
Novel Combinatorial Approaches to Tackle the Immunosuppressive Microenvironment of Prostate Cancer



 and
and
Abstract
:Simple Summary
Abstract
1. Prostate Cancer Background
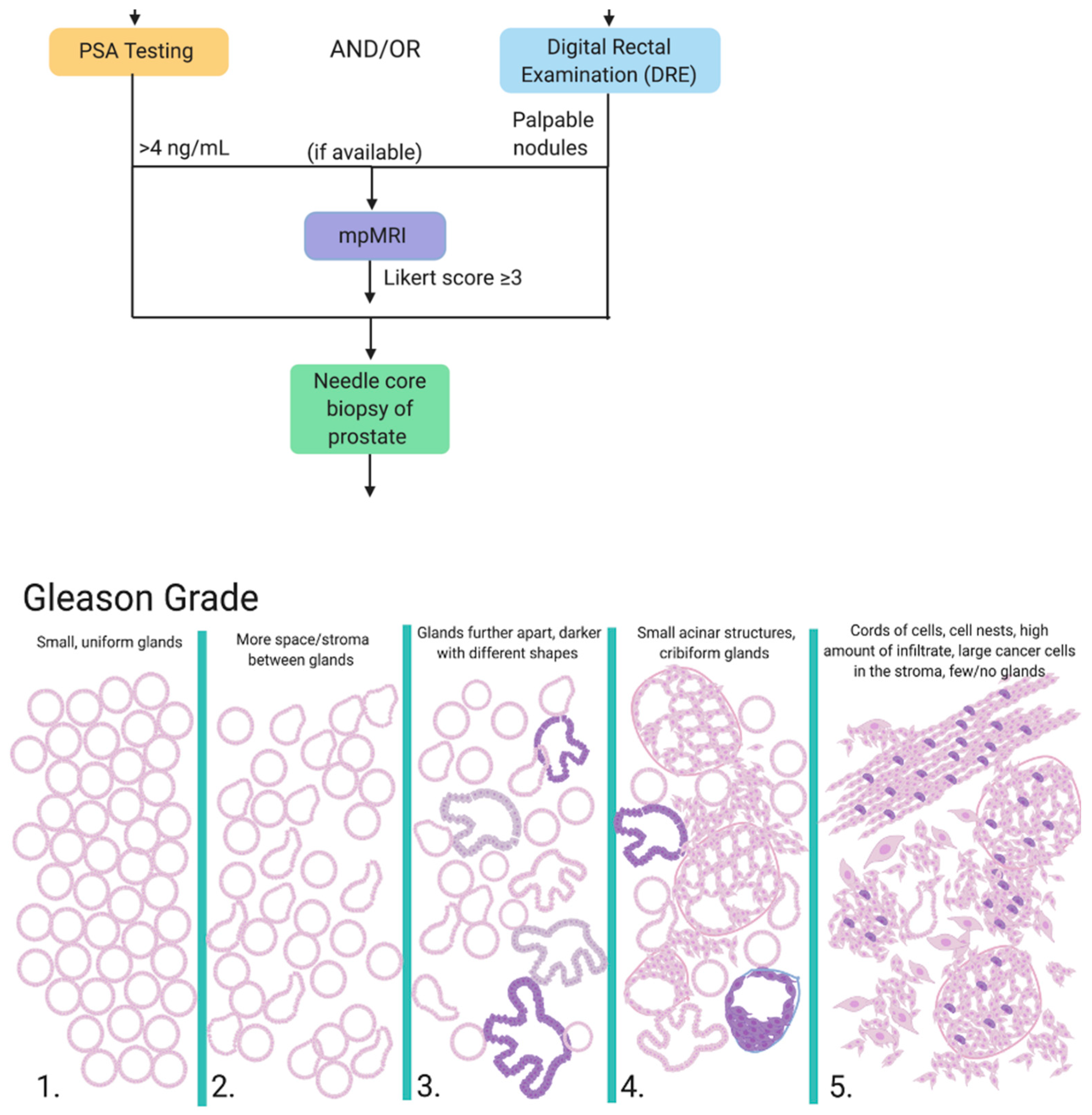
1.1. Diagnosis
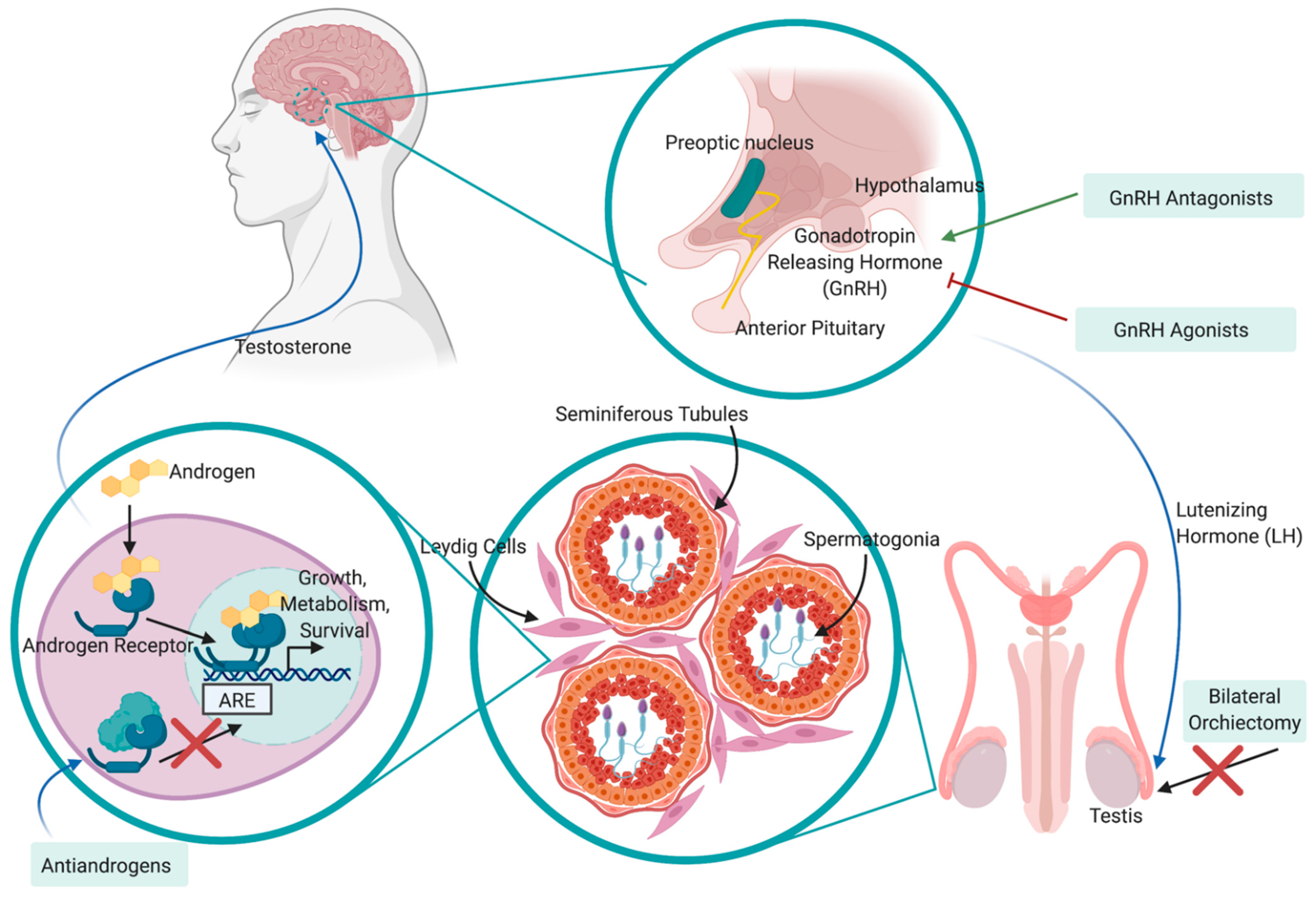
1.2. Treatment
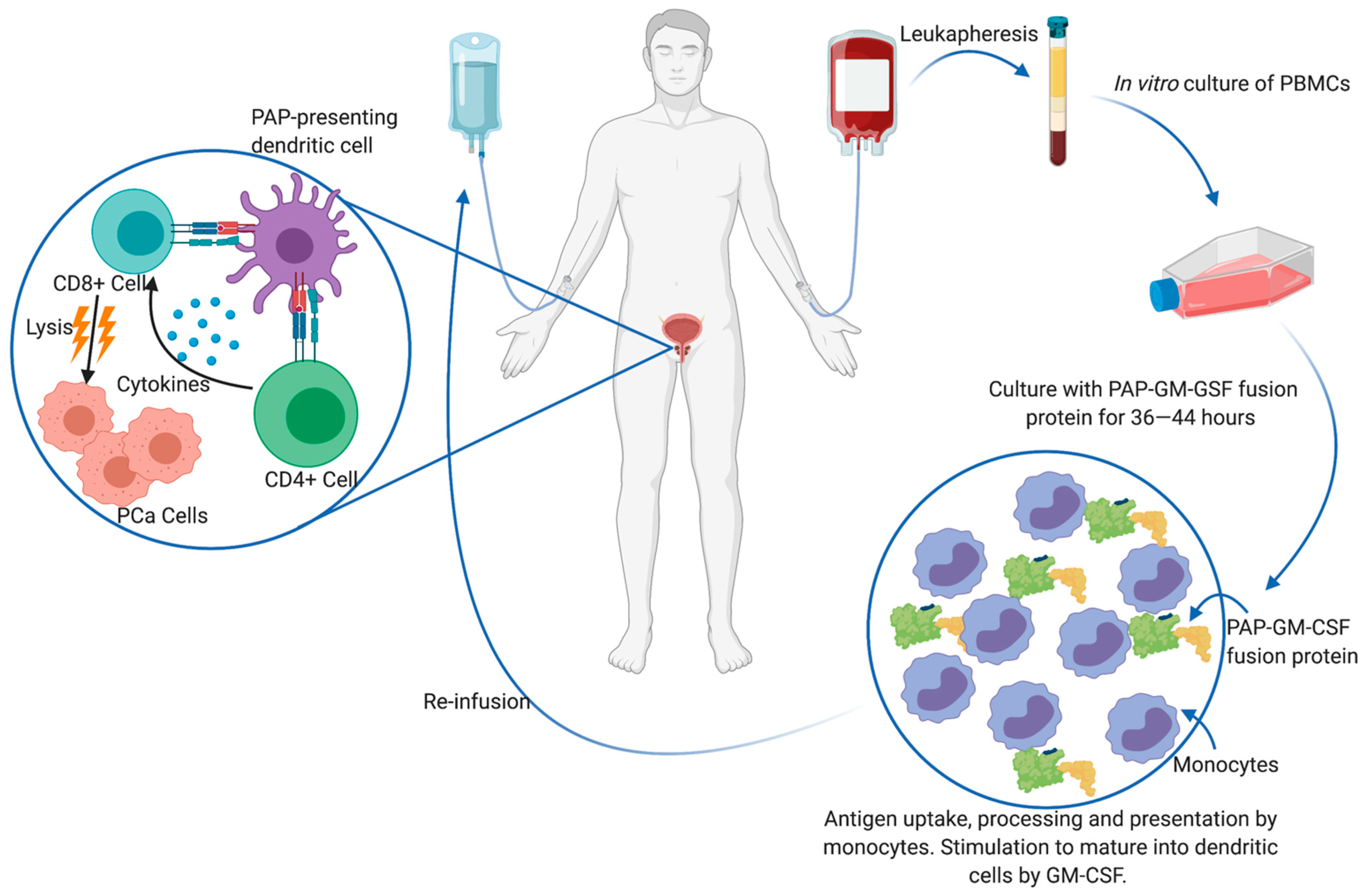
1.3. Immunotherapy
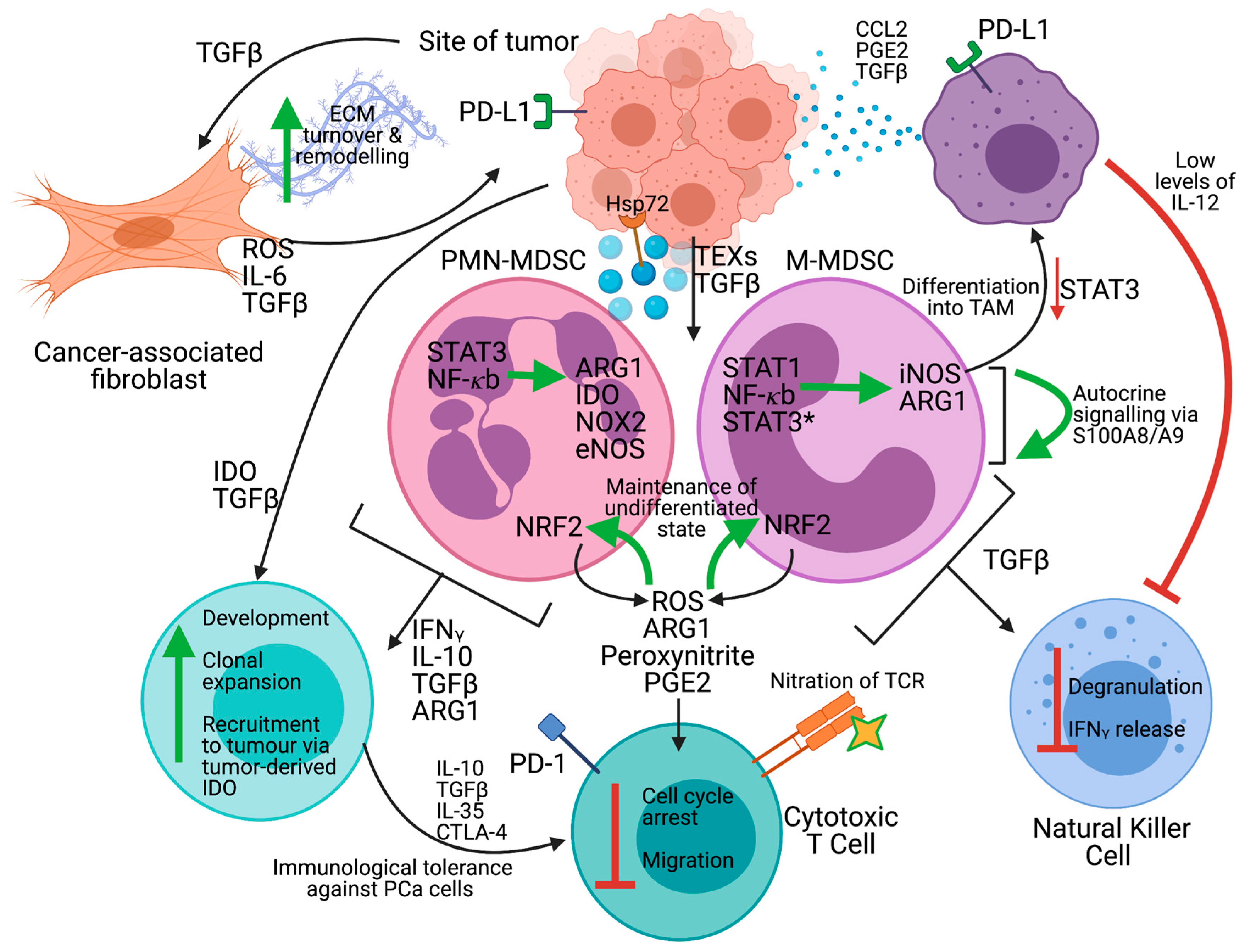
2. Myeloid-Derived Suppressor Cells
2.1. The Tumor Microenvironment
2.2. Subpopulations of Myeloid-Derived Suppressor Cells
2.3. Expansion and Activation
2.4. MDSCs in PCa Tumor Progression
2.5. Mechanisms of Immunosuppression
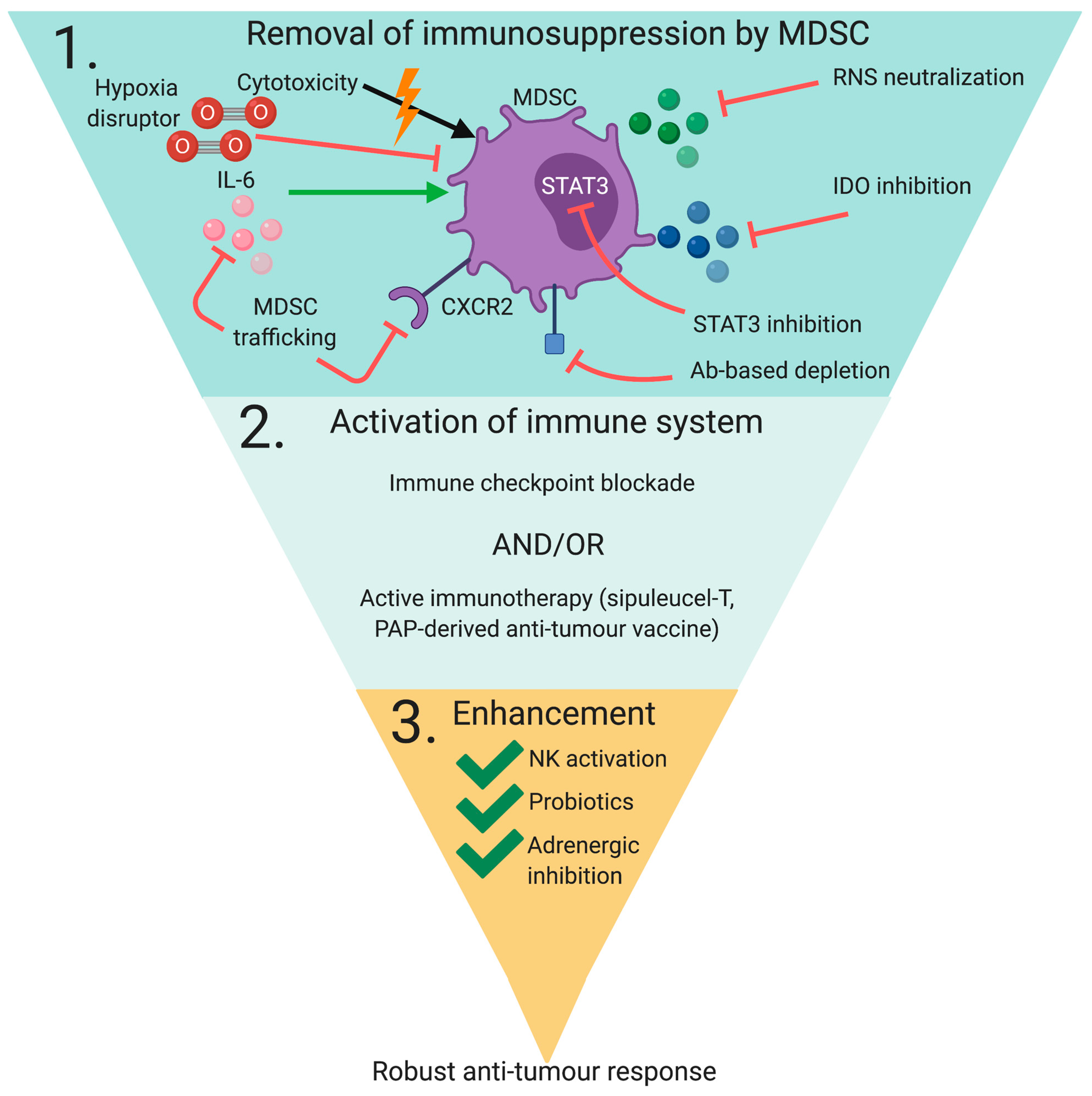
3. Targeting MDSC In Immunotherapy
3.1. Targeting MDSC Immune Regulatory Properties
3.2. Targeting MDSC Infiltration/Activation
3.3. Targeting MDSC Development/Maturation
3.4. Inducing MDSC Depletion
3.5. Combinatorial Strategies
4. Other Important Cells and Regulator Contributing to the Overall Immunosuppression or Lack of Antitumor Immune Response
4.1. ADRB2
4.2. Other Important Regulators of PCa Progression—Histone Deacetylase (HDAC)
5. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| 5-FU | 5-fluorouracil |
| Ad | Adrenaline |
| ADRB2 | β-adrenergic receptor |
| ADRB2i | ADRB2i inhibitor |
| ADT | Androgen deprivation therapy |
| APC | Antigen-presenting cell |
| AR | Androgen receptor |
| ARE | Antioxidant response element |
| ARG1 | Arginase 1 |
| ATRA | All transretinoic acid |
| CAB | Combined androgen blockade |
| CAF | Cancer-associated fibroblast |
| CREB | cAMP response element-binding protein |
| CRPC | Castration-resistant prostate cancer |
| CTL | Cytotoxic T-lymphocyte |
| DAMPs | Danger-associated molecular patterns |
| DC | Dendritic cell |
| DHEA | Dehydroepiandrosterone |
| DHT | Dihydrotestosterone |
| DRE | Digital rectal examination |
| ECM | Extracellular matrix |
| EMT | Epithelial–mesenchymal transition |
| EMC | Epithelial-mesenchymal cell |
| ER | Endoplasmic reticulum |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| G-CSF | Granulocyte CSF |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylases |
| HDACi | HDAC inhibitor |
| HMGB1 | High mobility group box 1 |
| ICB | Immune checkpoint blockade |
| IDO | Indoleamine 2,3-dioxygenase |
| IGRT | Image-guided radiation therapy |
| IL-[number] | Interleukin-[number] |
| IMRT | Intensity-modulated radiation therapy |
| LCK | Lymphocyte-specific protein tyrosine kinase |
| LH | Luteinizing hormone |
| LHRH | Luteinizing hormone-releasing hormone |
| M-CSF | Macrophage CSF |
| M-MDSC | Monocytic MDSC |
| MDSC | Myeloid-derived suppressor cell |
| MMP | Matrix metalloproteinase |
| MSC | Mesenchymal stem cell |
| NAd | Noradrenaline |
| NDRG1 | N-myc downstream-regulated gene |
| NEC | Neuroendocrine-like cell |
| NEPC | Neuroendocrine prostate cancer |
| NICE | National Institute for Health and Care Excellence |
| NK | Natural killer |
| NO | Nitric oxide |
| NOX2 | NADPH oxidase 2 |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PAMPs | Pathogen-associated molecular patterns |
| PAP | Prostatic acid phosphatase |
| PBMC | Peripheral blood mononuclear cell |
| PCa | Prostate cancer |
| PCNA | Proliferating cell nuclear antigen |
| PGE2 | Prostaglandin E2 |
| PIN | Prostatic intraepithelial neoplasia |
| PKA | Protein kinase A |
| PMN-MDSC | Polymorphonuclear MDSC |
| PNI | Perineural invasion |
| PSA | Prostate-specific antigen |
| RARP | Robot-assisted radical prostatectomy |
| RHAMM | Receptor for hyaluronan-mediated motility |
| RNS | Reactive nitrogen species |
| ROS | Reactive oxygen species |
| RP | Radical prostatectomy |
| RTK | Receptor tyrosine kinase |
| SAHA | Suberoylanilide hydroxamic acid |
| SCF | Stem cell factor |
| STAT3 | Signal transducer and activator of transcription 3 |
| TAM | Tumour-associated macrophage |
| TCR | T cell receptor |
| TGFβ | Transforming growth factor-beta |
| TI1IFN | Tumour intrinsic type 1 interferon |
| TIMP1 | Tissue inhibitor of metalloproteinase 1 |
| TLR9 | Toll-like receptor 9 |
| TME | Tumour microenvironment |
| TRAIL-R | TNF-related apoptosis-induced ligand receptor 2 |
| Treg | T regulatory cell |
| TRUS | Transrectal ultrasound |
| UA | Uric acid |
| UPR | Unfolded protein response |
| VEGF | Vascular endothelial growth factor |
| Zol | Zoledronic acid |
