory Multiple Myeloma. Blood 2019, 134, 204. [Google Scholar] [CrossRef]
- Kinneer, K.; Meekin, J.; Varkey, R.; Xiao, X.; Zhong, H.; Breen, S.; Hurt, E.; Thomas, S.; Flynn, M.; Hynes, P.; et al. Preclinical Evaluation of MEDI2228, a BCMA-Targeting Pyrrolobenzodiazepine-Linked Antibody Drug Conjugate for the Treatment of Multiple Myeloma. Blood 2017, 130, 3153. [Google Scholar] [CrossRef]
- Zah, E.; Nam, E.; Bhuvan, V.; Tran, U.; Ji, B.Y.; Gosliner, S.B.; Wang, X.; Brown, C.E.; Chen, Y.Y. Systematically optimized BCMA/CS1 bispecific CAR-T cells robustly control heterogeneous multiple myeloma. Nat. Commun. 2020, 11, 2283. [Google Scholar] [CrossRef]
- Kinneer, K.; Flynn, M.; Thomas, S.B.; Meekin, J.; Varkey, R.; Xiao, X.; Zhong, H.; Breen, S.; Hynes, P.G.; Fleming, R.; et al. Preclinical assessment of an antibody-PBD conjugate that targets BCMA on multiple myeloma and myeloma progenitor cells. Leukemia 2019, 33, 766–771. [Google Scholar] [CrossRef]
- Xing, L.; Lin, L.; Yu, T.; Li, Y.; Cho, S.F.; Liu, J.; Wen, K.; Hsieh, P.A.; Kinneer, K.; Munshi, N.; et al. A novel BCMA PBD-ADC with ATM/ATR/WEE1 inhibitors or bortezomib induce synergistic lethality in multiple myeloma. Leukemia 2020, 34, 2150–2162. [Google Scholar] [CrossRef]
- LeBlanc, R.; Hideshima, T.; Catley, L.P.; Shringarpure, R.; Burger, R.; Mitsiades, N.; Mitsiades, C.; Cheema, P.; Chauhan, D.; Richardson, P.G.; et al. Immunomodulatory drug costimulates T cells via the B7-CD28 pathway. Blood 2004, 103, 1787–1790. [Google Scholar] [CrossRef]
- Gorgun, G.; Calabrese, E.; Soydan, E.; Hideshima, T.; Perrone, G.; Bandi, M.; Cirstea, D.; Santo, L.; Hu, Y.; Tai, Y.T.; et al. Immunomodulatory effects of lenalidomide and pomalidomide on interaction of tumor and bone marrow accessory cells in multiple myeloma. Blood 2010, 116, 3227–3237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Walter, M.; Urak, R.; Weng, L.; Huynh, C.; Lim, L.; Wong, C.W.; Chang, W.C.; Thomas, S.H.; Sanchez, J.F.; et al. Lenalidomide Enhances the Function of CS1 Chimeric Antigen Receptor-Redirected T Cells Against Multiple Myeloma. Clin. Cancer Res. 2018, 24, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.J.; Minnema, M.C.; Lee, H.C.; Spencer, A.; Kapoor, P.; Madduri, D.; Larsen, J.; Ailawadhi, S.; Kaufman, J.L.; Raab, M.S.; et al. A Phase 1 First in Human (FIH) Study of AMG 701, an Anti-B-Cell Maturation Antigen (BCMA) Half-Life Extended (HLE) BiTE® (bispecific T-cell engager) Molecule, in Relapsed/Refractory (RR) Multiple Myeloma (MM). Blood 2020, 136, 28–29. [Google Scholar] [CrossRef]
- Jiang, H.; Acharya, C.; An, G.; Zhong, M.; Feng, X.; Wang, L.; Dasilva, N.; Song, Z.; Yang, G.; Adrian, F.; et al. SAR650984 directly induces multiple myeloma cell death via lysosomal-associated and apoptotic pathways, which is further enhanced by pomalidomide. Leukemia 2016, 30, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Perez, C.; Zabaleta, A.; Manrique, I.; Alignani, D.; Ajona, D.; Blanco, L.; Lasa, M.; Maiso, P.; Rodriguez, I.; et al. The Mechanism of Action of the Anti-CD38 Monoclonal Antibody Isatuximab in Multiple Myeloma. Clin. Cancer Res. 2019, 25, 3176–3187. [Google Scholar] [CrossRef] [Green Version]
- Abadier, M.; Estevam, J.; Berg, D.; Fedyk, E.R. Mezagitamab Induces Immunomodulatory Effect in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 9. [Google Scholar] [CrossRef]
- Krishnan, A.Y.; Patel, K.K.; Hari, P.; Jagannath, S.; Niesvizky, R.; Silbermann, R.W.; Berg, D.T.; Li, Q.; Allikmets, K.; Stockerl-Goldstein, K. A phase Ib study of TAK-079, an investigational anti-CD38 monoclonal antibody (mAb) in patients with relapsed/ refractory multiple myeloma (RRMM): Preliminary results. J. Clin. Oncol. 2020, 38, 8539. [Google Scholar] [CrossRef]
- Bruins, W.S.C.; Zheng, W.; Higgins, J.P.; Willert, E.K.; Newcomb, J.; Dash, A.B.; van de Donk, N.W.C.J.; Zweegman, S.; Mutis, T. TAK-169, a Novel Recombinant Immunotoxin Specific for CD38, Induces Powerful Preclinical Activity Against Patient-Derived Multiple Myeloma Cells. Blood 2020, 136, 11–12. [Google Scholar] [CrossRef]
- Vogl, D.T.; Kaufman, J.L.; Holstein, S.A.; Nadeem, O.; O’Donnell, E.; Suryanarayan, K.; Collins, S.; Parot, X.; Chaudhry, M. TAK-573, an Anti-CD38/Attenuated Ifnα Fusion Protein, Has Clinical Activity and Modulates the Ifnα Receptor (IFNAR) Pathway in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 37–38. [Google Scholar] [CrossRef]
