
Increased Replication Stress Determines ATR Inhibitor Sensitivity in Neuroblastoma Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Inhibitors and Chemotherapeutic Agents
2.3. Clonogenic Survival Assay
2.4. XTT Cell Proliferation Assay
2.5. Western Blotting
2.6. Immunofluorescence
2.7. DNA Fibre Analysis
2.8. COMET Assay
2.9. Cell Cycle Analysis
2.10. Live Cell Imaging
2.11. Statistical Analysis
3. Results
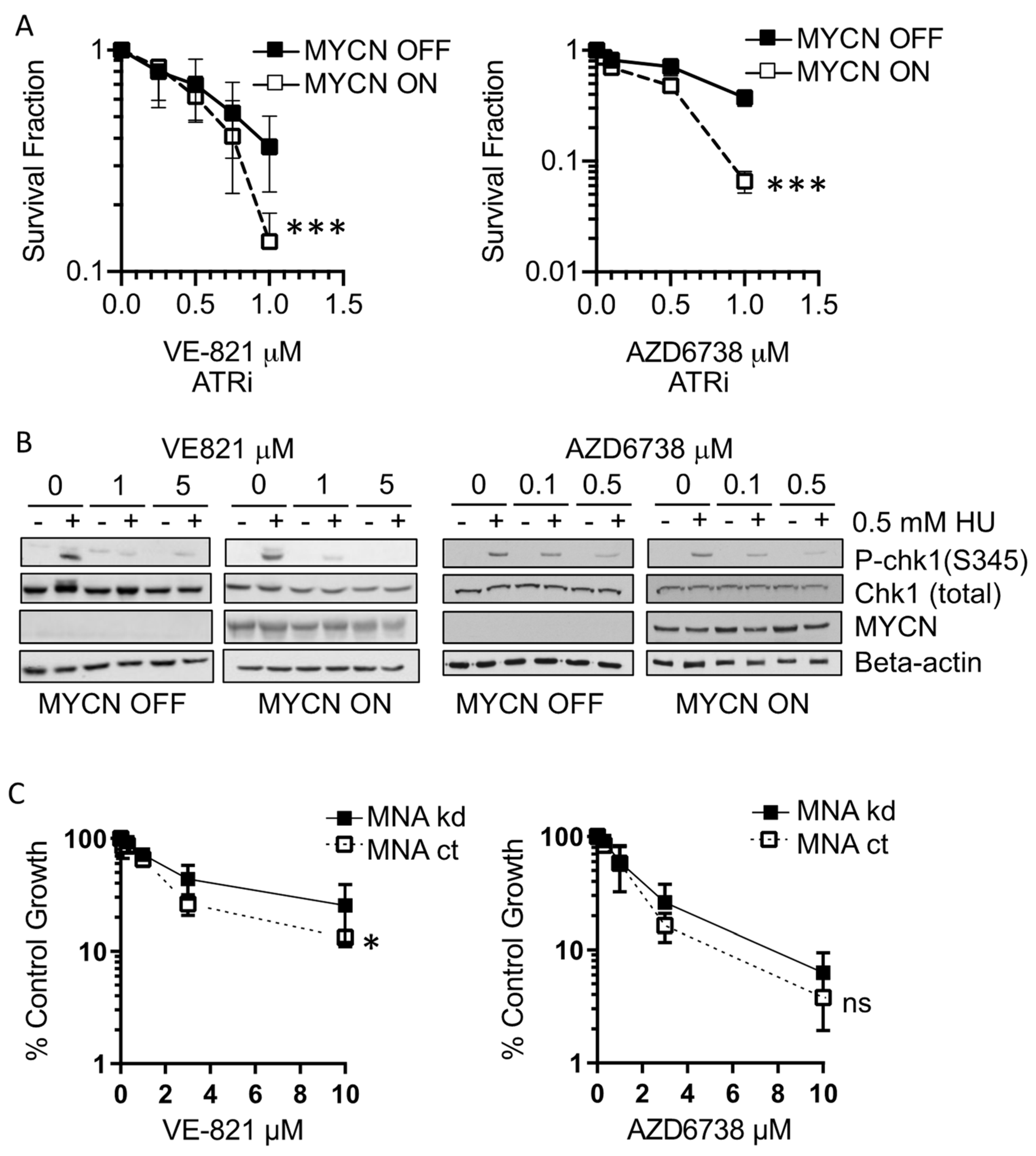
3.1. MYCN Expression Sensitises NB Cells to ATR Inhibition
3.2. MYCN-Induced Replication Stress Is Exacerbated by Inhibition of ATR
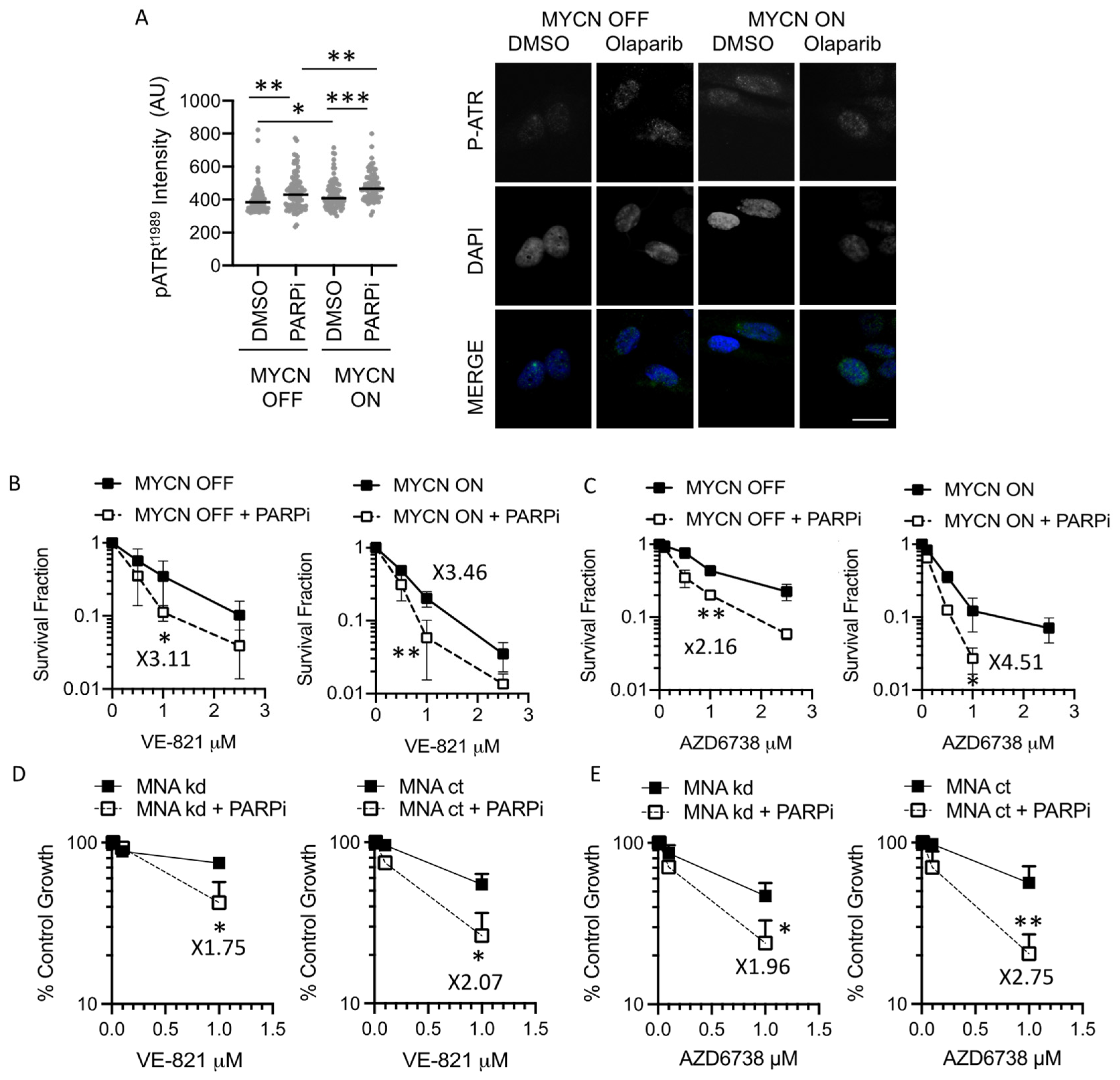
3.3. PARP Inhibition Sensitises NB Cells to ATR Inhibition Independent of MYCN Status
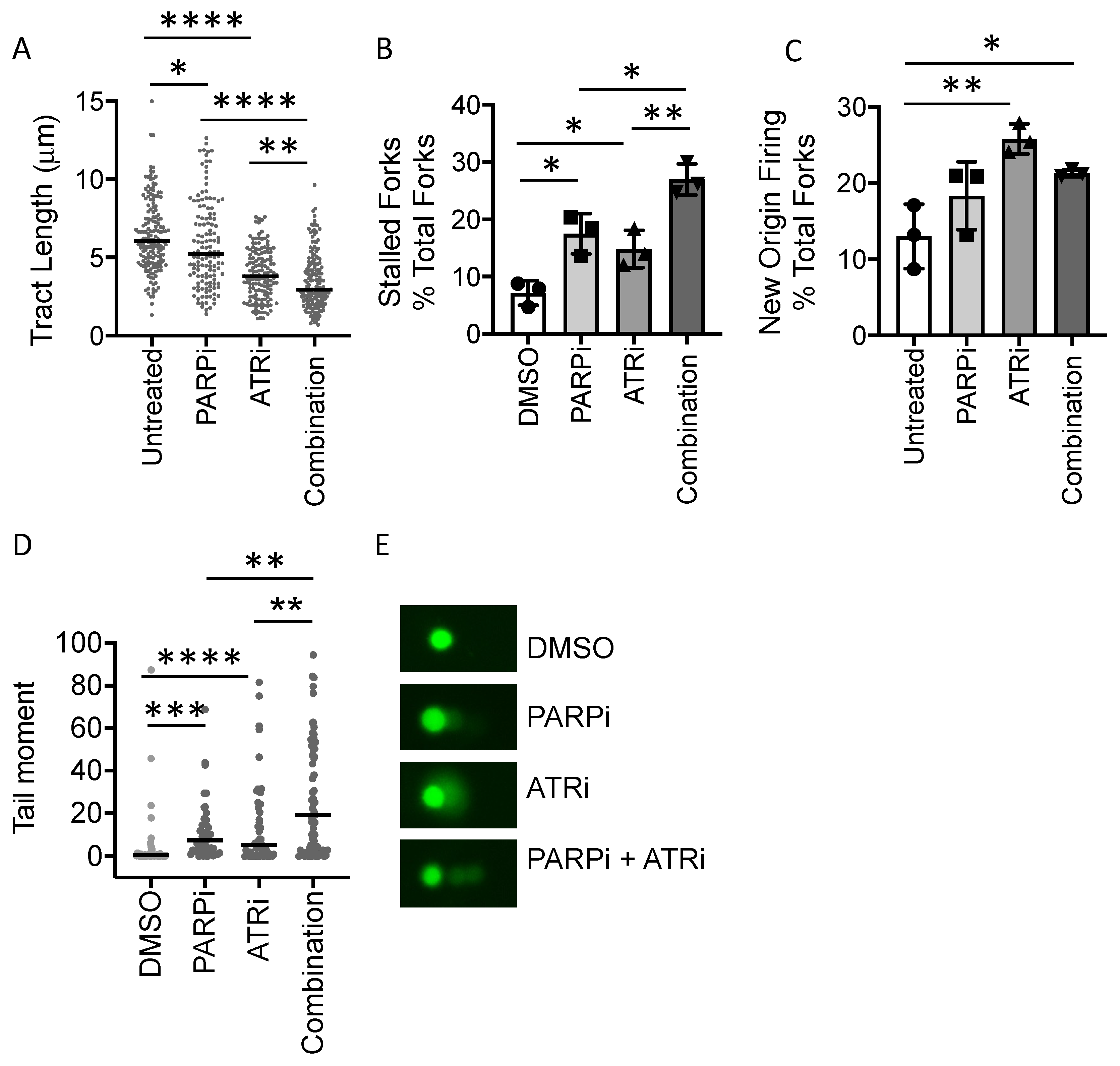
3.4. PARP Inhibitor-Induced Replication Fork Stalling and DNA Damage Is Exacerbated by ATR Inhibition
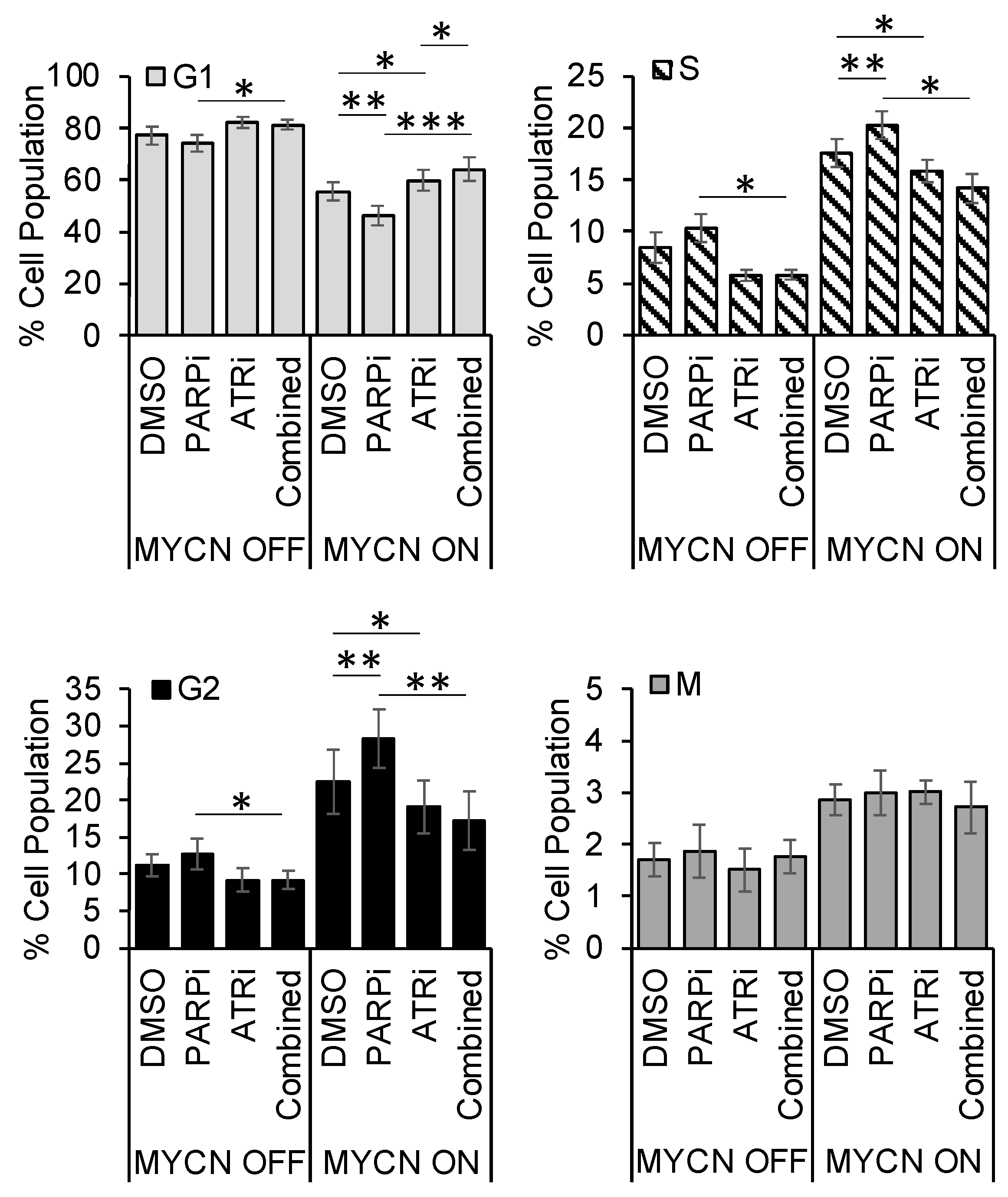
3.5. PARP Inhibitor-Induced Accumulation of Cells in S/G2 Is Overcome by ATR Inhibition
3.6. MYCN- or PARP Inhibitor-Induced Replication Stress Can Increase Mitotic Aberrance Which Is Exacerbated by ATR Inhibition
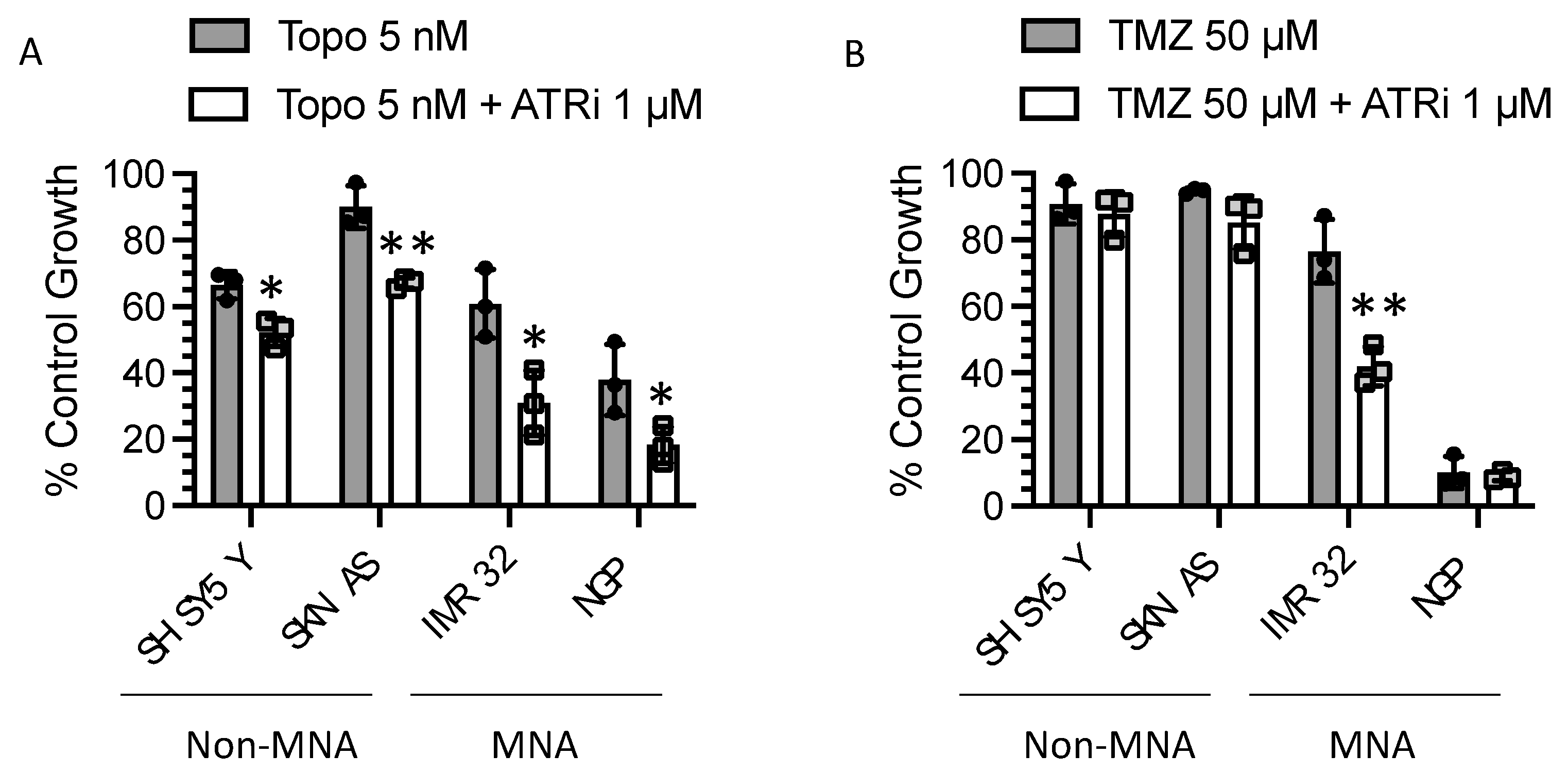
3.7. Replication Stress-Inducing Chemotherapeutic Agents also Sensitise to ATR Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The international neuroblastoma risk group (INRG) classification system: An INRG task force report. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N- myc in Untreated Human Neuroblastomas Correlates with Advanced Disease Stage. Science 1984, 224, 1121–1124. [Google Scholar] [CrossRef]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of Multiple Copies of the N-mycOncogene with Rapid Progression of Neuroblastomas. N. Engl. J. Med. 1985, 313, 1111–1116. [Google Scholar] [CrossRef]
- Basta, N.O.; Halliday, G.C.; Makin, G.; Birch, J.; Feltbower, R.; Bown, N.; Elliott, M.; Moreno, L.; Barone, G.; Pearson, A.D.; et al. Factors associated with recurrence and survival length following relapse in patients with neuroblastoma. Br. J. Cancer 2016, 115, 1048–1057. [Google Scholar] [CrossRef] [Green Version]
- Maris, J.M. Recent Advances in Neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.R.; Bagatell, R.; London, W.B.; Maris, J.M.; Cohn, S.L.; Mattay, K.K.; Hogarty, M. Children’s Oncology Group’s 2013 blueprint for research: Neuroblastoma. Pediatr Blood Cancer 2013, 60, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes. Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [Green Version]
- Rohban, S.; Campaner, S. Myc induced replicative stress response: How to cope with it and exploit it. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2015, 1849, 517–524. [Google Scholar] [CrossRef]
