
Exploiting Gangliosides for the Therapy of Ewing’s Sarcoma and H3K27M-Mutant Diffuse Midline Glioma

 , , , , , ,
, , , , , ,  and
and
Abstract
:Simple Summary
Abstract

1. Introduction
2. Results
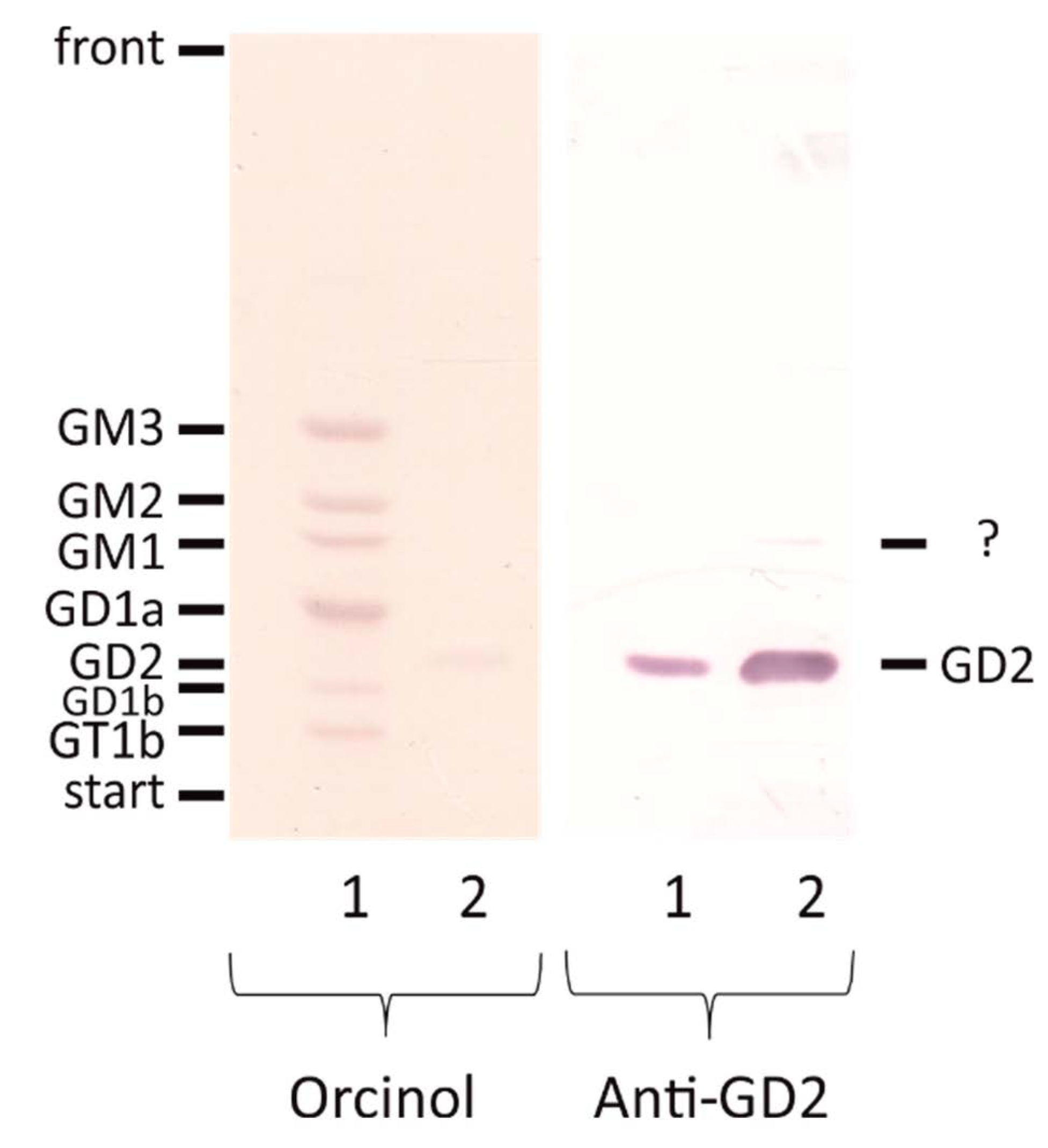
2.1. Validation of an Anti-GD2 Antibody
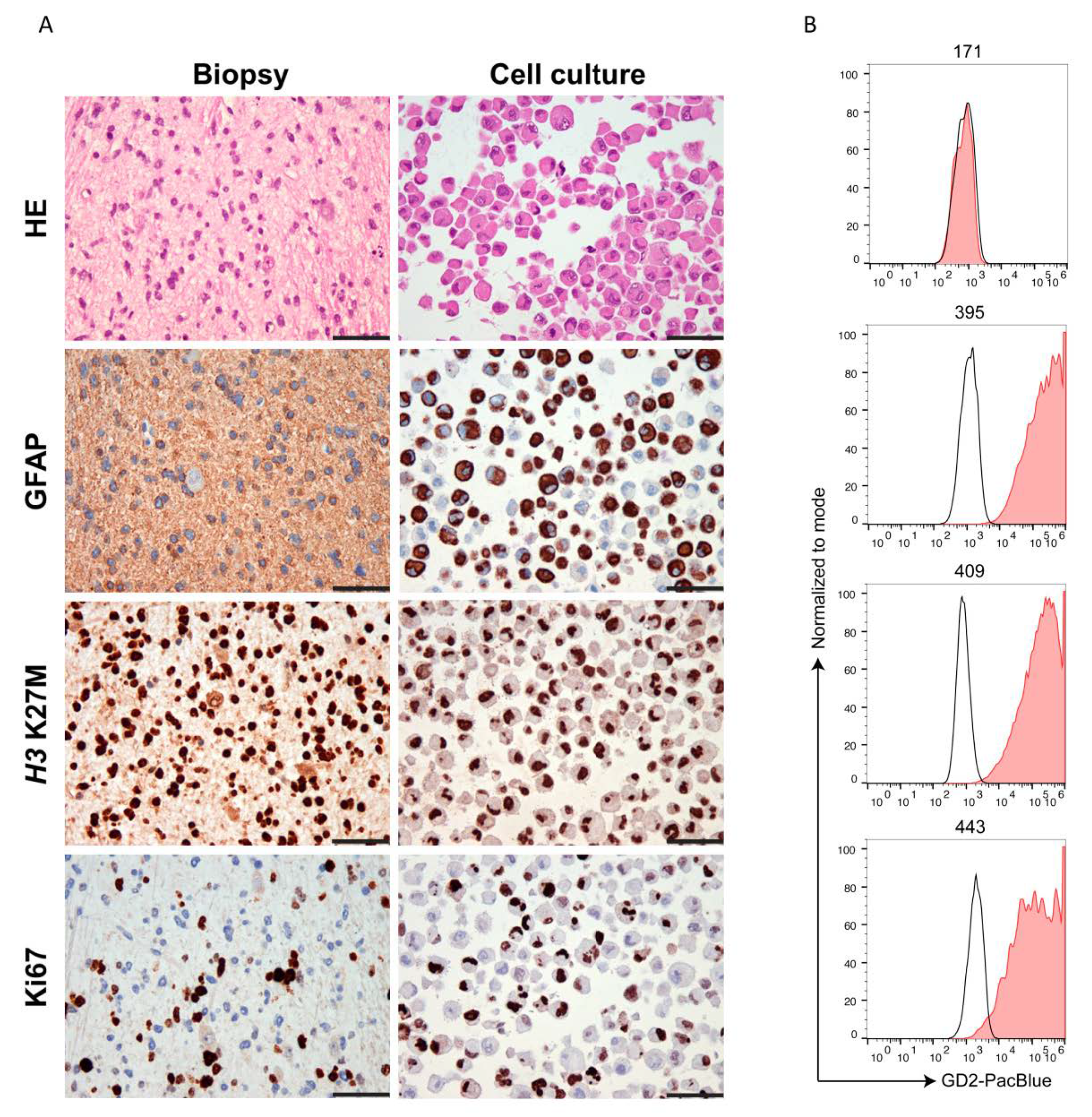
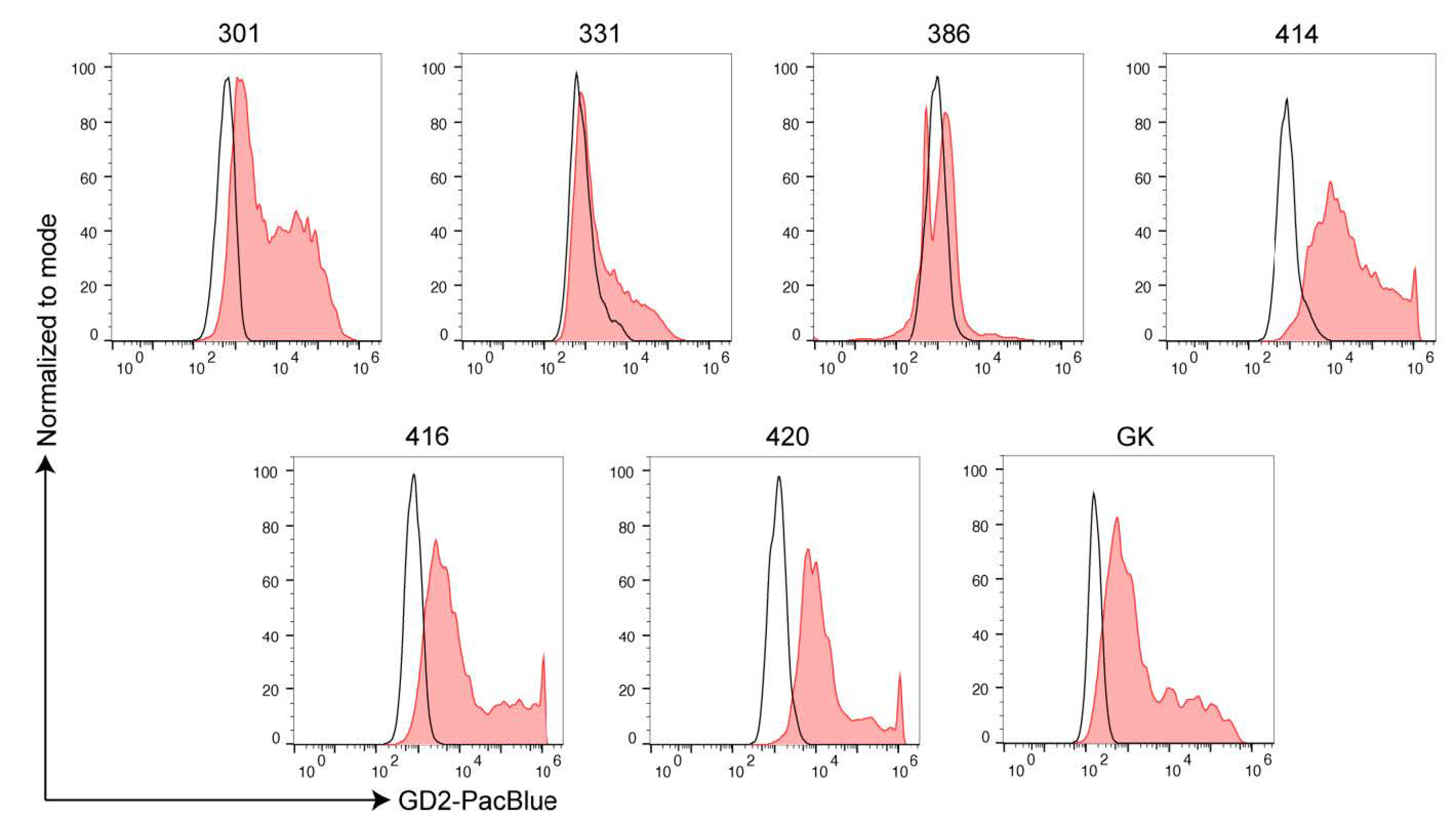
2.2. Ewing’s Sarcoma and H3K27M-Mutant DMG Highly Express GD2
2.3. Anti-GD2 Antibody Treatment in a Patient with Ewing´s Sarcoma
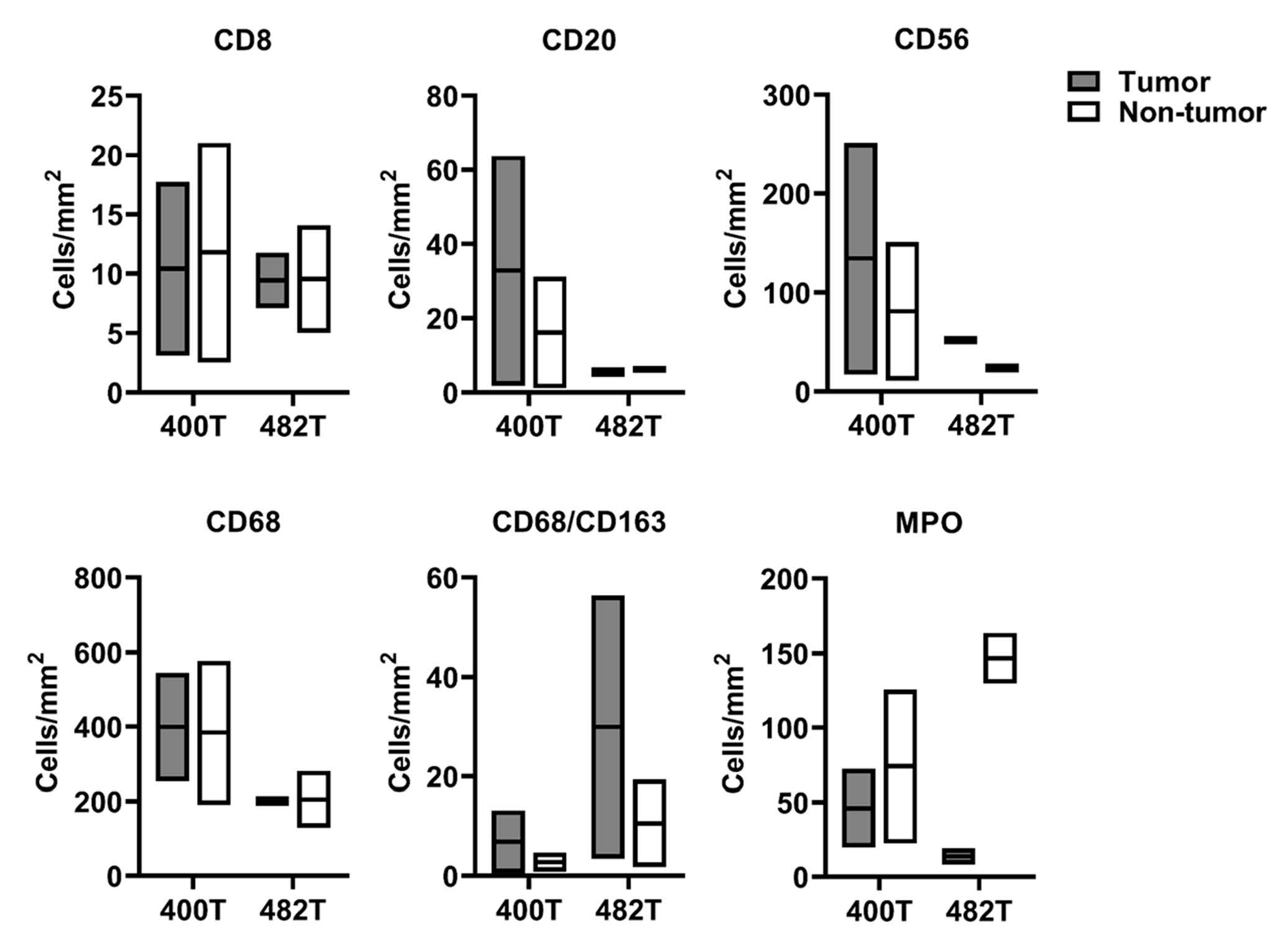
2.4. GD2 Expression and Immune Cells Infiltration in the Dinutuximab-Refractory Metastasis
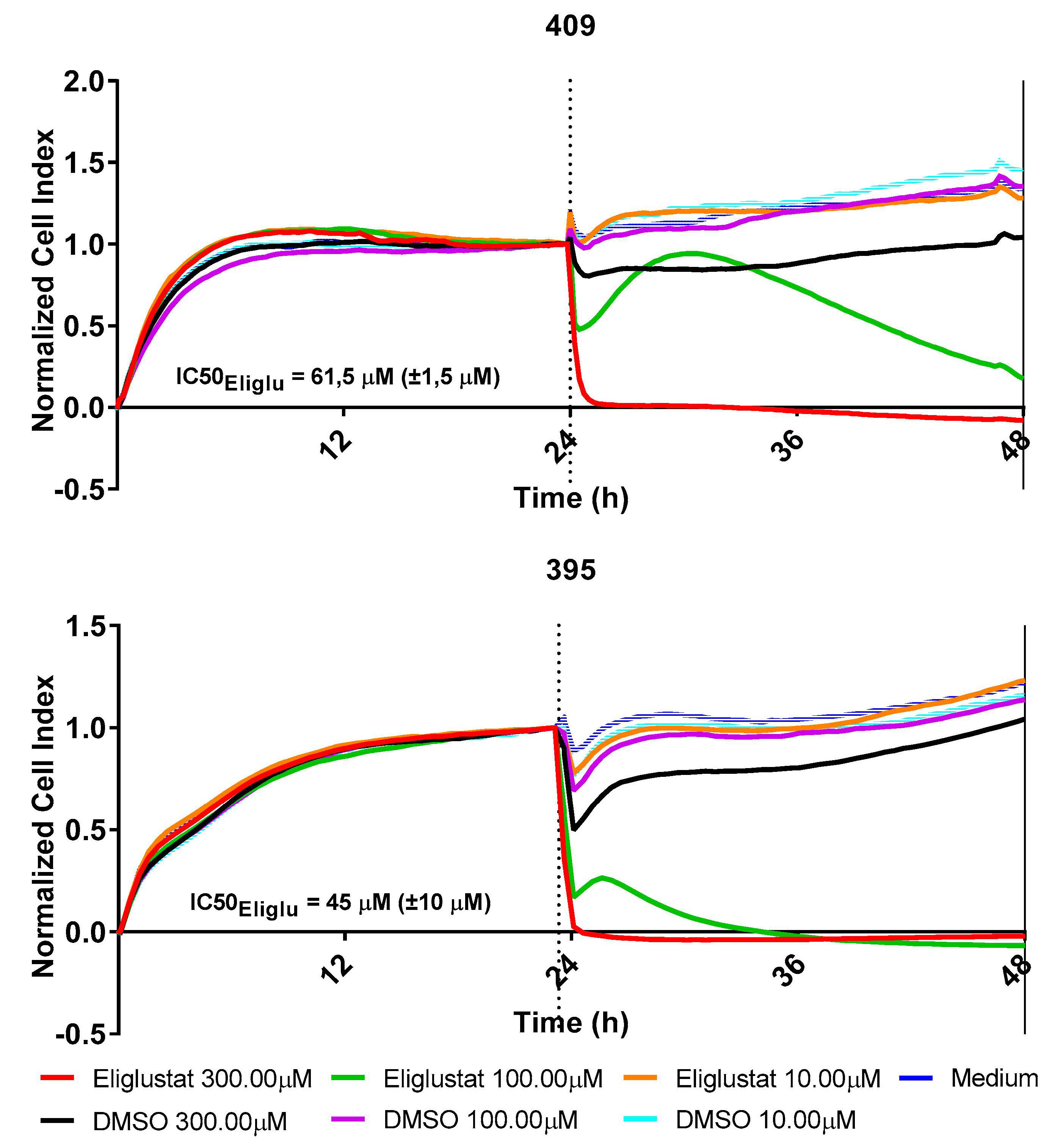
2.5. Perturbation of the Glycosphingolipid Metabolism Inhibits the Growth of H3K27M-Mutant DMG
3. Discussion
GD2 Expression and Immune Cells Infiltration in the Dinutuximab-Refractory Metastasis
4. Materials and Methods
4.1. Patients and Material
4.2. Isolation and Cultivation of Primary Tumor Cells
4.3. Flow Cytometric Analysis of GD2
4.4. Lipid Analysis
4.4.1. Lipid Extraction
4.4.2. Thin Layer Chromatography and Immune Overlay
4.4.3. HILIC-Coupled Tandem Mass Spectrometry of Gangliosides
4.5. Validation of Primary Cell Cultures
4.6. Immune Cells Infiltration
4.7. Cell Proliferation
4.8. Detection of the EWSR1-FLI1 Fusion
4.9. Patient History and Design of the Individual Treatment Protocol
4.10. Software
4.11. Statistical analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sandhoff, R.; Sandhoff, K. Emerging concepts of ganglioside metabolism. FEBS Lett. 2018, 592, 3835–3864. [Google Scholar] [CrossRef] [Green Version]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Navid, F.; Santana, V.M.; Barfield, R.C. Anti-GD2 antibody therapy for GD2-expressing tumors. Curr. Cancer Drug. Targets 2010, 10, 200–209. [Google Scholar] [CrossRef]
- Fredman, P.; Hedberg, K.; Brezicka, T. Gangliosides as therapeutic targets for cancer. BioDrugs 2003, 17, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Hero, B.; Schulte, J.H.; Deubzer, H.; Hundsdoerfer, P.; von Schweinitz, D.; Fuchs, J.; Schmidt, M.; Prasad, V.; Krug, B.; et al. 2017 GPOH guidelines for diagnosis and treatment of patients with neuroblastic tumors. Klin. Padiatr. 2017, 229, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.; Foster, J. High-risk neuroblastoma treatment review. Children 2018, 5, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mody, R.; Naranjo, A.; Van Ryn, C.; Yu, A.L.; London, W.B.; Shulkin, B.L.; Parisi, M.T.; Servaes, S.E.; Diccianni, M.B.; Sondel, P.M.; et al. Irinotecan-temozolomide with temsirolimus or dinutuximab in children with refractory or relapsed neuroblastoma (COG ANBL1221): An open-label, randomised, phase 2 trial. Lancet Oncol. 2017, 18, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; London, W.B.; Kreissman, S.G.; Chen, H.X.; Smith, M.; Anderson, B.; Villablanca, J.G.; Matthay, K.K.; et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010, 363, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotterill, S.J.; Ahrens, S.; Paulussen, M.; Jurgens, H.F.; Voute, P.A.; Gadner, H.; Craft, A.W. Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J. Clin. Oncol. 2000, 18, 3108–3114. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Meltzer, J.; Pscherer, S.; Luecke, A.; Dierkes, C.; Titze, U.; Leuchte, K.; Landmeier, S.; Hotfilder, M.; et al. The ganglioside antigen G(D2) is surface-expressed in Ewing sarcoma and allows for MHC-independent immune targeting. Br. J. Cancer 2012, 106, 1123–1133. [Google Scholar] [CrossRef]
- Casey, D.L.; Lin, T.Y.; Cheung, N.V. Exploiting signaling pathways and immune targets beyond the standard of care for ewing sarcoma. Front. Oncol. 2019, 9, 537. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.W.; Hutson, P.R.; Hank, J.A.; Sondel, P.M.; Furman, W.L.; Meagher, M.M.; Navid, F.; Santana, V.M. A Phase 1 and pharmacokinetic study evaluating daily or weekly schedules of the humanized anti-GD2 antibody hu14.18K322A in recurrent/refractory solid tumors. MAbs 2020, 12, 1773751. [Google Scholar] [CrossRef] [PubMed]
- Jansen, M.H.; van Vuurden, D.G.; Vandertop, W.P.; Kaspers, G.J. Diffuse intrinsic pontine gliomas: A systematic update on clinical trials and biology. Cancer Treat. Rev. 2012, 38, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M(+) diffuse midline gliomas. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Linkowski, M.; Tarim, J.; Piperdi, S.; Sowers, R.; Geller, D.; Gill, J.; Gorlick, R. Ganglioside GD2 as a therapeutic target for antibody-mediated therapy in patients with osteosarcoma. Cancer 2014, 120, 548–554. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.K.; Lazarus, H.; Miraldi, F.D.; Abramowsky, C.R.; Kallick, S.; Saarinen, U.M.; Spitzer, T.; Strandjord, S.E.; Coccia, P.F.; Berger, N.A. Ganglioside GD2 specific monoclonal antibody 3F8: A phase I study in patients with neuroblastoma and malignant melanoma. J. Clin. Oncol. 1987, 5, 1430–1440. [Google Scholar] [CrossRef]
- Murray, J.L.; Cunningham, J.E.; Brewer, H.; Mujoo, K.; Zukiwski, A.A.; Podoloff, D.A.; Kasi, L.P.; Bhadkamkar, V.; Fritsche, H.A.; Benjamin, R.S. Phase I trial of murine monoclonal antibody 14G2a administered by prolonged intravenous infusion in patients with neuroectodermal tumors. J. Clin. Oncol. 1994, 12, 184–193. [Google Scholar] [CrossRef]
- Cheung, N.K.; Sowers, R.; Vickers, A.J.; Cheung, I.Y.; Kushner, B.H.; Gorlick, R. FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J. Clin. Oncol. 2006, 24, 2885–2890. [Google Scholar] [CrossRef]
- Zeng, Y.; Fest, S.; Kunert, R.; Katinger, H.; Pistoia, V.; Michon, J.; Lewis, G.; Ladenstein, R.; Lode, H.N. Anti-neuroblastoma effect of ch14.18 antibody produced in CHO cells is mediated by NK-cells in mice. Mol. Immunol. 2005, 42, 1311–1319. [Google Scholar] [CrossRef]
- Razpotnik, R.; Novak, N.; Curin Serbec, V.; Rajcevic, U. Targeting malignant brain tumors with antibodies. Front. Immunol. 2017, 8, 1181. [Google Scholar] [CrossRef] [Green Version]