| β-AR | β-adrenergic receptors |
| cAMP | Cyclic adenosine monophosphate |
| eNOS | Endothelial nitric oxide synthase |
| iNOS | Inducible nitric oxide synthase |
| mCRPC | Metastatic castration-resistant prostate cancer |
| mpMRI | Multiparametric magnetic resonance imaging |
| MSMB | Microseminoprotein-β |
| tNEPC | Treatment-dependent NEPC |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 86, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawla, P. Epidemiology of prostate cancer. World J. Oncol. 2019, 10, 63–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilic, D.; Djulbegovic, M.; Jung, J.H.; Hwang, E.C.; Zhou, Q.; Cleves, A.; Agoritsas, T.; Dahm, P. Prostate cancer screening with prostate-specific antigen (PSA) test: A systematic review and meta-analysis. Br. Med. J. 2018, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loeb, S.; Bjurlin, M.; Nicholson, J.; Tammella, T.L.; Penson, D.; Carter, H.B.; Carrol, P.; Etzioni, R. Overdiagosis and overtreatment of prostate cancer. Eur. Urol. 2014, 65, 1046–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahn, J.L.; Giovannucci, E.L.; Stampfer, M.J. The high prevalence of undiagnosed prostate cancer at autopsy: Implications for epidemiology and treatment of prostate cancer in the Prostate-specific Antigen-era. Int. J. Cancer 2015, 137, 2795–2802. [Google Scholar]
- Jalloh, M.; Friebel, T.M.; Thiam, F.S.; Niang, L.; Sy, C.; Siby, T.; Fernandez, P.; Mapulanga, V.; Maina, S.; Doodu Mante, S.; et al. Evaluation of 4,672 routine prostate biopsies performed in six African countries. J. Afr. Cancer 2013, 5, 144–154. [Google Scholar] [CrossRef]
- Tindall, E.A.; Monare, L.R.; Petersen, D.C.; Zyl, S.V.; Hardie, R.A.; Segone, A.M.; Venter, P.A.; Bornman, M.S.R.; Hayes, V.M. Clinical presentation of prostate cancer in Black South Africans. Prostate 2014, 74, 880–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, D.C.; Jaratlerdsiri, W.; Van Wyk, A.; Chan, E.K.F.; Fernandez, P.; Lyons, R.J.; Mutambirw, S.B.A.; Van der Merwe, A.; Venter, P.A.; Bates, W.; et al. African KhoeSan ancestry linked to high-risk prostate cancer. BMC Med. Genom. 2019, 12, 82. [Google Scholar] [CrossRef]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Möller, S.; Unger, R.H.; et al. Familial risk and heritability of cancer among twins in Nordic countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, F.R.; Al Olama, A.A.; Berndt, S.I.; Benlloch, S.; Ahmed, M.; Saunders, E.J.; Dadaev, T.; Leongamornlert, D.; Anokian, E.; Cieza-Borrella, C.; et al. Association analyses of more than 140,000 men identify 63 new prostate cancer susceptibility loci. Nat. Genet. 2018, 50, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Maskarinec, G.; Noh, J.J. The effect of migration on cancer incidence among Japanese in Hawaii. Ethn. Dis. 2004, 14, 431–439. [Google Scholar] [PubMed]
- Lee, J.; Demissie, K.; Lu, S.; Rhoads, G.G. Cancer incidence among Korean-American immigrants in the United States and native Koreans in South Korea. Cancer Control 2007, 14, 78–85. [Google Scholar] [CrossRef]
- Hanley, A.J.; Choi, B.C.; Holowaty, E.J. Cancer mortality among Chinese migrants: A review. Int. J. Epidemiol. 1995, 24, 255–265. [Google Scholar] [CrossRef]
- Le, G.M.; Gomez, S.L.; Clarke, C.A.; Glaser, S.L.; West, D.W. Cancer incidence patterns among Vietnamese in the United States and Ha Noi, Vietnam. Int. J. Cancer 2002, 102, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Dickerman, B.A.; Torfadottir, J.E.; Valdimarsdottir, U.A.; Giovannucci, E.; Wilson, K.M.; Aspelund, T.; Tryggvadottir, L.; Sigurdardottir, L.G.; Harris, T.B.; Launer, L.J.; et al. Body fat distribution on computed tomography imagine and prostate cancer risk and mortality in the AGES-Reykjavik study. Cancer 2019, 125, 2730–2731. [Google Scholar]
- Dickerman, B.A.; Ahearn, T.U.; Giovannucci, E.; Stampfer, M.J.; Nguyen, P.L.; Mucci, L.A.; Wilson, K.M. Weight change, obesity and risk of prostate cancer aggression among men with clinically localized prostate cancer. Int. J. Cancer 2017, 141, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.; Tilling, K.; Turner, E.L.; Martin, R.M.; Lennon, R.; Lane, J.A.; Donovan, J.L.; Hamdy, F.C.; Neal, D.E.; Ruud Bosch, J.H.L.; et al. Systematic review and meta-analysis of the associations between body mass index, prostate cancer, advanced prostate cancer, and prostate-specific antigen. Cancer Causes Contol 2020, 31, 431–449. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Han, F.F.; Zeng, X.T.; Liu, T.Z.; Li, S.; Gao, Z.Y. Fat intake is not limited to prostate cancer: A systematic review and dose-response meta-analysis. PLoS ONE 2015, 10, e0131747. [Google Scholar]