- De Goeij, B.E.; Janmaat, M.L.; Andringa, G.; Kil, L.; van Kessel, B.; Frerichs, K.A.; Lingnau, A.; Freidig, A.; Mutis, T.; Sasser, A.K.; et al. Hexabody-CD38, a Novel CD38 Antibody with a Hexamerization Enhancing Mutation, Demonstrates Enhanced Complement-Dependent Cytotoxicity and Shows Potent Anti-Tumor Activity in Preclinical Models of Hematological Malignancies. Blood 2019, 134, 3106. [Google Scholar] [CrossRef]
- Macor, P.; Secco, E.; Mezzaroba, N.; Zorzet, S.; Durigutto, P.; Gaiotto, T.; de Maso, L.; Biffi, S.; Garrovo, C.; Capolla, S.; et al. Bispecific antibodies targeting tumor-associated antigens and neutralizing complement regulators increase the efficacy of antibody-based immunotherapy in mice. Leukemia 2015, 29, 406–414. [Google Scholar] [CrossRef]
- Vij, R.; Nath, R.; Afar, D.E.H.; Mateos, M.V.; Berdeja, J.G.; Raab, M.S.; Guenther, A.; Martinez-Lopez, J.; Jakubowiak, A.J.; Leleu, X.; et al. First-in-Human Phase I Study of ABBV-838, an Antibody-Drug Conjugate Targeting SLAMF7/CS1 in Patients with Relapsed and Refractory Multiple Myeloma. Clin. Cancer Res. 2020, 26, 2308–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.K.; Migkou, M.; Bhutani, M.; Spencer, A.; Ailawadhi, S.; Kalff, A.; Walcott, F.; Pore, N.; Gibson, D.; Wang, F.; et al. Phase 1, First-in-Human Study of MEDI2228, a BCMA-Targeted ADC in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Lam, N.; Trinklein, N.D.; Buelow, B.; Patterson, G.H.; Ojha, N.; Kochenderfer, J.N. Anti-BCMA chimeric antigen receptors with fully human heavy-chain-only antigen recognition domains. Nat. Commun. 2020, 11, 283. [Google Scholar] [CrossRef] [Green Version]
- Frigault, M.J.; Bishop, M.R.; O’Donnell, E.K.; Raje, N.S.; Mondillo, A.; Cook, D.; Daley, H.; Mangus, C.; LeFleur, D.W.; Buonato, J.M.; et al. Phase 1 Study of CART-Ddbcma, a CAR-T Therapy Utilizing a Novel Synthetic Binding Domain for the Treatment of Subjects with Relapsed and Refractory Multiple Myeloma. Blood 2020, 136, 2. [Google Scholar] [CrossRef]
- Finney, O.C.; Yeri, A.; Mao, P.; Pandya, C.; Alonzo, E.; Hopkins, G.; Hymson, S.; Hu, T.; Foos, M.; Bhadoriya, S.; et al. Molecular and Phenotypic Profiling of Drug Product and Post-Infusion Samples from CRB-402, an Ongoing: Phase I Clinical Study of bb21217 a BCMA-Directed CAR T Cell Therapy. Blood 2020, 136, 3–4. [Google Scholar] [CrossRef]
- Lin, L.; Cho, S.F.; Xing, L.; Wen, K.; Li, Y.; Yu, T.; Hsieh, P.A.; Chen, H.; Kurtoglu, M.; Zhang, Y.; et al. Preclinical evaluation of CD8+ anti-BCMA mRNA CAR T cells for treatment of multiple myeloma. Leukemia 2020, 35, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janakiram, M.; Vij, R.; Siegel, D.S.; Shih, T.; Weymer, S.; Valamehr, B.; Chu, Y.-W.; Miller, J.S. A Phase I Study of FT538, a First-of-Kind, Off-the-Shelf, Multiplexed Engineered, iPSC-Derived NK Cell Therapy As Monotherapy in Relapsed/Refractory Acute Myelogenous Leukemia and in Combination with Daratumumab or Elotuzumab in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 3. [Google Scholar] [CrossRef]
- Ghai, A.; Maji, D.; Cho, N.; Chanswangphuwana, C.; Rettig, M.; Shen, D.; DiPersio, J.; Akers, W.; Dehdashti, F.; Achilefu, S.; et al. Preclinical Development of CD38-Targeted [(89)Zr]Zr-DFO-Daratumumab for Imaging Multiple Myeloma. J. Nucl. Med. 2018, 59, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Ghai, A.; Zheleznyak, A.; Mixdorf, M.; O’Neal, J.; Ritchey, J.; Rettig, M.; DiPersio, J.; Shokeen, M.; Achilefu, S. Development of [(89)Zr]DFO-elotuzumab for immunoPET imaging of CS1 in multiple myeloma. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Pathobiological function of validated MM target antigens commonly used in current MM immunotherapies. (A) CD38, a receptor and an ectoenzyme, regulates the conversion of NAD+ to ADO, which contributes to the immunocompromised tumor microenvironment. IL-6, an important MM growth, survival, and immunosuppressive cytokine, mainly produced by non-myeloma BM cells (i.e., BMSCs, MDSC, OC, and TAM), binds to IL-6R, which interacts with gp130 to activate the JAK1/2-STAT1/3 signaling pathway and subsequently downregulates CD38 expression in MM cells. (B) BCMA is the cognate receptor for APRIL and BAFF, which are present in the BM microenvironment. APRIL, an important plasma cell factor, preferentially binds to BCMA