- Kramer, K.; Humm, J.L.; Souweidane, M.M.; Zanzonico, P.B.; Dunkel, I.J.; Gerald, W.L.; Khakoo, Y.; Yeh, S.D.; Yeung, H.W.; Finn, R.D.; et al. Phase I study of targeted radioimmunotherapy for leptomeningeal cancers using intra-Ommaya 131-I-3F8. J. Clin. Oncol. 2007, 25, 5465–5470. [Google Scholar] [CrossRef] [PubMed]
- Kramer, K.; Pandit-Taskar, N.; Humm, J.L.; Zanzonico, P.B.; Haque, S.; Dunkel, I.J.; Wolden, S.L.; Donzelli, M.; Goldman, D.A.; Lewis, J.S.; et al. A phase II study of radioimmunotherapy with intraventricular (131) I-3F8 for medulloblastoma. Pediatr. Blood Cancer 2018, 65. [Google Scholar] [CrossRef] [PubMed]
- Souweidane, M.M.; Kramer, K.; Pandit-Taskar, N.; Zhou, Z.; Haque, S.; Zanzonico, P.; Carrasquillo, J.A.; Lyashchenko, S.K.; Thakur, S.B.; Donzelli, M.; et al. Convection-enhanced delivery for diffuse intrinsic pontine glioma: A single-centre, dose-escalation, phase 1 trial. Lancet Oncol. 2018, 19, 1040–1050. [Google Scholar] [CrossRef]
- Birkle, S.; Zeng, G.; Gao, L.; Yu, R.K.; Aubry, J. Role of tumor-associated gangliosides in cancer progression. Biochimie 2003, 85, 455–463. [Google Scholar] [CrossRef]
- Vantaku, V.; Donepudi, S.R.; Ambati, C.R.; Jin, F.; Putluri, V.; Nguyen, K.; Rajapakshe, K.; Coarfa, C.; Battula, V.L.; Lotan, Y.; et al. Expression of ganglioside GD2, reprogram the lipid metabolism and EMT phenotype in bladder cancer. Oncotarget 2017, 8, 95620–95631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zheng, X.; Pang, X.; Li, L.; Wang, J.; Yang, C.; Du, G. Ganglioside GD3 synthase (GD3S), a novel cancer drug target. Acta Pharm. Sin. B 2018, 8, 713–720. [Google Scholar] [CrossRef]
- McEachern, K.A.; Fung, J.; Komarnitsky, S.; Siegel, C.S.; Chuang, W.L.; Hutto, E.; Shayman, J.A.; Grabowski, G.A.; Aerts, J.M.; Cheng, S.H.; et al. A specific and potent inhibitor of glucosylceramide synthase for substrate inhibition therapy of Gaucher disease. Mol. Genet. Metab. 2007, 91, 259–267. [Google Scholar] [CrossRef]
- Mujoo, K.; Kipps, T.J.; Yang, H.M.; Cheresh, D.A.; Wargalla, U.; Sander, D.J.; Reisfeld, R.A. Functional properties and effect on growth suppression of human neuroblastoma tumors by isotype switch variants of monoclonal antiganglioside GD2 antibody 14.18. Cancer Res. 1989, 49, 2857–2861. [Google Scholar]
- Vewinger, N.; Huprich, S.; Seidmann, L.; Russo, A.; Alt, F.; Bender, H.; Sommer, C.; Samuel, D.; Lehmann, N.; Backes, N.; et al. IGF1R is a potential new therapeutic target for HGNET-BCOR brain tumor patients. Int. J. Mol. Sci. 2019, 20, 3027. [Google Scholar] [CrossRef] [Green Version]