- Hayashi, T.; Fujita, K.; Nojima, S.; Hayashi, Y.; Nakano, K.; Ishizuya, Y.; Wang, C.; Yamamoto, Y.; Kinouchi, T.; Matsuzaki, K.; et al. High-fat diet-induced inflammation accelerates prostate cancer growth via IL6 signaling. Clin. Cancer Res. 2018, 24, 4309–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lophatananon, A.; Stewart-Brown, S.; Kote-Jarai, Z.; Al Olama, A.A.; Garcia, S.B.; Neal, D.E.; Hamdy, F.C.; Donovan, J.L.; Giles, G.G.; Fitzgerald, L.M.; et al. Height, selected genetic markers and prostate cancer risk: Results from the PRACTICAL consortium. Br. J. Cancer 2018, 118. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.; Lennon, R.; Holly, J.; Higgins, J.P.T.; Gardner, M.; Perks, C.; Gaunt, T.; Tan, V.; Borwick, C.; Emmet, P.; et al. Does milk intake promote prostate cancer initiation or progression via effects of insulin-like growth factors (IGFs)? A systematic review and meta-analysis. Cancer Causes Control 2017, 28, 297–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islami, F.; Moreira, D.M.; Boffetta, P.; Freedland, S.J. A systemaic review and meta-analysis of tobacco use and prostate cancer mortality and incidence in prospective cohort studies. Eur. Urol. 2014, 66, 1054–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arneth, B.M. Clinical significance of measuring prostate-specific antigen. Lab.Med. 2009, 40, 487–491. [Google Scholar] [CrossRef] [Green Version]
- Thompson, I.M.; Pauler, D.K.; Goodman, P.J.; Tangen, C.M.; Lucia, M.S.; Parnes, H.L.; Minasian, L.M.; Ford, L.G.; Lippman, S.M.; Crawford, E.D. Prevalence of prostate cancer among men with a prostate-specific antigen level ≤4.0ng per milliliter. N. Engl. J. Med. 2004, 350, 2239–2246. [Google Scholar] [CrossRef] [Green Version]
- Naji, L.; Randhawa, H.; Sohani, Z.; Dennis, B.; Lautenbach, D.; Kavanagh, O.; Bawor, M.; Banfield, L.; Profetto, J. Digital rectal examination for prostate cancer screening in primary care: A systematic review and meta-analysis. Ann. Fam. Med. 2018, 16, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Streicher, J.; Lee Myerson, B.; Karivedu, V.; Sidana, A. A review of optimal prostate biopsy: Indications and techniques. Ther. Adv. Urol. 2019, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- National Institute for Health and Care Excellence. Prostate Cancer: Diagnosis and Management. Available online: https://www.nice.org.uk/guidance/ng131/resources/prostate-cancer-diagnosis-and-management-pdf-66141714312133 (accessed on 26 February 2021).
- Litwin, M.S.; Tan, H.J. The diagnosis and treatment of prostate cancer: A review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef]
- Omer, A.; Lamb, A.D. Optimizing prostate biopsy techniques. Curr. Opin. Urol. 2019, 29, 578–586. [Google Scholar] [CrossRef]
- Kum, F.; Elhage, O.; Maliyil, J.; Wong, K.; Faure Walker, N.; Kulkarni, M.; Namdarian, B.; Callacombe, B.; Cathcart, P.; Popert, R. Initial outcomes of local anaesthetic freehand transperineal prostate biopsies in the outpatient setting. BJU Int. 2020, 125, 244–252. [Google Scholar] [CrossRef]
- Kamel, M.H.; Khalil, M.I.; Alobuia, W.M.; Su, J.; Davis, R. Incidence of metastasis and prostate-specific antigen levels at diagnosis in Gleason 3+4 versus 4+3 prostate cancer. Urol. Ann. 2018, 10, 203–208. [Google Scholar] [CrossRef]
- Bjurlin, M.A.; Carter, H.B.; Schellhammer, P.; Cookson, M.S.; Gomella, L.G.; Troyer, D.; Wheeler, T.M.; Schlossberg, S.; Penson, D.F.; Taneja, S.S. Optimization of initial prostate biopsy in clinical practice: Sampling, labelling and specimen processing. J. Urol. 2013, 189, 2039–2046. [Google Scholar] [CrossRef] [Green Version]
- Mottet, N.; Van den Bergh, R.C.N.; Briers, E.; Cornford, P.; De Santis, M.; Fanti, S.; Gillessen, J.; Grummet, A.M.; Henry, T.B.; Lam, M.D.; et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer. Available online: https://uroweb.org/wp-content/uploads/EAU-EANM-ESUR-ESTRO-SIOG-Guidelines-on-Prostate-Cancer-2019-1.pdf (accessed on 26 February 2021).