when compared with BAFF, to induce signaling pathways critical to promote survival, proliferation, and drug resistance, as well as immunosuppression of MM cells. BCMA expression is induced by BLIMP1, a key plasma cell transcriptional regulator, and BCMA protein is cleaved by gamma (γ)-secretase, resulting in the soluble form (sBCMA) that is detected in MM patient serum and correlated with disease advancement. (C) CD138, the transmembrane heparan sulfate proteoglycan syndecan-1, is overexpressed in malignant plasma cells and its levels are associated with increased proliferation and survival, as well as decreased apoptosis in MM cell lines and patient MM cells. Levels of shed CD138 (sCD138) cleaved by metalloproteinases and heparanase in MM patient serum samples are also linked to disease progression and sCD138 acts locally or distally with various effector molecules (i.e., IL-6, APRIL, and VEGF) to impact tumor progression. (D) SLAMF7, also named CS1, promotes adhesion of MM cells to BMSCs. The expression of SLAMF7 is regulated by the transcriptional factor IKZF1 (Ikaros), a MM-related target by lenalidomide and pomalidomide, in MM cells. Soluble SLAMF7 (sSLAMF7) is detected in patient serum and promotes MM cell growth via homophilic interaction with SLAMF7 on MM cells. Among these four MM antigens, BCMA has the most limited expression at the transcript and protein levels in plasma cells but no other normal tissues except a minute subset of plasmacytoid dendritic cell. ADO, adenosine; ADPR, ADP-ribose; APRIL, A proliferation inducing ligand; BAFF, B-cell activating factor; BMSC, bone marrow stromal cell; JAK, Janus kinase; MDSC, myeloid-derived suppressor cell; NAD+, nicotinamide adenine dinucleotide; OC, osteoclast; TAM, tumor-associated macrophages; VEGF, vascular endothelial growth factor.
Figure 1.
Pathobiological function of validated MM target antigens commonly used in current MM immunotherapies. (A) CD38, a receptor and an ectoenzyme, regulates the conversion of NAD+ to ADO, which contributes to the immunocompromised tumor microenvironment. IL-6, an important MM growth, survival, and immunosuppressive cytokine, mainly produced by non-myeloma BM cells (i.e., BMSCs, MDSC, OC, and TAM), binds to IL-6R, which interacts with gp130 to activate the JAK1/2-STAT1/3 signaling pathway and subsequently downregulates CD38 expression in MM cells. (B) BCMA is the cognate receptor for APRIL and BAFF, which are present in the BM microenvironment. APRIL, an important plasma cell factor, preferentially binds to BCMA when compared with BAFF, to induce signaling pathways critical to promote survival, proliferation, and drug resistance, as well as immunosuppression of MM cells. BCMA expression is induced by BLIMP1, a key plasma cell transcriptional regulator, and BCMA protein is cleaved by gamma (γ)-secretase, resulting in the soluble form (sBCMA) that is detected in MM patient serum and correlated with disease advancement. (C) CD138, the transmembrane heparan sulfate proteoglycan syndecan-1, is overexpressed in malignant plasma cells and its levels are associated with increased proliferation and survival, as well as decreased apoptosis in MM cell lines and patient MM cells. Levels of shed CD138 (sCD138) cleaved by metalloproteinases and heparanase in MM patient serum samples are also linked to disease progression and sCD138 acts locally or distally with various effector molecules (i.e., IL-6, APRIL, and VEGF) to impact tumor progression. (D) SLAMF7, also named CS1, promotes adhesion of MM cells to BMSCs. The expression of SLAMF7 is regulated by the transcriptional factor IKZF1 (Ikaros), a MM-related target by lenalidomide and pomalidomide, in MM cells. Soluble SLAMF7 (sSLAMF7) is detected in patient serum and promotes MM cell growth via homophilic interaction with SLAMF7 on MM cells. Among these four MM antigens, BCMA has the most limited expression at the transcript and protein levels in plasma cells but no other normal tissues except a minute subset of plasmacytoid dendritic cell. ADO, adenosine; ADPR, ADP-ribose; APRIL, A proliferation inducing ligand; BAFF, B-cell activating factor; BMSC, bone marrow stromal cell; JAK, Janus kinase; MDSC, myeloid-derived suppressor cell; NAD+, nicotinamide adenine dinucleotide; OC, osteoclast; TAM, tumor-associated macrophages; VEGF, vascular endothelial growth factor.