- Nagaraja, S.; Vitanza, N.A.; Woo, P.J.; Taylor, K.R.; Liu, F.; Zhang, L.; Li, M.; Meng, W.; Ponnuswami, A.; Sun, W.; et al. Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 2017, 31, 635–652.e6. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.Y.; Gardner, J.M.; Lucas, D.R.; McHugh, J.B.; Patel, R.M. Ewing sarcoma. Semin. Diagn. Pathol. 2014, 31, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Beck, O.; Paret, C.; Russo, A.; Burhenne, J.; Fresnais, M.; Steimel, K.; Seidmann, L.; Wagner, D.C.; Vewinger, N.; Lehmann, N.; et al. Safety and activity of the combination of ceritinib and dasatinib in osteosarcoma. Cancers 2020, 12, 793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawlor, E.R.; Sorensen, P.H. Twenty years on: What do we really know about ewing sarcoma and what is the path forward? Crit. Rev. Oncog. 2015, 20, 155–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worst, B.C.; van Tilburg, C.M.; Balasubramanian, G.P.; Fiesel, P.; Witt, R.; Freitag, A.; Boudalil, M.; Previti, C.; Wolf, S.; Schmidt, S.; et al. Next-generation personalised medicine for high-risk paediatric cancer patients—The INFORM pilot study. Eur. J. Cancer 2016, 65, 91–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.; Kopp, L.; Anderson, N.; Cornelius, K.; Herzog, C.; Hughes, D.; Huh, W. Novel bone cancer drugs: Investigational agents and control paradigms for primary bone sarcomas (Ewing’s sarcoma and osteosarcoma). Expert Opin. Investig. Drugs 2008, 17, 1703–1715. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.A.; Wexler, L.H.; Merchant, M.S.; Chou, A.J.; Merola, P.R.; Price, A.P.; Meyers, P.A. Irinotecan and temozolomide for Ewing sarcoma: The Memorial Sloan-Kettering experience. Pediatr. Blood Cancer 2009, 53, 1029–1034. [Google Scholar] [CrossRef]
- Salah, S.; To, Y.H.; Khozouz, O.; Ismail, T.; Yaser, S.; Alnsour, A.; Shahin, O.; Sultan, I.; Abuhijlih, R.; Halalsheh, H.; et al. Irinotecan and temozolomide chemotherapy in paediatric and adult populations with relapsed Ewing Sarcoma. Clin. Transl. Oncol. 2020. [Google Scholar] [CrossRef]
- Wagner, L.M.; McAllister, N.; Goldsby, R.E.; Rausen, A.R.; McNall-Knapp, R.Y.; McCarville, M.B.; Albritton, K. Temozolomide and intravenous irinotecan for treatment of advanced Ewing sarcoma. Pediatr. Blood Cancer 2007, 48, 132–139. [Google Scholar] [CrossRef]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [Green Version]
- Mujoo, K.; Cheresh, D.A.; Yang, H.M.; Reisfeld, R.A. Disialoganglioside GD2 on human neuroblastoma cells: Target antigen for monoclonal antibody-mediated cytolysis and suppression of tumor growth. Cancer Res. 1987, 47, 1098–1104. [Google Scholar]
- Alvarez-Rueda, N.; Desselle, A.; Cochonneau, D.; Chaumette, T.; Clemenceau, B.; Leprieur, S.; Bougras, G.; Supiot, S.; Mussini, J.M.; Barbet, J.; et al. A monoclonal antibody to O-acetyl-GD2 ganglioside and not to GD2 shows potent anti-tumor activity without peripheral nervous system cross-reactivity. PLoS ONE 2011, 6, e25220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sariola, H.; Terava, H.; Rapola, J.; Saarinen, U.M. Cell-surface ganglioside GD2 in the immunohistochemical detection and differential diagnosis of neuroblastoma. Am. J. Clin. Pathol. 