- King, D.; Li, X.D.; Almeida, G.S.; Kwok, C.; Gravells, P.; Harrison, D.; Burke, S.; Hallsworth, A.; Jamin, Y.; George, S.; et al. MYCN expression induces replication stress and sensitivity to PARP inhibition in neuroblastoma. Oncotarget 2020, 11, 2141–2159. [Google Scholar] [CrossRef]
- Curti, L.; Campaner, S. MYC-Induced Replicative Stress: A Double-Edged Sword for Cancer Development and Treatment. Int. J. Mol. Sci. 2021, 22, 6168. [Google Scholar] [CrossRef] [PubMed]
- Sala, A. Editorial: Targeting MYCN in Pediatric Cancers. Front. Oncol. 2015, 4, 330. [Google Scholar] [CrossRef] [Green Version]
- Cole, K.A.; Huggins, J.; Laquaglia, M.; Hulderman, C.E.; Russell, M.R.; Bosse, K.; Diskin, S.J.; Attiyeh, E.F.; Sennett, R.; Norris, G.; et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. 2011, 108, 3336–3341. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Chu, P.; Lingeman, R.; McDaniel, H.; Kechichian, S.; Hickey, R.J.; Liu, Z.; Yuan, Y.-C.; Sandoval, J.A.; Fields, G.B.; et al. The Mechanism by Which MYCN Amplification Confers an Enhanced Sensitivity to a PCNA-Derived Cell Permeable Peptide in Neuroblastoma Cells. EBioMedicine 2015, 2, 1923–1931. [Google Scholar] [CrossRef] [Green Version]
- Colicchia, V.; Petroni, M.; Guarguaglini, G.; Sardina, F.; Sahún-Roncero, M.; Carbonari, M.; Ricci, B.; Heil, C.; Capalbo, C.; Belardinilli, F.; et al. PARP inhibitors enhance replication stress and cause mitotic catastrophe in MYCN-dependent neuroblastoma. Oncogene 2017, 36, 4682–4691. [Google Scholar] [CrossRef]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.