Figure 2.
Multiple mechanisms of action and immunomodulatory effects of current targeted immunotherapies in MM. (A) Monoclonal antibodies (MoAbs) identify and bind to specific antigens to trigger MM cell lysis via multiple immune-dependent mechanisms, including ADCC, ADCP, and CDC. (B) The single chain fragment variant of the CAR-T cell first targets the MM-specific antigen and potent MM cell lysis is induced in a major histocompatibility complex-independent manner followed by increased proliferation and activation of T effector cells, including those with central and effector memory phenotypes. (C) BiAb or BiTE, off-shelf products different from autologous CAR-T cells, simultaneously binds to specific tumor antigen on MM cells and CD3 on T cells to trigger potent MM cell killing as well as proliferation of immune effector T cells. T cells with memory phenotypes (Tcm, Tem, Tscm) and the major cytolytic T cells (CD8+ cells) are increased significantly. (D) The MoAb portion of antibody drug conjugate (ADC) binds to tumor antigen on MM cell membrane and the ADC is endocytosed followed by the release of potent cytotoxic payloads from lysosomes to directly induce MM cell apoptosis. Payloads used in ADC are typically even more potent than those used in chemotherapies to improve superior specific killing of tumors cells but not the surrounding normal tissues. Depending on the design of the ADC, NK cells and macrophages are directly or indirectly activated to induce ADCC and phagocytosis, respectively, to further augment killing of tumor cells. ADCC, antibody-dependent cellular cytotoxicity (NK cells are the predominant effector cells. Other CD16-expressing effectors including monocytes and neutrophils are also capable to induce ADCC); ADCP, antibody-dependent cellular phagocytosis (macrophage (M), especially with the M1 characteristics, are the key effector cells whereas macrophages with M2 features (TAM in Figure 1) promote tumor growth); BiAb, bispecific antibody engaging with T cells; BiTE, bispecific T cell engager; CDC, complement-dependent cytotoxicity; Tcm, central memory T cell; Tem, effector memory T cell; Tscm, stem cell-like memory T cell.
Figure 2.
Multiple mechanisms of action and immunomodulatory effects of current targeted immunotherapies in MM. (A) Monoclonal antibodies (MoAbs) identify and bind to specific antigens to trigger MM cell lysis via multiple immune-dependent mechanisms, including ADCC, ADCP, and CDC. (B) The single chain fragment variant of the CAR-T cell first targets the MM-specific antigen and potent MM cell lysis is induced in a major histocompatibility complex-independent manner followed by increased proliferation and activation of T effector cells, including those with central and effector memory phenotypes. (C) BiAb or BiTE, off-shelf products different from autologous CAR-T cells, simultaneously binds to specific tumor antigen on MM cells and CD3 on T cells to trigger potent MM cell killing as well as proliferation of immune effector T cells. T cells with memory phenotypes (Tcm, Tem, Tscm) and the major cytolytic T cells (CD8+ cells) are increased significantly. (D) The MoAb portion of antibody drug conjugate (ADC) binds to tumor antigen on MM cell membrane and the ADC is endocytosed followed by the release of potent cytotoxic payloads from lysosomes to directly induce MM cell apoptosis. Payloads used in ADC are typically even more potent than those used in chemotherapies to improve superior specific killing of tumors cells but not the surrounding normal tissues. Depending on the design of the ADC, NK cells and macrophages are directly or indirectly activated to induce ADCC and phagocytosis, respectively, to further augment killing of tumor cells. ADCC, antibody-dependent cellular cytotoxicity (NK cells are the predominant effector cells. Other CD16-expressing effectors including monocytes and neutrophils are also capable to induce ADCC); ADCP, antibody-dependent cellular phagocytosis (macrophage (M), especially with the M1 characteristics, are the key effector cells whereas macrophages with M2 features (TAM in Figure 1) promote tumor growth); BiAb, bispecific antibody engaging with T cells; BiTE, bispecific T cell engager; CDC, complement-dependent cytotoxicity; Tcm, central memory T cell; Tem, effector memory T cell; Tscm, stem cell-like memory T cell.

Figure 3.