- Wilt, T.J.; Brawer, M.K.; Jones, K.M.; Barry, M.J.; Aronson, W.J.; Fox, S.; Gingrich, J.R.; Wei, J.T.; Gilhooly, P.; Grob, B.M. Radical prostatectomy versus observation for localized prostate cancer. N. Engl. J. Med. 2012, 367, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bill-Axelson, A.; Holmberg, L.; Garmo, H.; Rider, J.R.; Taari, K.; Busch, C.; Nordling, S.; Haggman, M.; Andersson, S.O.; Spangberg, A. Radical prostatectomy or watchful waiting in early prostate cancer. N. Engl. J. Med. 2014, 370, 932–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Violette, P.D.; Agoritsas, T.; Alexander, P.; Piikonen, J.; Santti, H.; Agarwal, A.; Bhatnagar, N.; Dahm, P.; Montori, V.; Guyatt, G.H.; et al. Decision aids for localized prostate cancer treatment choice: Systematic review and meta-analysis. CA Cancer J. Clin. 2015, 65, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Haglind, E.; Carlsson, S.; Strane, J.; Wallerstedt, A.; Wilderäng, U.; Thorsteindottie, T.; Lagerkvist, M.; Damber, J.E.; Bjartell, A.; Hugosson, J.; et al. Urinary incontinence and erectile dysfunction after robotic versus open radical prostatectomy: A prospective, controlled, nonrandomised trial. Eur. Urol. 2015, 68, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Minke, H.; Yaohui, L.; Zhuoyi, X.; Li-an, S.; Yanjun, Z.; Xiaoyi, H.; Jianming, G.; Hang, W. Short internval of biopsy to robotic-assisted laparoscopic radical prostatectomy does not render any adverse effects on the perioperative outcomes. Medicine 2018, 97, e11686. [Google Scholar]
- Cao, L.; Yang, Z.; Qi, L.; Chen, M. Robot-assisted and laparoscopic vs. open radical prostatectomy in clinically localized prostate cancer perioperative, function and oncological outcomes: A systematic review and meta-analysis. Medicine 2019, 98, e15770. [Google Scholar]
- Gao, L.; Yang, L.; Qian, S.; Tang, Z.; Qin, F.; Wei, Q.; Han, P.; Yuan, J. Cryosurgery would be an effect option for clinically localized prostate cancer: A meta-analysis and systematic review. Sci. Rep. 2016, 7, 27490. [Google Scholar] [CrossRef] [Green Version]
- Bonekamp, D.; Wolf, M.B.; Roethke, M.C.; Pahernik, S.; Hadaschik, B.A.; Hatiboglu, G.; Kuru, T.H.; Popeneciu, I.V.; Chin, J.L.; Billia, M.; et al. Tweleve-month prostate volume reduction after MRI-guided transurethral ultrasound ablation of the prostate. Eur. Radiol. 2019, 29, 299–308. [Google Scholar] [CrossRef]
- Van den Bos, W.; Scheltema, M.J.; Siriwardana, A.R.; Kalsbeek, A.M.F.; Thompson, J.E.; Ting, F.; Böhm, M.; Haynes, A.; Shnier, R.; Delprado, W.; et al. Focal irreversible electroporation as primary treatment for localized prostate cancer. BJU Int. 2018, 121, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Gill, I.S.; Azzouzi, A.R.; Emberton, M.; Coleman, J.A.; Coeytaux, E.; Scherz, A.; Scardino, P.T.; PCM301 Study, Group. Randomized trial of partial gland ablation with vascular targeted phototherapy versus active surveillance for low risk prostate cancer: Extended followup and analyses of effectiveness. J. Urol. 2018, 200, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer: I. the effect of castration, of estrogen, and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941, 1, 293–297. [Google Scholar]
- Crawford, E.D.; Heidenreich, A.; Lawrentschuk, N.; Tombal, B.; Pompeo, A.C.L.; Mendoza-Valdes, A.; Miller, K.; Debruyne, F.M.J.; Klotz, L. Androgen-targeted therapy in men with prostate cancer: Evolving practice and future considerations. Prosate Cancer Prostatic Dis. 2019, 22, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate-resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef]
- Mehtälä, J.; Zong, J.; Vassilev, Z.; Brobert, G.; Gabarró, M.S.; Stattin, P.; Khanfir, H. Overall survival and second primary malignancies in men with metastatic prostate cancer. PLoS ONE 2020, 15, e0227552. [Google Scholar] [CrossRef] [Green Version]
- Moreira, D.M.; Howard, L.E.; Sourbeer, K.N.; Amarasekara, H.S.; Chow, L.C.; Cockrell, D.C.; Pratson, C.L.; Hanyok, B.T.; Aronson, W.J.; Kane, C.J.; et al. Predicting time from metastasis to overall survival in castration-resistant prostate cancer: Results from SEARCH. Clin. Gent. Cancer 2017, 15, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; Oudard, S.; Ozguroglu, M.; Hansen, S.; Machiels, J.P.; Kocak, I.; Gravis, G.; Bodrogi, I.; Mackenzie, M.J.; Roessner, M.; et al. Prenisone plus cabazitaxel or mitoxanone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: A randomized open-label trial. Lancet 2010, 376, 1147–1154. [Google Scholar] [CrossRef]
- Parker, C.; Nilsson, S.; Heinrich, D.; Helle, S.I.; O’Sullivan, J.M.; Fosså, S.D.; Chodacki, A.; Wiechno, P.; Logue, J.; Seke, M. Alpha emitter radium-223 and survival in metastatic prostate cancer. N. Engl. J. Med. 2013, 369, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Caffo, O.; Wissing, M.; Bianchini, D.; Bergman, A.; Thomsen, F.B.; Schmid, S.; Yu, E.Y.; Bournakis, E.; Sella, A.; Zagonel, V.; et al. Survival outcomes from a cumulative analysis of worldwide observational studies on sequential use of new agents in metastatic castration-resistant prostate cancer. Clin. Gent. Cancer 2019, 18, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Lombard, A.P.; Liu, L.; Cucchiara, V.; Liu, C.; Armstrong, C.M.; Zhao, R.; Yang, J.C.; Evans, C.P.; Gao, A.C. Intra vs. inter cross resistance determines treatment sequence between taxane and AR-targeting therapies in advanced prostate cancer. Mol. Cancer Ther. 2018, 17, 2197–2205. [Google Scholar] [CrossRef] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.; Cowey, L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, K.; et al. Pembrolizumab for the treatment of non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Frontera, O.A.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, A.; Nanda, R. Immune checkpoint blockade for breast cancer. Cancer Treat. Res. 2018, 173, 155–165. [Google Scholar] [PubMed]
- Beer, T.M.; Kwon, E.D.; Drake, C.G.; Fizazi, K.; Logothetis, C.; Gravis, G.; Ganju, V.; Polikoff, J.; Saad, F.; Humanski, P.; et al. Randomized, double-blind, Phase II trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naïve castration-resistant prostate cancer. J. Clin. Oncol. 2017, 35, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kwon, E.D.; Drake, C.G.; Scher, H.I.; Fizazi, K.; Bossi, A.; Van den Eertwegh, A.J.; Krainer, M.; Houede, N.; Santos, R.; Mahammedi, H.; et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): A multicentre, randomised, double-blind, Phase 3 trial. Lancet Oncol. 2014, 15, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Hodi, S.H.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Cavajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Boudadi, K.; Suzman, D.L.; Luber, B.; Wang, H.; Silberstein, J.; Sullivan, R.; Dowling, D.; Harb, R.; Nirschl, T.; Dittamore, R.V.; et al. Phase 2 biomarker-driven study of ipilimumab plus nivolumab (Ipi/Nivo) for ARV7-positive metastatic castrate-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2017, 35, 5035. [Google Scholar] [CrossRef]
- Karzai, F.; VanderWeele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L. Activity of durvalumab plus Olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations. J. Immunother. Cancer 2018, 6. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: Multicohort, open label Phase II KEYNOTE-199 study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef]
- Sonpavde, G.; McMannis, J.D.; Bai, Y.; Seethammangari, M.R.; Bull, J.M.V.; Hawkins, V.; Dancsak, T.K.; Lapteva, N.; Levitt, J.M.; Moseley, A.; et al. Phase I trial of antigen-targeted autologous dendritic cell-based vaccine with in vivo activation of inducible CD40 for advanced prostate cancer. Cancer Immunol. Immunother. 2017, 66, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Lilleby, W.; Gaudernack, G.; Brunsvig, P.F.; Vlatkovic, L.; Schulz, M.; Mills, K.; Hole, K.H.; Inderberg, E.M. Phase I/IIa clinical trial of novel hTERT peptide vaccine in men with metastatic hormone-naïve prostate cancer. Cancer Immunol. Immunother. 2017, 66, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Arai, G.; Egawa, S.; Ohyama, C.; Naito, S.; Matsumoto, K.; Uemura, H.; Nakagawa, M.; Nasu, Y.; Eto, M.; et al. Mixed 20-peptide cancer vaccine in combination with docetaxel and dexamethasone for castration-resistant prostate cancer: A randomized phase II trial. Cancer Immunol. Immunother. 2020, 69, 847–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeel, D.G.; Eickhoff, J.C.; Johnson, L.E.; Roth, A.R.; Perk, T.G.; Fong, L.; Antonarakis, E.S.; Wargowski, E.; Jarej, R.; Liu, G. Phase II trial of a DNA vaccine encoding prostatic acid phosphatase (pTVG-HP [MVI-816]) in patients with progressive, nonmetastatic, castration-sensitive prostate cancer. J. Clin. Oncol. 2019, 37, 3507–3517. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T cell dysfunction and exhaustion in cancer. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy in castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Wang, F.; Li, H.; Ji, J.; Cao, Z.; Lyu, J.; Shi, X.; Zhu, Y.; Zhang, C.; Guo, F.; et al. Prostatic acid phosphatase (PAP) predicts prostate cancer progress in a population-based study: The renewal of PAP? Dis. Markers 2019, 2019, 7090545. [Google Scholar] [CrossRef]
- Fong, L.; Carroll, P.; Weinberg, V.; Chan, S.; Lewis, J.; Corman, J.; Amling, C.L.; Stephenson, R.A.; Simko, J.; Sheikh, N.A.; et al. Activated lymphocyte recruitment into the tumor microenvironment following preoperative sipuleucel-T for localized prostate cancer. J. Natl. Cancer Inst. 2014, 10611. [Google Scholar] [CrossRef]
- Hagihara, K.; Chan, S.; Zhang, L.; Oh, D.Y.; Wei, X.X.; Simko, J.; Fong, L. Neoadjuvant sipuleucel-T induces both Th1 activation and immune regulation in localized prostate cancer. Oncoimmunology 2019, 8, e1486953. [Google Scholar] [CrossRef]
- Sheikh, N.; Cham, J.; Zhang, L.; DeVries, T.; Letarte, S.; Pufnock, J.; Hamm, D.; Trager, J.; Fong, L. Clonotypic diversification of intratumoral T cells following sipuleucel-T treatment in prostate cancer subjects. Cancer Res. 2016, 76, 3711–3718. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Kibel, A.S.; Yu, E.Y.; Karsh, L.I.; Elfiky, A.; Shore, N.D.; Vozelgang, N.J.; Corman, J.M.; Millard, F.E.; Maher, J.C.; et al. Sequencing of sipuleucel-T and androgen deprivation therapy in men with hormone-sensitive biochemically recurrent prostate cancer: A phase II randomized trial. Clin. Cancer Res. 2017, 23, 2451–2459. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, N.A.; Patrylak, D.; Kantoff, P.W.; Dela Rosa, C.; Stewart, F.P.; Kuan, L.; Whitmore, J.B.; Trager, J.B.; Poehlein, C.H.; Frohlich, M.W.; et al. Sipuleucel-T immune parameters correlate with survival: An analysis of the randomized phase 3 clinical trials in men with castration-resistant prostate cancer. Cancer Immunol. Immunother. 2013, 62, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, E.J.; Reese, D.M.; Whisenant, D.; Dixon, S.C.; Figg, W.D. Therapy of advanced prostate cancer with granulocyte macrophage colony-stimulating factor. Clin. Cancer. Res. 1999, 5, 1738–1744. [Google Scholar] [PubMed]