-S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaillard, H.; García-Muse, T.; Aguilera, A. Replication stress and cancer, Nature reviews. Cancer 2015, 15, 276–289. [Google Scholar] [PubMed]
- Yazinski, S.A.; Zou, L. Functions, Regulation, and Therapeutic Implications of the ATR Checkpoint Pathway. Annu. Rev. Genet. 2016, 50, 155–173. [Google Scholar] [CrossRef] [PubMed]
- Ronson, G.; Piberger, A.L.; Higgs, M.R.; Olsen, A.L.; Stewart, G.; McHugh, P.J.; Petermann, E.; Lakin, N.D. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zou, L.; Lu, T.; Bao, S.; Hurov, K.E.; Hittelman, W.N.; Elledge, S.J.; Li, L. Rad17 Phosphorylation Is Required for Claspin Recruitment and Chk1 Activation in Response to Replication Stress. Mol. Cell 2006, 23, 331–341. [Google Scholar] [CrossRef]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [Green Version]
- Saldivar, J.; Cortez, D.; Cimprich, K.A. The essential kinase ATR: Ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol. 2017, 18, 622–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Southgate, H.E.D.; Chen, L.; Tweddle, D.A.; Curtin, N.J. ATR Inhibition Potentiates PARP Inhibitor Cytotoxicity in High Risk Neuroblastoma Cell Lines by Multiple Mechanisms. Cancers 2020, 12, 1095. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Levin, K.; Rader, J.; Belcastro, L.; Li, Y.; Martinez, D.; Pawel, B.; Shumway, S.D.; Maris, J.M.; Cole, K.A. Combination Therapy Targeting the Chk1 and Wee1 Kinases Shows Therapeutic Efficacy in Neuroblastoma. Cancer Res. 2013, 73, 776–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, C.D.; VanWye, A.B.; Dowless, M.; Blosser, W.; Falcon, B.L.; Stewart, J.; Stephens, J.; Beckmann, R.P.; Lin, A.B.; Stancato, L.F. The Checkpoint Kinase 1 Inhibitor Prexasertib Induces Regression of Preclinical Models of Human Neuroblastoma. Clin. Cancer Res. An Off. J. Am. Assoc. Cancer Res. 2017, 23, 4354–4363. [Google Scholar] [CrossRef] [Green Version]
- Dreidax, D.; Bannert, S.; Henrich, K.-O.; Schröder, C.; Bender, S.; Oakes, C.C.; Lindner, S.; Schulte, J.H.; Duffy, D.; Schwarzl, T.; et al. Westermann, p19-INK4d inhibits neuroblastoma cell growth, induces differentiation and is hypermethylated and downregulated in MYCN-amplified neuroblastomas. Hum. Mol. Genet. 2014, 23, 6826–6837. [Google Scholar] [CrossRef] [Green Version]
- Lutz, W.; Stöhr, M.; Schürmann, J.; Wenzel, A.; Löhr, A.; Schwab, M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene 1996, 13, 803–812. [Google Scholar]
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Littler, S.; Sloss, O.; Geary, B.; Pierce, A.; Whetton, A.D.; Taylor, S.S. Oncogenic MYC amplifies mitotic perturbations. Open Biol. 2019, 9, 190136. [Google Scholar] [CrossRef] [Green Version]