Molecular and cellular mechanisms for enhanced MM cell killing by simultaneously targeting BCMA and CD38. Combination BCMA- and CD38-directed therapies hold great promises to improve the potency and durability of current targeted immunotherapies in MM. For instance, as shown here, the additional immunomodulatory effects and molecular mechanisms of the BCMA-specific ADC, MEDI2228, could augment NK cell killing of drug-resistant MM cells in the presence or absence of daratumumab (dara) via enhanced MM cell recognition and destruction by effector cells. Although only showing NK cells here, T and macrophages also will be activated by inflammatory cytokines (i.e., IFN I molecules) and chemokines secreted from MM cells, to further boost the anti-MM immune function of this ADC. Significantly, MEDI2228 activates DNA-damage-related ATM/ATR-CHK1/2 and cGAS/STING/TBK1/IRF3, as well as STAT1/IRF1 signaling cascades followed by the induction of downstream type I IFN and IFN-stimulated genes in MM cells (right panel). Cell membrane expression of CD38, an IFN-associated gene, and NKG2D ligands (MICA/B, ULBP2/3/5) are elevated in MEDI2228-treated MM cells, thereby further restoring CD38 targeting by Dara.
Figure 3.
Molecular and cellular mechanisms for enhanced MM cell killing by simultaneously targeting BCMA and CD38. Combination BCMA- and CD38-directed therapies hold great promises to improve the potency and durability of current targeted immunotherapies in MM. For instance, as shown here, the additional immunomodulatory effects and molecular mechanisms of the BCMA-specific ADC, MEDI2228, could augment NK cell killing of drug-resistant MM cells in the presence or absence of daratumumab (dara) via enhanced MM cell recognition and destruction by effector cells. Although only showing NK cells here, T and macrophages also will be activated by inflammatory cytokines (i.e., IFN I molecules) and chemokines secreted from MM cells, to further boost the anti-MM immune function of this ADC. Significantly, MEDI2228 activates DNA-damage-related ATM/ATR-CHK1/2 and cGAS/STING/TBK1/IRF3, as well as STAT1/IRF1 signaling cascades followed by the induction of downstream type I IFN and IFN-stimulated genes in MM cells (right panel). Cell membrane expression of CD38, an IFN-associated gene, and NKG2D ligands (MICA/B, ULBP2/3/5) are elevated in MEDI2228-treated MM cells, thereby further restoring CD38 targeting by Dara.

Figure 4.