1991, 96, 248–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, V.I.; Roth, M.; Piperdi, S.; Geller, D.; Gill, J.; Rudzinski, E.R.; Hawkins, D.S.; Gorlick, R. Ganglioside GD2 expression is maintained upon recurrence in patients with osteosarcoma. Clin. Sarcoma Res. 2015, 5, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terzic, T.; Cordeau, M.; Herblot, S.; Teira, P.; Cournoyer, S.; Beaunoyer, M.; Peuchmaur, M.; Duval, M.; Sartelet, H. Expression of Disialoganglioside (GD2) in neuroblastic tumors: A prognostic value for patients treated with anti-GD2 immunotherapy. Pediatr. Dev. Pathol. 2018, 21, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Kailayangiri, S.; Altvater, B.; Lesch, S.; Balbach, S.; Gottlich, C.; Kuhnemundt, J.; Mikesch, J.H.; Schelhaas, S.; Jamitzky, S.; Meltzer, J.; et al. EZH2 inhibition in ewing sarcoma upregulates GD2 expression for targeting with gene-modified T cells. Mol. Ther. 2019, 27, 933–946. [Google Scholar] [CrossRef]
- Richter, G.H.; Plehm, S.; Fasan, A.; Rossler, S.; Unland, R.; Bennani-Baiti, I.M.; Hotfilder, M.; Lowel, D.; von Luettichau, I.; Mossbrugger, I.; et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 5324–5329. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.P.; Flutter, B.; Wesemann, F.; Frosch, J.; Rossig, C.; Gustafsson, K.; Anderson, J. Effective combination treatment of GD2-expressing neuroblastoma and Ewing’s sarcoma using anti-GD2 ch14.18/CHO antibody with Vgamma9Vdelta2+ gammadeltaT cells. Oncoimmunology 2016, 5, e1025194. [Google Scholar] [CrossRef] [Green Version]
- Nowak, A.K.; Robinson, B.W.; Lake, R.A. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res. 2003, 63, 4490–4496. [Google Scholar]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef]
- Hernandez-Marques, C.; Lassaletta-Atienza, A.; Ruiz Hernandez, A.; Blumenfeld Olivares, J.A.; Arce Abaitua, B.; Cormenzana Carpio, M.; Madero Lopez, L. [Irinotecan plus temozolomide in refractory or relapsed pediatric solid tumors]. An. Pediatr. (Barc) 2013, 79, 68–74. [Google Scholar] [CrossRef]
- McCabe, M.G.; Kirton, L.; Khan, M.; Fenwick, N.; Dirksen, U.; Gaspar, N.; Kanerva, J.; Kuehne, T.; Longhi, A.; Luksch, R.; et al. Results of the second interim assessment of rEECur, an international randomized controlled trial of chemotherapy for the treatment of recurrent and primary refractory Ewing sarcoma (RR-ES). J. Clin. Oncol. 2020, 38, 11502. [Google Scholar] [CrossRef]
- Bailey, K.; Cost, C.; Davis, I.; Glade-Bender, J.; Grohar, P.; Houghton, P.; Isakoff, M.; Stewart, E.; Laack, N.; Yustein, J.; et al. Emerging novel agents for patients with advanced Ewing sarcoma: A report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force. F1000Res 2019, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hank, J.A.; Robinson, R.R.; Surfus, J.; Mueller, B.M.; Reisfeld, R.A.; Cheung, N.K.; Sondel, P.M. Augmentation of antibody dependent cell mediated cytotoxicity following in vivo therapy with recombinant interleukin 2. Cancer Res. 1990, 50, 5234–5239. [Google Scholar] [PubMed]