- Dréan, A.; Lord, C.J.; Ashworth, A. PARP inhibitor combination therapy. Crit. Rev. Oncol. 2016, 108, 73–85. [Google Scholar] [CrossRef]
- Daniel, R.A.; Rozanska, A.L.; Thomas, H.D.; Mulligan, E.A.; Drew, Y.; Castelbuono, D.J.; Hostomsky, Z.; Plummer, E.R.; Boddy, A.; Tweddle, D.A.; et al. Inhibition of Poly(ADP-Ribose) Polymerase-1 Enhances Temozolomide and Topotecan Activity against Childhood Neuroblastoma. Clin. Cancer Res. An Off. J. Am. Assoc. Cancer Res. 2009, 15, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Sanmartín, E.; Muñoz, L.; Piqueras, M.; Sirerol, J.A.; Berlanga, P.; Cañete, A.; Castel, V.; De Mora, J.F. Deletion of 11q in Neuroblastomas Drives Sensitivity to PARP Inhibition. Clin. Cancer Res. An Off. J. Am. Assoc. Cancer Res. 2017, 23, 6875–6887. [Google Scholar] [CrossRef] [Green Version]
- Moiseeva, T.; Hood, B.; Schamus, S.; O’Connor, M.J.; Conrads, T.P.; Bakkenist, C.J. ATR kinase inhibition induces unscheduled origin firing through a Cdc7-dependent association between GINS and And-1. Nat. Commun. 2017, 8, 1392. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.; Dixit, S.; Somyajit, K.; Nagaraju, G. ATR Signaling Uncouples the Role of RAD51 Paralogs in Homologous Recombination and Replication Stress Response. Cell Rep. 2019, 29, 551–559.e4. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Saleh-Gohari, N.; Bryant, H.E.; Schultz, N.; Parker, K.M.; Cassel, T.N.; Helleday, T. Spontaneous Homologous Recombination Is Induced by Collapsed Replication Forks That Are Caused by Endogenous DNA Single-Strand Breaks. Mol. Cell. Biol. 2005, 25, 7158–7169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-Dependent Degradation of Stalled DNA Replication Forks Is Prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [Green Version]
- Petermann, E.; Orta, M.L.; Issaeva, N.; Schultz, N.; Helleday, T. Hydroxyurea-Stalled Replication Forks Become Progressively Inactivated and Require Two Different RAD51-Mediated Pathways for Restart and Repair. Mol. Cell 2010, 37, 492–502. [Google Scholar] [CrossRef] [Green Version]
- Zellweger, R.; Dalcher, D.; Mutreja, K.; Berti, M.; Schmid, J.; Herrador, R.; Vindigni, A.; Lopes, M. Rad51-mediated replication fork reversal is a global response to genotoxic treatments in human cells. J. Cell Biol. 2015, 208, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Yoshida, M.; Nemoto, Y.; Tamaichi, H.; Tsuchida, R.; Seki, M.; Uryu, K.; Hoshino, N.; Nishii, R.; Miyamoto, S.; et al. Loss of DNA Damage Response in Neuroblastoma and Utility of a PARP Inhibitor. J. Natl. Cancer Inst. 2017, 109, djx062. [Google Scholar] [CrossRef] [Green Version]