- Small, E.J.; Fratesi, P.; Reese, D.M. Immunotherapy for hormone-refractory prostate cancer with antigen-loaded dendritic cells. J. Clin. Oncol. 2000, 18, 3894–3903. [Google Scholar] [CrossRef] [PubMed]
- Burch, P.A.; Breen, J.K.; Buckner, J.C.; Gastineau, D.A.; Kaur, J.A.; Laus, R.L.; Padley, D.J.; Peshwa, M.V.; Pitot, H.C.; Richardson, R.L.; et al. Priming tissue-specific cellular immunity in a phase I trial of autologous dendritic cells for prostate cancer. Clin. Cancer Res. 2000, 6, 2175–2182. [Google Scholar]
- Antonarakis, E.S.; Small, E.J.; Petrylak, D.P.; Quinn, D.I.; Kibel, A.S.; Chang, N.N.; Dearstyne, E.; Harmon, M.; Campogan, D.; Haynes, H.; et al. Antigen-specific CD8 lytic phenotype induced by sipuleucel-T in hormone-sensitive or castration-resistant prostate cancer and association with overall survival. Clin. Cancer Res. 2018, 24, 4662–4671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saif, J.M.; Vadakekolathu, J.; Rane, S.S.; McDonald, D.; Ahmad, M.; Mathieu, M.; Pockley, A.G.; Durrent, L.; Metheringham, R.; Rees, R.C.; et al. Novel prostate acid phosphatase-based peptide vaccination strategy induces antigen-specific T-cell responses and limits tumour growth in mice. Eur. J. Immunol. 2014, 44, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Garzala, F.F.; McCabe, A.; Santos, E.J.; Jones, J.; Takeshita, L.Y.; Ortega-Rivera, N.D.; Del Cid-Pavon, G.M.; Ramsbottom, K.; Ghattaoraya, G.S.; Alfirevic, A.; et al. Allele frequency net database (AFND) 2020 update: Gold-standard data classification, open access genotype data and new query tools. Nucleic Acid Res. 2020, 48, D783–D788. [Google Scholar]
- Speetjens, F.M.; Kuppen, P.J.; Welters, M.J.; Essahsah, F.; Voet van den Brink, A.M.; Lantrua, M.G.; Valentijn, A.R.; Oostendorp, J.; Fathers, L.M.; Nijman, H.W.; et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin. Cancer Res. 2009, 15, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Vu, P.; Vadakekolathu, J.; Nicholls, H.; Christensen, D.; Durrant, L.; Pockley, A.; McArdle, S.E. Novel PAP-derived vaccine for the treatment of advanced prostate cancer. Eur. J. Cancer 2018, 92, S18. [Google Scholar] [CrossRef]
- Bonollo, F.; Thalmann, G.N.; Kruithof-de Julio, M.; Karampouna, S. The role of cancer-associated fibroblasts in prostate cancer tumorigenesis. Cancers 2020, 12, 1887. [Google Scholar] [CrossRef]
- Kreuger, T.E.; Thorek, D.J.L.; Meeker, A.K.; Issacs, J.T.; Brennen, W.N. Tumor-infiltrating mesenchymal stem cells: Drivers of the immunosuppressive tumor microenvironment in prostate cancer? Prostate 2018, 79. [Google Scholar] [CrossRef] [PubMed]
- Yen, B.L.; Yen, M.L.; Hsu, P.; Liu, K.; Wang, C.; Bai, C.; Sytwu, H. Multipotent human mesenchymal stromal cells mediate expansion of myeloid-derived suppressor cells via hepatocyte growth factor/c-met and STAT3. Stem Cell Rep. 2013, 1, 139–151. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chen, H.; Wang, L.; Wang, F.; Fang, L.; Lai, H.; Chen, H.; Lu, J.; Hung, M.; Cheng, Y.; et al. Mesenchymal stem cells tune the development of monocyte-derived dendritic cells toward a myeloid-derived suppressive phenotype through growth-regulated oncogene chemokines. J. Immunol. 2013, 190, 5065–5077. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D. I Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Maolake, A.; Izumi, K.; Shigehara, K.; Natsagdorj, A.; Iwamoto, H.; Kadamoto, S.; Takezawa, Y.; Machioka, K.; Narimotoa, K.; Namiki, M.; et al. Tumor-associated macrophages promote prostate cancer migration through activation of the CCL22-CCR4 axis. Oncotarget 2017, 8, 9739–9751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhary, B.; Elkord, E. Regulatory T cells in the tumor microenvironment and cancer progression: Role and therapeutic targeting. Vaccines 2016, 4, 28. [Google Scholar] [CrossRef] [Green Version]
- Parker, K.H.; Beury, D.W.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: Critical cells driving immune suppression in the tumor microenvironment. In Advances in Cancer Research; Wang, X., Fisher, P.B., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 128, pp. 95–127. [Google Scholar]
- Condamine, T.; Dominguez, G.A.; Youn, J.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.K.; Kortylewski, M. Breaking bad habits: Targeting MDSCs to alleviate immunosuppression in prostate cancer. Oncoimmunology 2016, 5, e1078060. [Google Scholar] [CrossRef] [Green Version]
- Jian, S.; Chen, W.; Su, Y.; Su, Y.; Chuang, T.; Hsu, S.; Huang, L. Glycolysis regulates the expansion of myeloid-derived suppressor cells in tumor-bearing hosts through prevention of ROS-mediated apoptosis. Cell Death Dis. 2017, 8, e2779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohl, K.; Fragoulis, A.; Klemm, P.; Baumeister, J.; Klock, W.; Verjans, E.; Böll, S.; Möllmann, J.; Lehrke, M.; Costa, I.; et al. Nrf2 is a central regulator of metabolic reprogramming of myeloid-derived suppressor cells in a steady state and sepsis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Kusmartsev, S.; Gabrilovich, D.I. Inhibition of myeloid cell differentiation in cancer: The role of reactive oxygen species. J. Leukoc. Biol. 2003, 74, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Vuk-Pavlović, S.; Bulur, P.A.; Lin, Y.; Qin, R.; Szumlanski, C.L.; Zhao, X.; Dietz, A.B. Immunosuppressive CD14+ HLA-DRlow/- monocytes in prostate cancer. Prostate 2010, 70, 443–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brusa, D.; Simone, M.; Gontero, P.; Spadi, R.; Racca, P.; Micari, J.; Degiuli, M.; Carletto, S.; Tizzani, A.; Matera, L. Circulating immunosuppressive cells of prostate cancer patients before and after radical prostatectomy: Profile comparison. Int. J. Urol. 2013, 20, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Idorn, M.; Køllgaard, T.; Kongsted, P.; Sengeløv, L.; Thor Straten, P. Correlation between frequencies of blood monocytic myeloid-derived suppressor cells, regulatory T cells and negative prognostic markers in patients with castration-resistant metastatic prostate cancer. Cancer Immunol. Immunother. 2014, 63, 1177–1187. [Google Scholar] [CrossRef]
- Hellsten, R.; Lilljebörn, L.; Johansson, M.; Leandersson, K.; Bjartell, A. The STAT3 inhibitor galiellalactone inhibits the generation of MDSC-like monocytes by prostate cancer cells and decreases immunosuppressive and tumorigenic factors. Prostate 2019, 79, 1611–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, N.; Tan, Z.; Ma, K.; Bao, L.; Yun, Z. Increased circulating myeloid-derived suppressor cells correlate with cancer stages, interleukin-8 and -6 in prostate cancer. Int. J. Clin. Exp. Med. 2014, 7, 3181–3192. [Google Scholar] [PubMed]
- Wang, G.; Lu, X.; Dey, P.; Deng, P.; Chin Wu, C.; Jiang, S.; Fang, Z.; Zhao, K.; Konaparthi, R.; Hua, S.; et al. Targeting YAP-dependent MDSC infiltration impairs tumor progression. Cancer Discov. 2016, 6, 80–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, D.M.S.; Pal, S.K.; Moreira, D.; Duttagupta, P.; Zhang, Q.; Won, H.; Jones, J.; D’Apuzzo, M.; Forman, S.; Kortylewski, M. TLR9-targeted STAT3 silencing abrogates immunosuppressve activity of myeloid-derived suppressor cells from prostate cancer patients. Clin. Cancer. Res. 2015, 21, 3771–3782. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Huang, G.; Liu, S.; Wan, J.; Wang, X.; Zhu, Y.; Kaliney, W.; Zhang, C.; Cheng, L.; Wen, X.; et al. Polymorphonuclear MDSCs are enriched in the stroma and expanded metastases of prostate cancer. J. Pathol. Clin. Res. 2020, 6, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Dong, H.; Kwon, E.; Karnes, J. Positive pelvic lymph nodes in prostate cancer harbour immune suppressor cells to impair tumor-reactive T cells. Eur. Urol. Focus 2018, 4, 75–79. [Google Scholar] [CrossRef]
- Movahedi, K.; Guilliams, M.; Van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; Van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef] [PubMed]
- Jachetti, E.; Cancila, V.; Rigoni, A.; Bongiovanni, L.; Cappetti, B.; Belmonte, B.; Enriquez, C.; Casalini, P.; Ostano, P.; Frossi, B.; et al. Cross-talk between myeloid-derived suppressor cells and mast cells mediates tumor-specific immunosuppression in prostate cancer. Cancer Immunol. Res. 2018, 6, 552–565. [Google Scholar] [CrossRef] [Green Version]
- Raber, P.L.; Thevenot, P.; Sierra, R.; Wyczechowska, D.; Halle, D.; Ramirez, M.E.; Ochoa, A.; Fletcher, M.; Velasco, C.; Wilk, A.; et al. Subpopulations of myeloid-derived suppressor cells (MDSC) impair T cell responses through nitric oxide-related pathways. Int. J. Cancer 2015, 134, 2853–2864. [Google Scholar] [CrossRef]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, G.; Cheng, W.A.; Dong, Z.; Sun, H.; Lee, V.Y.; Cha, S.; Smith, D.L.; Kwak, L.W.; Qin, H. Targeting myeloid-derived suppressor cells for cancer immunotherapy. Cancer Immunol. Immunother. 2018, 67, 1181–1195. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Lim, B.; Kim, S.; Sohn, H.; Kim, S.; Kim, T. GM-CSF promotes the expansion and differentiation of cord blood myeloid-derived suppressor cells, which attenuate xenogenic graft-vs.-host disease. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Tomić, S.; Joksimović, B.; Bekić, M.; Vasiljević, M.; Milanović, M.; Čolić, M.; Vučević, D. Prostaglandin-E2 potentiates the suppressive functions of human mononuclear myeloid-derived suppressor cells and increases their capacity to expand IL-10-producing regulatory T cell subsets. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Srikrishna, G. S100A8 and S100A9: New insights into their roles in malignancy. J. Innate Immun. 2012, 4, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Bujanda, Z.A.; Haffner, M.C.; Chaimowitz, M.G.; Chowdhury, N.; Venturini, N.J.; Obradovic, A.; Hansen, C.S.; Jacków, J.; Sfanos, K.S.; Bieberich, C.J.; et al. Castration-mediated IL-8 promotes myeloid infiltration and prostate cancer progression. BioRxiv 2019, 651083. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, J.R.; Mohamed, E.; Rodriguez, P.C. Unfolding anti-tumor immunity: ER stress responses sculpt tolergenic myeloid cells in cancer. J. Immunother. Cancer 2017, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.; Celis, E.; Finnberg, N.; El-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [Green Version]
- Minciacchi, V.R.; Zijlstra, A.; Rubin, M.A.; De Vizio, D. Extracellular vesicles for liquid biopsy in prostate cancer: Where are we and where are we headed? Prostate Cancer Prostatic Dis. 2017, 20, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-assosciated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [PubMed]