- Masucci, G.; Wersall, P.; Ragnhammar, P.; Mellstedt, H. Granulocyte-monocyte-colony-stimulating factor augments the cytotoxic capacity of lymphocytes and monocytes in antibody-dependent cellular cytotoxicity. Cancer Immunol. Immunother. 1989, 29, 288–292. [Google Scholar] [CrossRef]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorokin, M.; Kholodenko, I.; Kalinovsky, D.; Shamanskaya, T.; Doronin, I.; Konovalov, D.; Mironov, A.; Kuzmin, D.; Nikitin, D.; Deyev, S.; et al. RNA Sequencing-Based Identification of Ganglioside GD2-Positive Cancer Phenotype. Biomedicines 2020, 8, 142. [Google Scholar] [CrossRef] [PubMed]
- Xenaki, K.T.; Oliveira, S.; van Bergen En Henegouwen, P.M.P. Antibody or Antibody Fragments: Implications for Molecular Imaging and Targeted Therapy of Solid Tumors. Front. Immunol. 2017, 8, 1287. [Google Scholar] [CrossRef]
- Tabrizi, M.; Bornstein, G.G.; Suria, H. Biodistribution mechanisms of therapeutic monoclonal antibodies in health and disease. AAPS J. 2010, 12, 33–43. [Google Scholar] [CrossRef]
- Triulzi, T.; Regondi, V.; De Cecco, L.; Cappelletti, M.R.; Di Modica, M.; Paolini, B.; Lollini, P.L.; Di Cosimo, S.; Sfondrini, L.; Generali, D.; et al. Early immune modulation by single-agent trastuzumab as a marker of trastuzumab benefit. Br. J. Cancer 2018, 119, 1487–1494. [Google Scholar] [CrossRef] [Green Version]
- Varchetta, S.; Gibelli, N.; Oliviero, B.; Nardini, E.; Gennari, R.; Gatti, G.; Silva, L.S.; Villani, L.; Tagliabue, E.; Menard, S.; et al. Elements related to heterogeneity of antibody-dependent cell cytotoxicity in patients under trastuzumab therapy for primary operable breast cancer overexpressing Her2. Cancer Res. 2007, 67, 11991–11999. [Google Scholar] [CrossRef] [Green Version]
- Matlung, H.L.; Babes, L.; Zhao, X.W.; van Houdt, M.; Treffers, L.W.; van Rees, D.J.; Franke, K.; Schornagel, K.; Verkuijlen, P.; Janssen, H.; et al. Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Rep. 2018, 23, 3946–3959.e3946. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, X.H. Tumor-Associated Neutrophils and Macrophages-Heterogenous but Not Chaotic. Front. Immunol. 2020, 11, 553967. [Google Scholar] [CrossRef] [PubMed]
- Tsao, L.C.; Crosby, E.J.; Trotter, T.N.; Agarwal, P.; Hwang, B.J.; Acharya, C.; Shuptrine, C.W.; Wang, T.; Wei, J.; Yang, X.; et al. CD47 blockade augmentation of trastuzumab antitumor efficacy dependent on antibody-dependent cellular phagocytosis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Jiang, Z.; Mortenson, E.D.; Deng, L.; Radkevich-Brown, O.; Yang, X.; Sattar, H.; Wang, Y.; Brown, N.K.; Greene, M.; et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010, 18, 160–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol 2018, 9, 2380. [Google Scholar] [CrossRef] [Green Version]