Statuses of various targeting immunotherapeutic agents under development in MM. Statuses of different immunotherapeutic formats based on the indicated MM target antigens were summarized from the ClinicalTrials.gov website (data cut-off: 14 October 2021). * CAR T therapy has moved to allogeneic strategies, including BCMA CAR T (NCT05000450 (ALLO-605), NCT04093596 (ALLO-647), NCT04960579 (P-BCMA-ALLO1)) and CS1/SLAMF7 CAR T (NCT04142619 (UCARTCS1A)). ** The BCMA CAR-NK trials are ongoing, including NCT03940833 in phase 1/2 and NCT05008536 in phase 1.
Figure 4.
Statuses of various targeting immunotherapeutic agents under development in MM. Statuses of different immunotherapeutic formats based on the indicated MM target antigens were summarized from the ClinicalTrials.gov website (data cut-off: 14 October 2021). * CAR T therapy has moved to allogeneic strategies, including BCMA CAR T (NCT05000450 (ALLO-605), NCT04093596 (ALLO-647), NCT04960579 (P-BCMA-ALLO1)) and CS1/SLAMF7 CAR T (NCT04142619 (UCARTCS1A)). ** The BCMA CAR-NK trials are ongoing, including NCT03940833 in phase 1/2 and NCT05008536 in phase 1.


{kind=link}
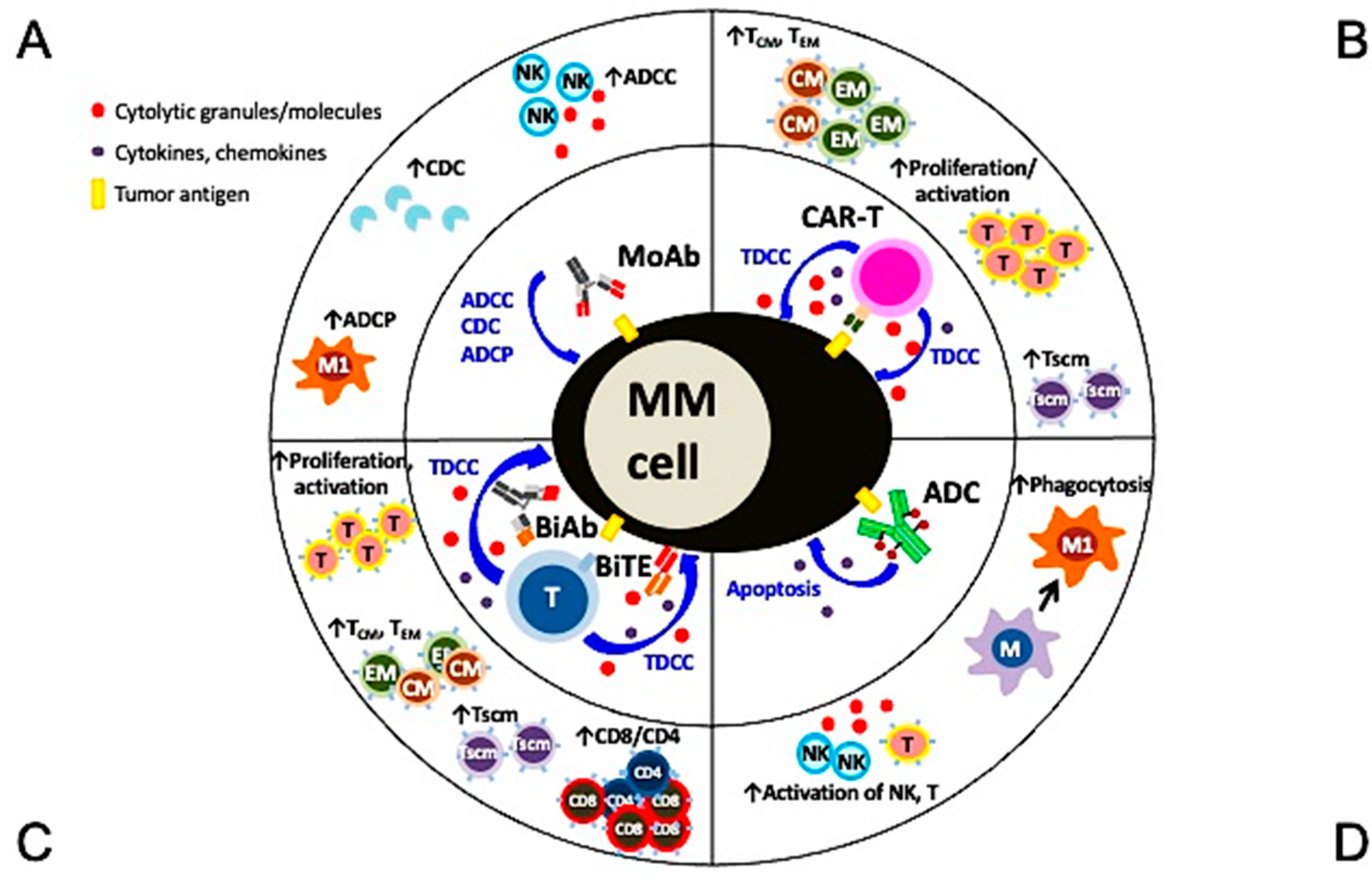{kind=link}
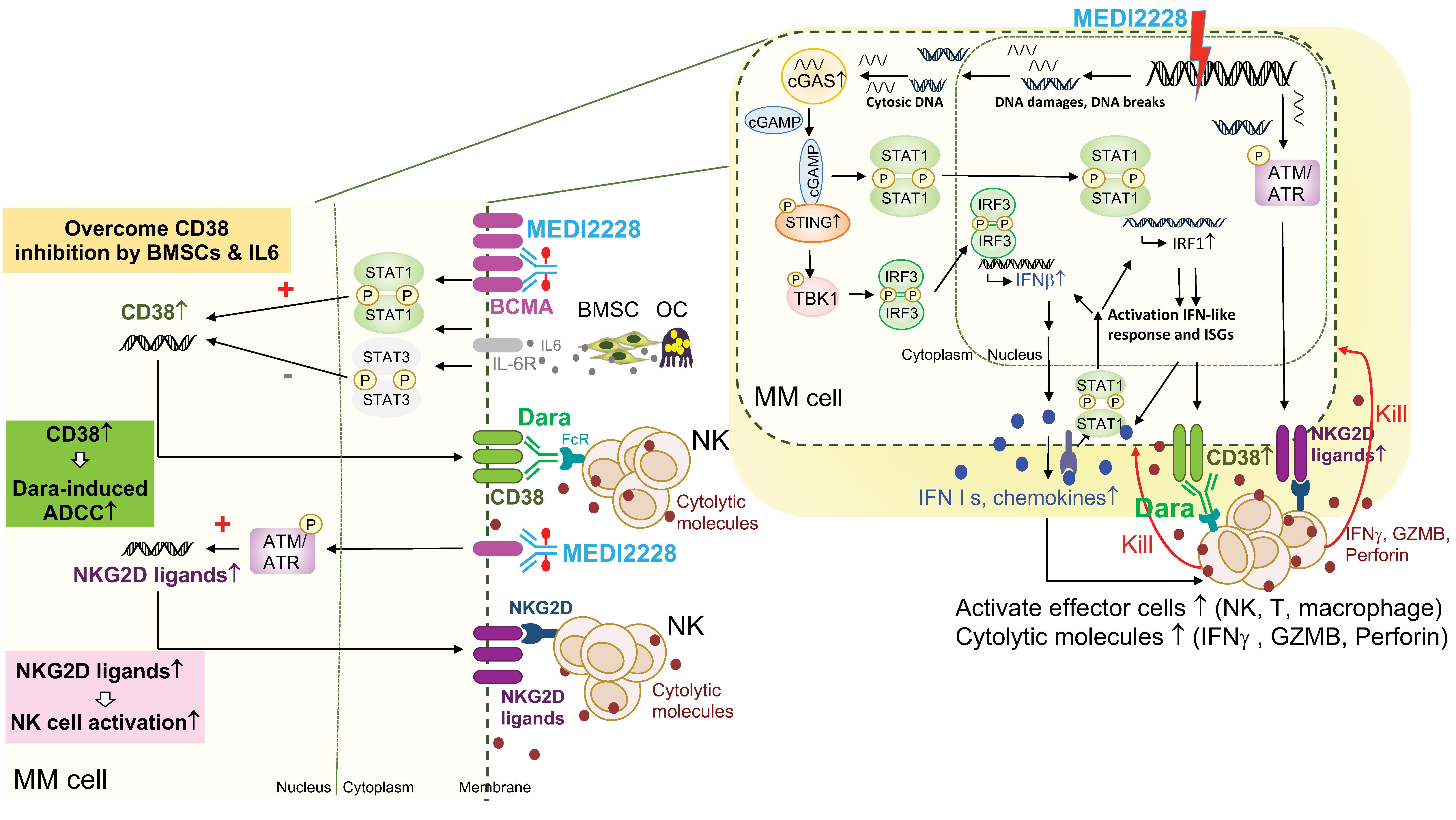{kind=link}
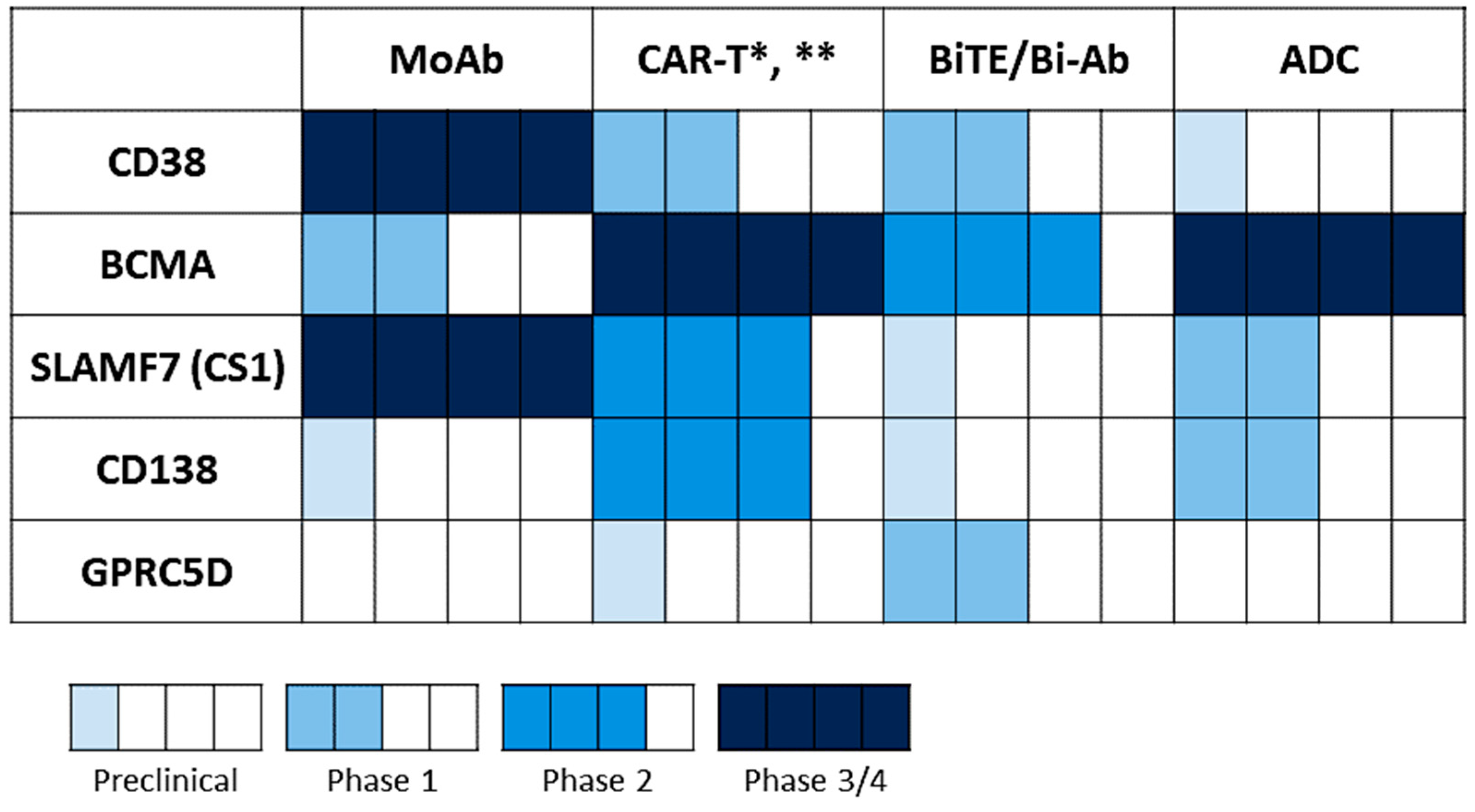{kind=link}
Table 1.