- Hurwitz, M.D.; Kaur, P.; Nagaraja, G.M.; Bausero, M.A.; Manola, J.; Asea, A. Radiation theraoy induces circulating serum Hsp72 in patients with prostate cancer. Radiother. Oncol. 2010, 95, 350–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Qiu, W.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Xue, H.; Li, G. Immunosuppresisve effects of hypoxia-induced glioma exosomes through myeloid-derived suppressor cells via the miR-10a/Rora and miR-21/Pten pathways. Oncogene 2018, 37, 4239–4259. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Zhang, X.; Li, W.; Feng, Q.; Feng, H.; Tong, Y.; Rong, H.; Wang, W.; Zhang, D.; Zhang, Z.; et al. Exosomal miRNA-107 induces myeloid-derived suppressor cell expansion in gastric cancer. Cancer Manag. Res. 2019, 11, 4032–4040. [Google Scholar] [CrossRef]
- Bruns, H.; Böttcher, M.; Qorraj, M.; Fabri, M.; Jitschin, S.; Dindorf, J.; Busch, L.; Jitschin, R.; Mackensen, A.; Mougiakakos, D. CLL-cell-mediated MDSC induction by exosomal miR-155 transfer is disrupted by vitamin D. Leukemia 2017, 31, 985–988. [Google Scholar] [CrossRef]
- Basso, D.; Gnatta, E.; Padoan, A.; Fogar, P.; Furlanello, S.; Aita, A.; Bozzato, D.; Zambon, C.F.; Arrigoni, G.; Frasson, C.; et al. PDAC-derived exosomes enrich the microenvironment in MDSCs in a SMAD4-dependent manner through a new calcium related axis. Oncotarget 2017, 8, 84928–84944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, L.; Mu, S.; Wang, Y.; Wang, H.; Cai, L.; Li, W.; Hu, Y. Prognostic role of myeloid-derived suppressor cells in cancers: A systematic review and meta-analysis. BMC Cancer 2018, 18, 1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Fu, X.; Li, T.; Yan, H. The prognostic value of myeloid derived suppressor cell level in hepatocellular carcinoma: A systematic review and meta-analysis. PLoS ONE 2019, 14, e0225327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirbod-Mobarakeh, A.; Mirghorbani, M.; Hajiju, F.; Marvi, M.; Bashiri, K.; Rezaei, N. Myeloid-derived suppressor cells in gastrointestinal cancers: A systematic review. J. Gastroentrol. Hepatol. 2016, 31, 1246–1256. [Google Scholar] [CrossRef]
- Chang, G.; Kang, I.; Shahabi, A.; Nadadur, M.; Athreya, K.; SuerDavid Canter, E.; Chen, M.; Martin, S.E.; Aron, M.; Groshen, S.G.; et al. MDSC clinical assay for disease surveillance in prostate cancer. J. Clin. Oncol. 2015, 33, e16091. [Google Scholar] [CrossRef]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-expressed IDO recruits and activates MDSCs in a Treg-dependent manner. Cell Rep. 2015, 13, 412–424. [Google Scholar] [CrossRef] [Green Version]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL23 secreted by myeloid cells drives castration resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Deng, Z.; Rong, Y.; Teng, Y.; Zhuang, X.; Samykutty, A.; Mu, J.; Zhang, L.; Cao, P.; Yan, J.; Miller, D.; et al. Exosome miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene 2017, 36, 639–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, I.; Hammes, S.R. Neutrophil elastase in the tumor microenvironment. Steroids 2018, 133, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Pivetta, E.; Danussi, C.; Wassermann, B.; Modica, T.M.E.; Del Bel Belluz, L.; Canzonieri, V.; Colombatti, A.; Spessotto, P. Neutrophil elastase-dependent cleavage compromises the tumor suppressor role of EMILIN1. Matrix Biol. 2014, 34, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhong, L.; Xiong, L.; Dan, W.; Li, J.; Ye, J.; Wan, P.; Luo, X.; Chu, X.; Liu, C.; et al. Neutrophil elastase-mediated proteolysis of the tumor suppressor p200 CUX1 promotes cell proliferation and inhibits cell differentiation in APL. Life Sci. 2020, 242, 117229. [Google Scholar] [CrossRef] [PubMed]
- Lerman, I.; Garcia-Hernandez, M.; Rangel-Moreno, J.; Chiriboga, L.; Pan, C.; Nastiuk, K.L.; Krolewski, J.J.; Sen, A.; Hammes, S.R. Infiltrating myeloid cells exert protumoirgenic actions via neutrophil elastase. Mol. Cancer Res. 2017, 15, 1138–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebelt, K.; Babaryka, G.; Frankenberger, B.; Stief, C.G.; Eisenmenger, W.; Kirchner, T.; Schendel, D.J.; Noessner, E. Prostate cancer lesions are surrounded by Foxp3+, PD-1+ and B7-H1+ lymphocyte clusters. Eur. J. Cancer 2009, 45, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a novel mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, S.; Schrum, A.G.; Cho, H.; Celis, E.; Gabrilovich, D.I. Mechanism of T-cell tolerance induced by myeloid-derived suppressor cells. J. Immunol. 2010, 184, 3106–3116. [Google Scholar] [CrossRef]
- Lu, T.; Ramakrishnan, R.; Altiok, S.; Youn, J.; Cheng, P.; Celis, E.; Pisarev, V.; Sherman, S.; Sporn, M.; Gabrilovich, D. Tumor-infiltrating myeloid cells induce tumor cell resistance to cytotoxic T cells in mice. J. Clin. Investig. 2011, 121, 4015–4029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, S.; Cheng, X.; Zhang, L.; Lu, X.; Chaudhary, S.; Teng, R.; Frederickson, C.; Champion, M.M.; Zhao, R.; Cheng, L.; et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 10094–10099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molon, B.; Ugel, S.; Del Pozzo, F.; Soldani, C.; Zilio, S.; Avella, D.; De Palma, A.; Mauri, P.; Monegal, A.; Rescigno, M.; et al. Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J. Exp. Med. 2011, 208, 1949–1962. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, S.K.; Bunt, S.M.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity towards a type 2 response. J. Immunol. 2007, 179, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid derived suppressor cells interactions with natural killer cells and pro-angiogenic activities: Roles in tumor progression. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Lu, C.; Redd, P.S.; Lee, J.R.; Savage, N.; Liu, K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1247135. [Google Scholar] [CrossRef] [Green Version]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Moreira, D.; Adamus, T.; Zhao, X.; Su, Y.; Zhang, Z.; White, S.V.; Swiderski, P.; Lu, P.; DePinho, R.A.; Pal, S.K.; et al. STAT3 inhibition combined with CpG immunostimulation activates antitumor immunity to eradicate genetically distinct castration-resistant prostate cancers. Clin. Cancer. Res. 2018, 24, 5948–5962. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef]
- Dufait, I.; Valckenborgh, E.V.; Menu, E.; Escors, D.; De Ridder, M.; Breckpot, K. Signal transducer and activator of transcription 3 in myeloid-derived suppressor cells: An opportunity for cancer therapy. Oncotarget 2016, 7, 42698–42715. [Google Scholar] [CrossRef] [Green Version]
- De Sanctis, F.; Solito, S.; Ugel, S.; Molon, B.; Bronte, V.; Marigo, I. MDSCs in cancer: Conceiving new prognostic and therapeutic targets. Biochim. Biophys. Acta. 2016, 1865, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Baba, T.; Hamanishi, J.; Yamaguchi, K.; Murakami, R.; Taki, M.; Ukita, M.; Hosoe, Y.; et al. Anti-VEGF therapy resistance in ovarian cancer is caused by GM-CSF-induced myeloid derived suppressor cell recruitment. Br. J. Cancer 2020, 122, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Highfill, S.L.; Cui, Y.; Giles, A.J.; Smith, J.P.; Zhang, H.; Morse, E.; Kaplan, R.N.; Mackall, C.L. Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD-1 efficacy. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting myeloid-drived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4, e128653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nefedova, Y.; Fishman, M.; Sherman, S.; Wang, X.; Beg, A.A.; Gabrilovich, D.I. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007, 67, 11021–11028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Yi, H.; Li, J. Response to: ‘issues with anti-Gr1 antibody-mediated myeloid-derived suppressor cell depletion’ by Xing et al. Ann. Rheum. Dis. 2016, 75, e50. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, G.A.; Condamine, T.; Mony, S.; Hashimoto, A.; Wang, F.; Liu, Q.; Forero, A.; Bendell, J.; Witt, R.; Hockstein, N.; et al. Selective targeting of myeloid-derived suppressor cells in cancer patients using DS-8273a, an agonistic TRAIL-R2 antibody. Clin. Cancer Res. 2017, 23, 2942–2950. [Google Scholar] [CrossRef] [Green Version]
- Hellsten, R.; Johansson, M.; Dahlman, A.; Sterner, O.; Bjartell, A. Galiellalactone inhibits stem cell-like ALDH-positive prostate cancer cells. PLoS ONE 2011, 6, e22118. [Google Scholar] [CrossRef] [Green Version]
- Siegel, J.I.; Heimall, J.; Freeman, A.F.; Hsu, A.P.; Brittain, E.; Brenchley, J.M.; Douek, D.C.; Fahle, G.H.; Cohen, J.I.; Holland, S.M.; et al. A critical role for STAT3 transcription factor signalling in the development and maintenance of human T cell memory. Immunity 2011, 35, 806–818. [Google Scholar] [CrossRef] [Green Version]
- Cui, W.; Liu, Y.; Weinstein, J.S.; Craft, J.; Kaech, S.M. An interleukin-21-interleukin-10-STAT3 pathway is critical for functional maturation of memory CD8+ T cells. Immunity 2011, 35, 792–805. [Google Scholar] [CrossRef] [Green Version]
- Manogue, C.; Cotogno, P.; Moses, M.M.; Barata, P.C.; Layton, J.L.; Sartor, A.O.; Lewis, B.E. Continuous infusion 5-fluorouracil (5FU) as a novel treatment for heavily pretreated prostate cancer patients: An update. J. Clin. Oncol. 2019, 37, 319. [Google Scholar] [CrossRef]
- Kuzel, T.M.; Tallman, M.S.; Shervin, D.; Braud, E.; Kilton, L.; Johnson, P.; Kozlowski, J.; Vogelzang, N.J.; Blough, R.; Benson, A.B. A phase II study of continuous infusion 5-fluorouracil in advanced hormone refactory prostate cancer. An Illinois cancer centre study. Cancer 1993, 72, 1965–1968. [Google Scholar] [CrossRef]
- Mehta, A.R.; Armstrong, A.J. Tasquinimod in the treatment of castrate-resistant prostate cancer-current status and future prospects. Ther. Adv. Urol. 2016, 8, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rébé, C.; Ghiringhelli, F. 5-fluorouracil selectively kills tumor-assosciated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [Green Version]
- Fultang, L.; Panetti, S.; Ng, M.; Collins, P.; Graef, S.; Rizkalla, N.; Booth, S.; Lenton, R.; Noyvert, B.; Shannon-Lowe, C.; et al. MDSC targeting with gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. Lancet 2019, 47, P235–P246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karkera, J.; Steiner, H.; Li, W.; Skradski, V.; Moser, P.L.; Riethdorf, S.; Reddy, M.; Puchalski, T.; Safer, K.; Prabhakar, U.; et al. The anti-interleukin-6 antibody siltuximab down-regulates genes implicated in tumorigenesis in prostate cancer patients from a phase I study. Prostate 2011, 71, 1455–1465. [Google Scholar] [CrossRef] [PubMed]