- Richman, S.A.; Nunez-Cruz, S.; Moghimi, B.; Li, L.Z.; Gershenson, Z.T.; Mourelatos, Z.; Barrett, D.M.; Grupp, S.A.; Milone, M.C. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol. Res. 2018, 6, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Weber, E.W.; Lynn, R.C.; Xu, P.; Mackall, C.L. Neurotoxicity Associated with a High-Affinity GD2 CAR-Letter. Cancer Immunol. Res. 2018, 6, 494–495. [Google Scholar] [CrossRef] [Green Version]
- Richman, S.A.; Milone, M.C. Neurotoxicity Associated with a High-Affinity GD2 CAR-Response. Cancer Immunol. Res. 2018, 6, 496–497. [Google Scholar] [CrossRef] [Green Version]
- Yeh, S.C.; Wang, P.Y.; Lou, Y.W.; Khoo, K.H.; Hsiao, M.; Hsu, T.L.; Wong, C.H. Glycolipid GD3 and GD3 synthase are key drivers for glioblastoma stem cells and tumorigenicity. Proc. Natl. Acad. Sci. USA 2016, 113, 5592–5597. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Koodie, L.; Jacobsen, K.; Hanzawa, K.; Miyamoto, Y.; Yamamoto, M. B4GALNT1 induces angiogenesis, anchorage independence growth and motility, and promotes tumorigenesis in melanoma by induction of ganglioside GM2/GD2. Sci. Rep. 2020, 10, 1199. [Google Scholar] [CrossRef] [Green Version]
- Vykoukal, J.; Fahrmann, J.F.; Gregg, J.R.; Tang, Z.; Basourakos, S.; Irajizad, E.; Park, S.; Yang, G.; Creighton, C.J.; Fleury, A.; et al. Caveolin-1-mediated sphingolipid oncometabolism underlies a metabolic vulnerability of prostate cancer. Nat. Commun. 2020, 11, 4279. [Google Scholar] [CrossRef] [PubMed]
- Morales, L.E. Gaucher’s disease: A review. Ann. Pharmacother. 1996, 30, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treiber, A.; Morand, O.; Clozel, M. The pharmacokinetics and tissue distribution of the glucosylceramide synthase inhibitor miglustat in the rat. Xenobiotica 2007, 37, 298–314. [Google Scholar] [CrossRef]
- Cabrera-Salazar, M.A.; Deriso, M.; Bercury, S.D.; Li, L.; Lydon, J.T.; Weber, W.; Pande, N.; Cromwell, M.A.; Copeland, D.; Leonard, J.; et al. Systemic delivery of a glucosylceramide synthase inhibitor reduces CNS substrates and increases lifespan in a mouse model of type 2 Gaucher disease. PLoS ONE 2012, 7, e43310. [Google Scholar] [CrossRef] [Green Version]
- Jennemann, R.; Rodden, A.; Bauer, B.L.; Mennel, H.D.; Wiegandt, H. Glycosphingolipids of human gliomas. Cancer Res. 1990, 50, 7444–7449. [Google Scholar]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Leavey, P.J.; Mascarenhas, L.; Marina, N.; Chen, Z.; Krailo, M.; Miser, J.; Brown, K.; Tarbell, N.; Bernstein, M.L.; Granowetter, L.; et al. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2008, 51, 334–338. [Google Scholar] [CrossRef] [Green Version]
- Ladenstein, R.; Potschger, U.; Valteau-Couanet, D.; Luksch, R.; Castel, V.; Yaniv, I.; Laureys, G.; Brock, P.; Michon, J.M.; Owens, C.; et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): A multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1617–1629. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Gender | Age | Diagnosis | Primary Tumor or Metastasis | Localization | [%] |