Selected clinical trials of combination therapy or multi-target immunotherapy.
| Clinical Trials | Agents | Format | Status |
|---|---|---|---|
| Anti-CD19/BCMA Bispecific CAR-T Cell Therapy for R/R MM (Phase 1, NCT03706547) | BCMA/CD19 CAR-T | CAR-T | Active, not recruiting |
| Anti-BCMA or/and Anti-CD19 CART Cells Treatment of Relapsed Multiple Myeloma (Phase 1, NCT03767725) | BCMA/CD19 CAR-T | CAR-T | Recruiting |
| Targeting CD19 and BCMA CAR-T Cells Immunotherapy in Patients with Relapsed or Refractory Multiple Myeloma (Phase 1+2, NCT04714827) | BCMA/CD19 CAR-T | CAR-T | Recruiting |
| A New Study Evaluating the Activity of Modular CAR T for mYeloma (MCARTY) (Phase 1, NCT04795882) | BCMA/CD19 CAR-T | CAR-T | Recruiting |
| A Feasibility and Safety Study of Dual Specificity CD38 and BCMA CAR-T Cell Immunotherapy for Relapsed or Refractory Multiple Myeloma (Phase 1+2, NCT03767751) | BCMA/CD38 CAR-T | CAR-T | Recruiting |
| BCMA-CS1 Compound CAR (cCAR) T Cells for Relapsed/Refractory Multiple Myeloma (Phase 1, NCT04156269) | BCMA-CS1 cCAR T | CAR-T | Recruiting |
| Safety and Efficiency Study of BCMA-PD1-CART Cells in Relapsed/Refractory Multiple Myeloma (Phase 2, NCT04162119) | BCMA-PD1-CART | CAR-T | Recruiting |
| BCMA-CD19 cCAR in Multiple Myeloma and Plasmacytoid Lymphoma (Phase 1, NCT04162353) | BCMA-CD19 cCAR | CAR-T | Recruiting |
| A Study of BCMA/CD19 Dual-Target CAR-T Cell Immunotherapy for Relapsed or Refractory Multiple Myeloma (Phase 1, NCT04182581) | BCMA/CD19 CAR-T | CAR-T | Recruiting |
| Humanized CAR-T Cells of Anti-BCAM and Anti-CD19 Against Relapsed and Refractory Multiple Myeloma (Phase 1, NCT04194931) | BCMA/CD19 CAR-T | CAR-T | Recruiting |
| A Study of BCMA/CD19 Dual-Target CAR-T Cell Immunotherapy for Relapsed or Refractory MM (Phase 1, NCT04412889) | BCMA/CD19 CAR-T | CAR-T | Recruiting |
| Up-front CART-BCMA with or without huCART19 in High-risk Multiple Myeloma (Phase 1, NCT03549442) | CART-BCMA, huCART19 | CAR-T | Recruiting |
| Study of T Cells Targeting CD19/BCMA (CART-19/BCMA) for High-Risk Multiple Myeloma Followed with Auto-HSCT (Phase 1+2, NCT03455972) | BCMA/CD19 CAR-T | CAR-T | Recruiting |
| CAR-T Cells Combined with Dasatinib for Patients with Relapsed and/or Refractory B-cell Hematological Malignancies (Phase 1, NCT04603872) | BCMA/CD19 CAR-T | CAR-T | Recruiting |
| A Feasibility and Safety Study of Dual Specificity CD38 and BCMA CAR-T Cell Immunotherapy for Relapsed or Refractory Multiple Myeloma (Phase 1/2, NCT03767751) | CD38/BCMA CAR-T | CAR-T | Recruiting |
| BCMA-CS1 Compound CAR (cCAR) T Cells for Relapsed/Refractory Multiple Myeloma (Phase 1, NCT04156269) | BCMA-CS1 CAR-T | CAR-T | Recruiting |
| Study of T Cells Targeting CD138/BCMA/CD19/More Antigens (CART-138/BCMA/19/More) for Chemotherapy Refractory and Relapsed Multiple Myeloma (Phase 1, NCT03196414) | Multiple targets | CAR-T | Recruiting |
The status of each trial is based on the description on the Clinicaltrials.gov website by 20 October 2021.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cho, S.-F.; Xing, L.; Anderson, K.C.; Tai, Y.-T. Promising Antigens for the New Frontier of Targeted Immunotherapy in Multiple Myeloma. Cancers 2021, 13, 6136. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236136
AMA Style
Cho S-F, Xing L, Anderson KC, Tai Y-T. Promising Antigens for the New Frontier of Targeted Immunotherapy in Multiple Myeloma. Cancers. 2021; 13(23):6136. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236136
Chicago/Turabian StyleCho, Shih-Feng, Lijie Xing, Kenneth C. Anderson, and Yu-Tzu Tai. 2021. "Promising Antigens for the New Frontier of Targeted Immunotherapy in Multiple Myeloma" Cancers 13, no. 23: 6136. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13236136
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.