- Dorff, T.B.; Goldman, B.; Pinski, J.K.; Mack, P.C.; Lara, P.N., Jr.; Van Veldhuizen, P.J., Jr.; Quinn, D.I.; Vogelzang, N.J.; Thompson, I.M., Jr.; Hussain, M.H.A. Clinical and correlative results of SWOG S0354: A phase II trials of CNT0328 (Siltuximab), a monoclonal antibody against interleukin-6, in chemotherapy-pretreated patients with castration-resistant prostate cancer. Clin. Cancer Res. 2010, 16, 3028–3934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fizazi, K.; De Bono, J.S.; Flechon, A.; Heidenreich, A.; Voog, E.; Davis, N.B.; Qi, M.; Bandekar, R.; Vermeulen, J.T.; Cornfeld, M.; et al. Randomised phase II study of siltuximab (CNTO 328), an anti-IL-6 monoclonal antibody, in combination with mitoxantrone/prednisone versus mitoxantrone/prednisone alone in metastatic castration-resistant prostate cancer. Eur. J. Cancer 2012, 48, 85–93. [Google Scholar] [CrossRef]
- Liss, M.A.; Rickborn, L.; DiGiovanni, J.; Bacich, D.; DeGraffenried, L.A.; Parihar, M.; Thompson, I.M.; Sharp, Z.D. mTOR inhibitors for treatment of low-risk prostate cancer. Med. Hypotheses 2018, 117, 63–68. [Google Scholar] [CrossRef]
- Gu, L.; Liao, Z.; Hoang, D.T.; Dagvadorj, A.; Gupta, S.; Blackmon, S.; Ellsworth, E.; Talati, P.; Leiby, B.; Zinda, M.; et al. Pharmacologic inhibition of Jak2-Stat5 signaling by Jak2 inhibitor AZD1480 potently suppresses growth of both primary and castrate-resistant prostate cancer. Clin Cancer Res. 2013, 19, 5658–5674. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Talati, O.; Vogiatzi, P.; Romero-Weaver, A.L.; Abdulghani, J.; Liao, Z.; Leiby, B.; Hoang, D.T.; Mirtti, T.; Alanen, K.; et al. Pharmacologic suppression of JAK1/2 by JAK1/2 inhibitor AZD1480 potently inhibits IL-6-induced experimental prostate cancer metastases formation. Mol. Cancer Ther. 2014, 13, 1246–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, W.; Choo, D.W.; Wu, Y.J.; Chan, T.F.; Lin, Z.F. Statins use and the risk of prostate cancer in ischemic heart disease patients in Taiwan. Clin. Pharmacol. Ther. 2019, 106, 458–466. [Google Scholar] [CrossRef]
- Van Rompay, M.I.; Solomon, K.R.; Nickel, J.C.; Ranganathan, G.; Kantoff, P.W.; McKinlay, J.B. Prostate cancer incidence and mortality among men using statins and non-statin lipid-lowering medications. Eur. J. Cancer 2019, 112, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommagio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Babcook, M.A.; Sramkoski, R.M.; Fujioka, H.; Daneshgari, F.; Almasan, A.; Shukla, S.; Nanavaty, R.R.; Gupta, S. Combination simvastatin and metforming induces G1-phase cell cycle arrest and Ripk1- and Ripk3-dependent necrosis in C4-2B osseous metastatic castration-resistant prostate cancer cells. Cell Death Dis. 2014, 5, e1536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, S.; Wang, C.; Chen, F.; Yu, C.; Hong, J.; Chiang, C. Sunitinib treatment-elicited distinct tumor microenvironment dramatically compensated the reduction of myeloid-derived suppressor cells. In Vivo 2020, 34, 1141–1152. [Google Scholar] [CrossRef]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef] [Green Version]
- Jayaprakash, P.; Ai, M.; Liu, A.; Budhani, P.; Bartkowiak, T.; Sheng, J.; Ager, C.; Nicholas, C.; Jaiswal, A.R.; Shah, K.; et al. Targeted hypoxia reduction restores T cell infiltration and sensitizes prostate cancer to immunotherapy. J. Clin. Investig. 2018, 128, 5137–5149. [Google Scholar] [CrossRef] [PubMed]
- Redman, J.M.; Steinberg, S.M.; Gulley, J.L. Quick efficacy seeking trial (QuEST1): A novel combination immunotherapy study designed for rapid clinical signal assessment metastatic castration-resistant prostate cancer. J. Immunother. Cancer 2018, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, D.H.; David, J.M.; Dominguez, C.; Palena, C. Development of cancer vaccines targeting brachyury, a transcription factor associated with EMT. Cells Tissues Organs 2017, 203, 128–138. [Google Scholar] [CrossRef] [Green Version]
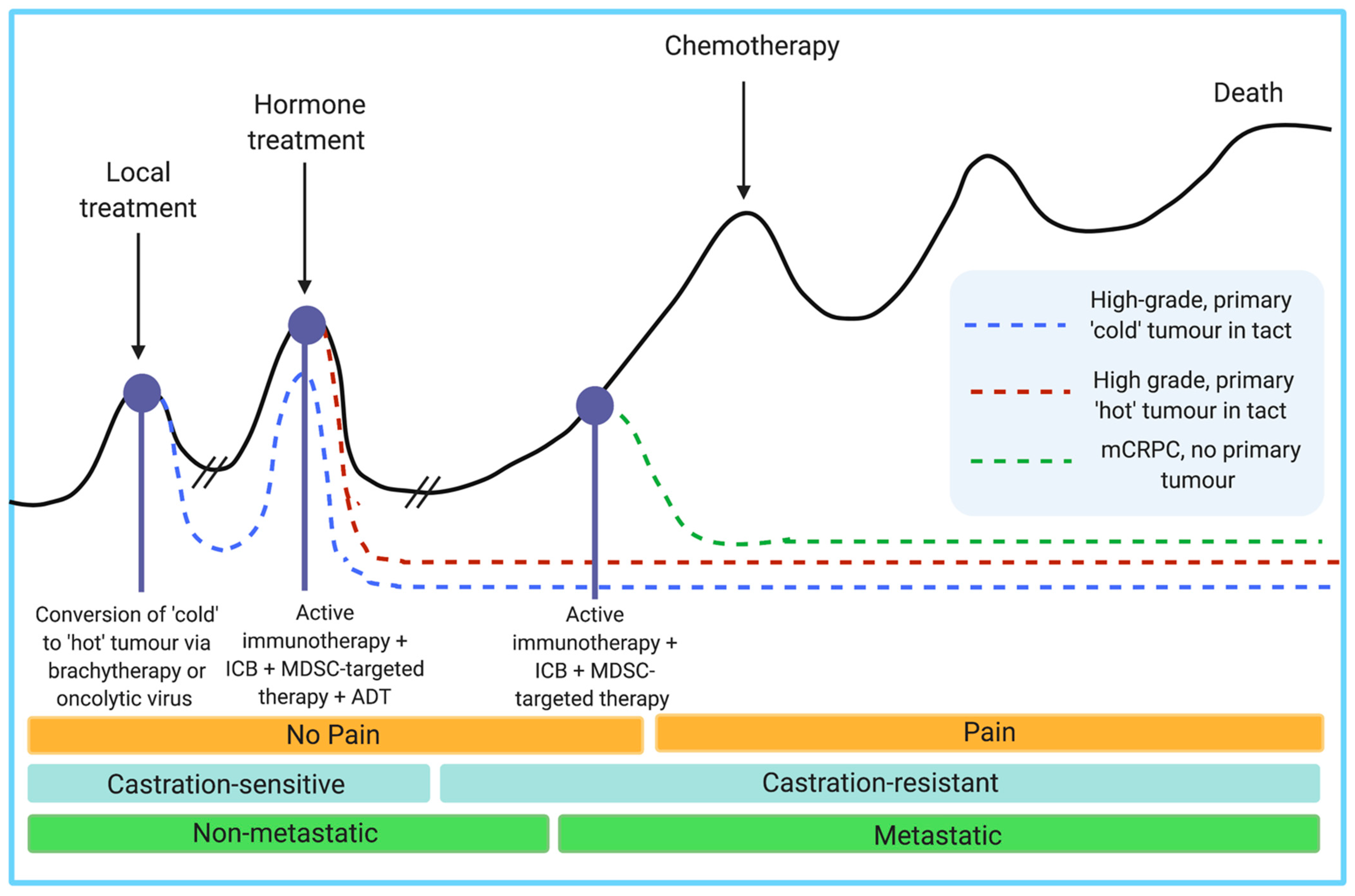
- Keam, S.P.; Halse, H.; Nguyen, T.; Wang, M.; Van Klooten Losio, N.; Mitchell, C.; Caramia, F.; Bryne, D.J.; Haupt, S.; Ryland, G.; et al. High dose-rate brachytherapy of localized prostate cancer converts tumors from cold to hot. J. Immunother. Cancer 2020, 8, e000792. [Google Scholar]
- Lee, P.; Gujar, S. Potentiating prostate cancer immunotherapy with oncolytic viruses. Nat. Rev. Urol. 2018, 15, 235–250. [Google Scholar] [CrossRef]
- Davies, A.H.; Beltran, H.; Zoubeidi, A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat. Rev. Urol. 2018, 15, 271–286. [Google Scholar] [CrossRef]
- Lin, P.; Liu, J.; Hsu, R.; Chuang, H.; Chang, S.; Pang, S.; Chang, Y.; Chuang, C.; Lin, S. Depression negatively impacts survival of patients with metastatic prostate cancer. Int. J. Environ. Res. Public Health 2018, 15, 2148. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.; Karpova, Y.; Baiz, D.; Yancey, D.; Pullikuth, A.; Flores, A.; Register, T.; Cline, J.M.; D’Agostino, R.; Danial, N.; et al. Behavioural stress accelerates prostate cancer development in mice. J. Clin. Investig. 2013, 123, 874–886. [Google Scholar] [PubMed] [Green Version]
- Cheng, Y.; Tang, X.; Li, Y.; Zhao, D.; Cao, Q.; Wu, H.; Yang, H.; Hao, K.; Yang, Y. Depression-induced neuropeptide Y secretion promotes prostate cancer growth by recruiting myeloid cells. Clin. Cancer Res. 2019, 25, 2621–2632. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, W. Beta-Adrenergic Signaling on Neuroendocrine Differentiation, Angiogenesis, and Metastasis in Prostate Cancer Progression. Asian J. Androl. 2019. [Google Scholar] [CrossRef]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Taskén, K.A. Î2-Adrenergic Receptor Signaling in Prostate Cancer. Front. Oncol. 2015, 4. [Google Scholar] [CrossRef] [Green Version]
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Urbanucci, A.; Nielsen, H.K.; Guldvik, I.J.; Engedal, A.; Ketola, K.; Wang, W.; Svindland, A.; et al. The Β2-Adrenergic Receptor Is a Molecular Switch for Neuroendocrine Transdifferentiation of Prostate Cancer Cells. Mol. Cancer Res. 2019, 17, 2154–2168. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liu, X.; Guo, F.; Tan, S.; Wang, G.; Liu, H.; Wang, J.; He, X.; Mo, Y.; Shi, B. Impact of beta-blockers on prostate cancer mortality: A meta-analysis of 16,825 patients. Onco Targets Ther. 2015, 8, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Ventura, S.; Evans, B.A. Does the Autonomic Nervous System Contribute to the Initiation and Progression of Prostate Cancer? Asian J. Androl. 2013, 15, 715–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulik, G. ADRB2-Targeting Therapies for Prostate Cancer. Cancers 2019, 11, 358. [Google Scholar] [CrossRef] [Green Version]
- Nagmani, R.; Pasco, D.S.; Salas, R.D.; Feller, D.R. Evaluation of β-Adrenergic Receptor Subtypes in the Human Prostate Cancer Cell Line-LNCaP. Biochem. Pharmacol. 2003, 65, 1489–1494. [Google Scholar] [CrossRef]
- Magnon, C.; Hall, S.J.; Lin, J.; Xue, X.; Gerber, L.; Freedland, S.J.; Frenette, P.S. Autonomic nerve development contributes to prostate cancer progression. Science 2013, 341, 1236361. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.E.; Deeble, P.D.; Lakhani, S.; Parsons, S.J. Acquisition of Neuroendocrine Characteristics by Prostate Tumor Cells Is Reversible: Implications for Prostate Cancer Progression. Cancer Res. 1999, 59, 3821–3830. [Google Scholar] [PubMed]
- Kwon, D.H.; Zhang, L.; Quigley, D.A.; Foye, A.; Chen, W.S.; Wong, C.K.; Feng, F.Y.; Bailey, A.; Huang, J.; Stuart, J.M.; et al. Down-Regulation of ADRB2 Expression Is Associated with Small Cell Neuroendocrine Prostate Cancer and Adverse Clinical Outcomes in Castration-Resistant Prostate Cancer. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 931-e9. [Google Scholar] [CrossRef]
- Kaushik, D.; Vashistha, V.; Isharwal, S.; Sediqe, S.A.; Lin, M.F. Histone Deacetylase Inhibitors in Castration-Resistant Prostate Cancer: Molecular Mechanism of Action and Recent Clinical Trials. Ther. Adv. Urol. 2015, 7, 388–395. [Google Scholar] [CrossRef] [Green Version]
- Shabbeer, S.; Kortenhorst, M.S.Q.; Kachhap, S.; Galloway, N.; Rodriguez, R.; Carducci, M.A. Multiple Molecular Pathways Explain the Anti-Proliferative Effect of Valproic Acid on Prostate Cancer Cells in Vitro and in Vivo. Prostate 2007. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Shen, L.; Ciesielski, M.; Ramakrishnan, S.; Miles, K.M.; Ellis, L.; Sotomayor, P.; Shrikant, P.; Fenstermaker, R.; Pili, R. Class I Histone Deacetylase Inhibitor Entinostat Suppresses Regulatory T Cells and Enhances Immunotherapies in Renal and Prostate Cancer Models. PLoS ONE 2012, 7, e30815. [Google Scholar] [CrossRef]
- Jung-Hynes, B.; Nihal, M.; Zhong, W.; Ahmad, N. Role of Sirtuin Histone Deacetylase SIRT1 in Prostate Cancer: A Target for Prostate Cancer Management via Its Inhibition? J. Biol. Chem. 2009, 284, 3823–3832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.J.; Demierre, M.F.; Kim, E.J.; Rook, A.H.; Lerner, A.; Duvic, M.; Scarisbrick, J.; Reddy, S.; Robak, T.; Becker, J.C.; et al. Final results from a multicenter, international, pivotal study of romidepsin in refractory cutaneous T-cell lymphoma. J. Clin. Oncol. 2010, 28, 4485–4491. [Google Scholar] [CrossRef]
- Raedler, L.A. Farydak (Panobinostat): First HDAC Inhibitor Approved for Patients with Relapsed Multiple Myeloma. Am. Health Drug Benefits 2016, 9, 84–87. [Google Scholar]
- Poole, R.M. Belinostat: First global approval. Drugs 2014, 74, 1543–1554. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weichert, W.; Röske, A.; Gekeler, V.; Beckers, T.; Stephan, C.; Jung, K.; Fritzsche, F.R.; Niesporek, S.; Denkert, C.; Dietel, M.; et al. Histone Deacetylases 1, 2 and 3 Are Highly Expressed in Prostate Cancer and HDAC2 Expression Is Associated with Shorter PSA Relapse Time after Radical Prostatectomy. Br. J. Cancer 2008. [Google Scholar] [CrossRef] [Green Version]
- De, U.; Kundu, S.; Patra, N.; Ahn, M.Y.; Ahn, J.H.; Son, J.Y.; Yoon, J.H.; Moon, H.R.; Lee, B.M.; Kim, H.S. A New Histone Deacetylase Inhibitor, MHY219, Inhibits the Migration of Human Prostate Cancer Cells via HDAC1. Biomol. Ther. 2015, 23, 434–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, L.M.; Agus, D.B.; Scher, H.I.; Higgins, B.; Rose, A.; Cordon-Cardo, C.; Thaler, H.T.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. Suberoylanilide Hydroxamic Acid, an Inhibitor of Histone Deacetylase, Suppresses the Growth of Prostate Cancer Cells in Vitro and in Vivo. Cancer Res. 2000, 60, 5165–5170. [Google Scholar]
- Hwang, J.J.; Kim, Y.S.; Kim, M.J.; Kim, D.E.; Jeong, I.G.; Kim, C.S. Histone Deacetylase Inhibitor Potentiates Anticancer Effect of Docetaxel via Modulation of Bcl-2 Family Proteins and Tubulin in Hormone Refractory Prostate Cancer Cells. J. Urol. 2010. [Google Scholar] [CrossRef]