|---|---|---|---|---|---|---|
| 301 | m | 13 | OS | Primary tumor | Distal femur | 43.5 |
| 331 | m | 18 | OS | Metastasis | Lung | 10.9 |
| 386 | f | 16 | OS | Metastasis | Lung | 2.65 |
| 414 | m | 15 | OS | Primary tumor | Proximal tibia | 32.7 |
| 1416 | m | 15 | OS | Primary tumor | Proximal tibia | 24.9 |
| 1420 | m | 15 | OS | Metastasis | Spine | 17.4 |
| GK | m | 11 | OS | Primary tumor | Proximal tibia | 26.6 |
| 393 | m | 16 | ES | Primary tumor | Clavicle | 5.83 |
| 411 | m | 20 | ES | Metastasis | Liver | 57.5 |
| 2408 | f | 5 | ES | Metastasis | Spine | 91 |
| 2482 | f | 6 | ES | Metastasis | Intracranial | 59.3 |
| WiFi7 | m | 28 | ES | Primary tumor | Femur | 98.4 |
| SHG | m | 50 | ES | Metastasis | pleural effusion | 0.9 |
| 443 | m | 9 | DNET | Primary tumor | CNS | 96.4 |
| 171 | m | 7 | HGNET-BCOR | Metastasis | Skull | 0 |
| 395 | m | 16 | H3K27M-mutant DMG | Primary tumor | CNS | 98.1 |
| 409 | m | 7 | H3K27M-mutant DMG | Primary tumor | CNS | 99 |
| Antibody | Dilution | Host | Clone | Provider | No. | Incubation Time (min) |
|---|---|---|---|---|---|---|
| anti-GFAP | RTU | rabbit | Pk | Dako | IR524 | 20 |
| anti-Ki-67 | RTU | mouse | MIB-1 | Dako | IR626 | 20 |
| anti-Synaptophysin | RTU | mouse | DAK-Synap | Dako | IR660 | 20 |
| anti-Neurofilament | RTU | mouse | 2F-11 | Dako | IR607 | 20 |
| anti-H3-K27-M | 1/500 | rabbit | Pk | Millipore | ABE419 | 20 |
| Antibody | Dilution | Host | Clone | Provider | No. | Incubation Time (min) |
|---|---|---|---|---|---|---|
| CD8 | RTU | mouse | C8/144B | DAKO | IR623 | 20 |
| CD20 | RTU | mouse | L26 | DAKO | IR604 | 20 |
| MPO | RTU | rabbit | polyclonal | DAKO | IR511 | 20 |
| CD68 | RTU | mouse | PG-M1 | DAKO | IR613 | 20 |
| CD163 | 1/200 | mouse | 10D6 | Leica | CD163-L-CE | 20 |
| CD56 | RTU | mouse | 123C3 | DAKO | IR628 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wingerter, A.; El Malki, K.; Sandhoff, R.; Seidmann, L.; Wagner, D.-C.; Lehmann, N.; Vewinger, N.; Frauenknecht, K.B.M.; Sommer, C.J.; Traub, F.; et al. Exploiting Gangliosides for the Therapy of Ewing’s Sarcoma and H3K27M-Mutant Diffuse Midline Glioma. Cancers 2021, 13, 520. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13030520
Wingerter A, El Malki K, Sandhoff R, Seidmann L, Wagner D-C, Lehmann N, Vewinger N, Frauenknecht KBM, Sommer CJ, Traub F, et al. Exploiting Gangliosides for the Therapy of Ewing’s Sarcoma and H3K27M-Mutant Diffuse Midline Glioma. Cancers. 2021; 13(3):520. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13030520
Chicago/Turabian StyleWingerter, Arthur, Khalifa El Malki, Roger Sandhoff, Larissa Seidmann, Daniel-Christoph Wagner, Nadine Lehmann, Nadine Vewinger, Katrin B. M. Frauenknecht, Clemens J. Sommer, Frank Traub, and et al. 2021. "Exploiting Gangliosides for the Therapy of Ewing’s Sarcoma and H3K27M-Mutant Diffuse Midline Glioma" Cancers 13, no. 3: 520. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13030520