- George, S.L.; Lorenzi, F.; King, D.; Hartlieb, S.; Campbell, J.; Pemberton, H.; Toprak, U.H.; Barker, K.; Tall, J.; da Costa, B.M.; et al. Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. EBioMedicine 2020, 59, 102971. [Google Scholar] [CrossRef] [PubMed]
- Zeineldin, M.; Federico, S.; Chen, X.; Fan, Y.; Xu, B.; Stewart, E.; Zhou, X.; Jeon, J.; Griffiths, L.; Nguyen, R.; et al. MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat. Commun. 2020, 11, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, D.; Southgate, H.E.D.; Roetschke, S.; Gravells, P.; Fields, L.; Watson, J.B.; Chen, L.; Chapman, D.; Harrison, D.; Yeomanson, D.; et al. Increased Replication Stress Determines ATR Inhibitor Sensitivity in Neuroblastoma Cells. Cancers 2021, 13, 6215. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13246215
King D, Southgate HED, Roetschke S, Gravells P, Fields L, Watson JB, Chen L, Chapman D, Harrison D, Yeomanson D, et al. Increased Replication Stress Determines ATR Inhibitor Sensitivity in Neuroblastoma Cells. Cancers. 2021; 13(24):6215. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13246215
Chicago/Turabian StyleKing, David, Harriet E. D. Southgate, Saskia Roetschke, Polly Gravells, Leona Fields, Jessica B. Watson, Lindi Chen, Devon Chapman, Daniel Harrison, Daniel Yeomanson, and et al. 2021. "Increased Replication Stress Determines ATR Inhibitor Sensitivity in Neuroblastoma Cells" Cancers 13, no. 24: 6215. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13246215