- Chen, L.; Meng, S.; Wang, H.; Bali, P.; Bai, W.; Li, B.; Atadja, P.; Bhalla, K.N.; Wu, J. Chemical Ablation of Androgen Receptor in Prostate Cancer Cells by the Histone Deacetylase Inhibitor LAQ824. Mol. Cancer Ther. 2005, 4, 1311–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; Kristeleit, R.; Tolcher, A.; Fong, P.; Pacey, S.; Karavasilis, V.; Mita, M.; Shaw, H.; Workman, P.; Kaye, S.; et al. Phase I Pharmacokinetic and Pharmacodynamic Study of LAQ824, a Hydroxamate Histone Deacetylase Inhibitor with a Heat Shock Protein-90 Inhibitory Profile, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2008, 14, 6663–6673. [Google Scholar] [CrossRef] [Green Version]
- Bradley, D.; Rathkopf, D.; Dunn, R.; Stadler, W.M.; Liu, G.; Smith, D.C.; Pili, R.; Zwiebel, J.; Scher, H.; Hussain, M. Vorinostat in Advanced Prostate Cancer Patients Progressing on Prior Chemotherapy (National Cancer Institute Trial 6862): Trial Results and Interleukin-6 Analysis: A Study by the Department of Defense Prostate Cancer Clinical Trial Consortium and Universi. Cancer 2009. [Google Scholar] [CrossRef] [Green Version]
- Patra, N.; De, U.; Kim, T.H.; Lee, Y.J.; Ahn, M.Y.; Kim, N.D.; Yoon, J.H.; Choi, W.S.; Moon, H.R.; Lee, B.M.; et al. A Novel Histone Deacetylase (HDAC) Inhibitor MHY219 Induces Apoptosis via up-Regulation of Androgen Receptor Expression in Human Prostate Cancer Cells. Biomed. Pharmacother. 2013. [Google Scholar] [CrossRef] [PubMed]
- Bruzzese, F.; Pucci, B.; Milone, M.R.; Ciardiello, C.; Franco, R.; Chianese, M.I.; Rocco, M.; Di Gennaro, E.; Leone, A.; Luciano, A.; et al. Panobinostat Synergizes with Zoledronic Acid in Prostate Cancer and Multiple Myeloma Models by Increasing ROS and Modulating Mevalonate and P38-MAPK Pathways. Cell Death Dis. 2013, 4, e878-10. [Google Scholar] [CrossRef]
- Rathkopf, D.; Wong, B.Y.; Ross, R.W.; Anand, A.; Tanaka, E.; Woo, M.M.; Hu, J.; Dzik-Jurasz, A.; Yang, W.; Scher, H.I. A Phase I Study of Oral Panobinostat Alone and in Combination with Docetaxel in Patients with Castration-Resistant Prostate Cancer. Cancer Chemother. Pharmacol. 2010, 66, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.E.; Picus, J.; Hussain, A.; Ellard, S.; Chi, K.N.; Nydam, T.; Allen-Freda, E.; Mishra, K.K.; Porro, M.G.; Scher, H.I.; et al. A Phase 2 Study of Intravenous Panobinostat in Patients with Castration-Resistant Prostate Cancer. Cancer Chemother. Pharmacol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Park, S.E.; Kim, H.G.; Kim, D.E.; Jung, Y.J.; Kim, Y.; Jeong, S.Y.; Choi, E.K.; Hwang, J.J.; Kim, C.S. Combination Treatment with Docetaxel and Histone Deacetylase Inhibitors Downregulates Androgen Receptor Signaling in Castration-Resistant Prostate Cancer. Investig. New Drugs 2018, 36, 195–205. [Google Scholar] [CrossRef]
- Lee, J.E.; Kim, J.H. Valproic Acid Inhibits the Invasion of PC3 Prostate Cancer Cells by Upregulating the Metastasis Suppressor Protein NDRG1. Genet. Mol. Biol. 2015, 38, 527–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, K.L.; Gearing, L.J.; Zanker, D.J.; Brockwell, N.K.; Khoo, W.H.; Roden, D.L.; Cmero, M.; Mangiola, S.; Hong, M.K.; Spurling, A.J.; et al. Prostate Cancer Cell-intrinsic Interferon Signaling Regulates Dormancy and Metastatic Outgrowth in Bone. EMBO Rep. 2020, 21, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Camphausen, K.; Burgan, W.; Cerra, M.; Oswald, K.A.; Trepel, J.B.; Lee, M.J.; Tofilon, P.J. Enhanced Radiation-Induced Cell Killing and Prolongation of ΓH2AX Foci Expression by the Histone Deacetylase Inhibitor MS-275. Cancer Res. 2004, 64, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camphausen, K.; Scott, T.; Sproull, M.; Tofilon, P.J. Enhancement of Xenograft Tumor Radiosensitivity by the Histone Deacetylase Inhibitor MS-275 and Correlation with Histone Hyperacetylation. Clin. Cancer Res. 2004, 10, 6066–6071. [Google Scholar] [CrossRef] [Green Version]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.M.; Patterson, S.; Riggs, C.E.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, Two-Stage, Single-Arm Trial of the Histone Deacetylase Inhibitor (HDACi) Romidepsin in Metastatic Castration-Resistant Prostate Cancer (CRPC). Ann. Oncol. 2009, 21, 109–113. [Google Scholar] [CrossRef]
- Eigl, B.J.; North, S.; Winquist, E.; Finch, D.; Wood, L.; Sridhar, S.S.; Powers, J.; Good, J.; Sharma, M.; Squire, J.A.; et al. A Phase II Study of the HDAC Inhibitor SB939 in Patients with Castration Resistant Prostate Cancer: NCIC Clinical Trials Group Study IND195. Investig. New Drugs 2015. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial Identifier | Compound(s) | Mechanism of Action | Phase Reached | Cohort | Therapy Type | Outcome |
|---|---|---|---|---|---|---|
| IL-6-Mediated Pathways | ||||||
| N/A [Karkera] | CNTO 328/Siltuximab | IL-6 chimeric monoclonal antibody | Phase I | Organ-confined PCa, n = 20 | Monotherapy | Favorable preliminary safety profile with evidence of decreased activation of IL-6-mediated signalling pathways. |
| NCT00401765 | Phase I | mCRPC, n = 40 | Polytherapy (+docetaxel) | Promising PSA results but high toxicity. | ||
| NCT00433446 | Phase II | mCRPC, prior taxane therapy, n = 53 | Monotherapy | Poor efficacy, IL-6 increased dramatically post-treatment. | ||
| NCT00385827 | Phase II | mCRPC, prior docetaxel therapy, n = 106 | Polytherapy (+ mitoxantrone/prednisone) | Well tolerated, but no improvement in survival outcome. | ||
| CXCL5/CXCR2-Mediated Pathways | ||||||
| NCT03177187 | AZD5069 | CXCR2 antagonist | Phase I/II | mCRPC | Polytherapy (+enzalutamide) | Currently recruiting. |
| Targeting S100A9 | ||||||
| NCT01234311 | Tasquinimod | S100A9 inhibitor | Phase III | mCRPC, n = 1245 | Monotherapy | No improvement in survival outcome. |
| Combinatorial Immunotherapeutic Approaches | ||||||
| NCT03098160 | Ipilipmumab/anti-CTLA4, Evofosamide | ICB, Hypoxia disrupting prodrug | Phase I | Prostate and other cancers | Combinatorial | Recruiting. Current recruitment status unknown. |
| NCT03689699 | Nivolumab/anti-PD-1, BMS-986253/anti IL-8 | ICB, fully monoclonal antibody against IL-8 | Phase I/II | Hormone-sensitive PCa | Currently recruiting. | |
| NCT03493945 | BN-Brachyury, M7824, ALT-803, Epacadostat | Anti-tumour vaccine, TGFβ TRAP/PD-L1, IL-15 agonist, IDO inhibitor | Phase I/II | CRPC | Currently recruiting. | |
| NCT02159950 | Tasquinimod, Sipuleucel T | S100A9 inhibitor, active immunotherapy | Phase II | mCRPC | Not completed due to lack of funding. | |
| HDACi | Targeted HDAC | Test Model | Strategies and Results | Reference |
|---|---|---|---|---|
| Dacinostat (LAQ824) | Pan (Class I, II) | Androgen-sensitive and -independent cell line | Acetylation of HSP-90 and concurrent suppression in ATP binding leads to proteasomal degradation of AR by dissociation of AR and HSP-90. HDACi is believed to target either HDAC6 or HDAC10. | [193] |
| Phase I trial | Reduced levels of HSP-90 protein as an indication of the inhibition by the HDACi. | [194] | ||
| Suberoylanilide hydroxamic acid (SAHA)/Vorinostat | Pan | cell lines and xenografts (CWR22 nude mice) | Acetylation of HDAC 3 and 4 resulted in tumour regression, apoptosis, and growth arrest in cancer cells. | [191] |
| Phase II | No significant outcome, progression-free disease in 2 patients out of 27 patients (NCT00330161). | [195] | ||
| Belinostat (PXD101) | Pan | Combination treatment with docetaxel on xenograft mice | Inhibition of HDAC 6, stabilizing tubulin acetylation, regulating antiapoptotic proteins including BclXL to induce death of hormone-refractory cancer cells resulting reduced tumour volume. | [192] |
| SAHA derivative (MHY219) | HDAC 1, 2, 3 and 4 | metastatic in vitro tissues | Increased apoptosis of PCa | [196] |
| Increased tissue inhibitor of metalloproteinase-1 (TIMP1) and associated mRNA levels. Reduced levels of MMP1 and MMP2. The regulator of MMPs, HDAC1 activity was also inhibited and limited cell migration. | [190] | |||
| Panobinostat (LBH589) | Pan | In vitro and in vivo studies with Zoledronic acid (Zol) | Panobinostat relieves drug resistance to Zol and results in cell arrest and apoptosis of tumour cells. | [197] |
| Phase I trial, oral administration to CRPC patients along with docetaxel | ≥50% drop of PSA in 5 out of 8 patients. | [198] | ||
| Phase II trial, IV administration to CRPC patients after receiving chemotherapy. | No significant outcome (NCT00667862). | [199] | ||
| Ivaltinostat(CG200745) | Pan | Combination treatment with docetaxel in vitro and in vivo models | The inhibition through entrapping ARs in microtubules [tubulin acetylation] and stabilize the microtubule. Results include downregulated full-length AR and AR splice variants, PSA, Bcl2 proteins and reduced cell viability. | [200] |
| Nicotinamide | Sirt1 | In vitro and in vivo studies | Silencing of SIRT1 with nicotinamide and genetical suppression by sirtinol result in growth arrest and apoptosis of PCa cells | [183] |
| Sirtinol | Sirt1/2 | |||
| VPA | Pan Class I, IIa HDACs | Administrated with drinking water in xenograft mice model | Cell cycle arrest, apoptosis and reduced angiogenesis and reduced levels of proliferating cell nuclear antigen(PCNA). | [180] |
| Metastatic PCa cell lines | Only altered metastasis tissue, increased metastasis suppressor gene N-myc downstream-regulated gene (NDRG1), reduced metastasis | [201] | ||
| Entinostat (MS275) | Class I HDACs | RM1-CRPC rodent modal (Resemble bone metastasis) | Induction of tumour intrinsic type 1 interferon (TI1IFN) correlated with T cell responses displaying the increased immunogenicity | [202] |
| In vitro analysis and DU145 xenograft mice | Inhibition of HDAC 4, Attenuation of DNA damage repair, enhanced radiosensitivity and tumour delay. | [203,204] | ||
| CRPC-MyCaP mice were administrated SurVaxM | Reduced FOXP3 expression and expansion of CD8+ T cells. Un changed Treg levels. | [182] | ||
| Romidepsin | Phase II trial | In a small portion, RECIST and ≥ 50% drop of PSA (NCT00106418). | [205] | |
| Pracinostat (SB939) | Pan Class-I, -II and -IV | Oral administration, Phase II trial mCRPC patients | Low drug toxicity, no significant outcome (NCT01075308). | [206] |
| Tasquinimod | HDAC3/4 | Phase II and III trials | Angiogenesis, MDSC suppression which shown progression-free disease but no improvement in survival of the patients (NCT01234311). | [207,208] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shackleton, E.G.; Ali, H.Y.; Khan, M.; Pockley, G.A.; McArdle, S.E. Novel Combinatorial Approaches to Tackle the Immunosuppressive Microenvironment of Prostate Cancer. Cancers 2021, 13, 1145. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13051145
Shackleton EG, Ali HY, Khan M, Pockley GA, McArdle SE. Novel Combinatorial Approaches to Tackle the Immunosuppressive Microenvironment of Prostate Cancer. Cancers. 2021; 13(5):1145. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13051145
Chicago/Turabian StyleShackleton, Erin G., Haleema Yoosuf Ali, Masood Khan, Graham A. Pockley, and Stephanie E. McArdle. 2021. "Novel Combinatorial Approaches to Tackle the Immunosuppressive Microenvironment of Prostate Cancer" Cancers 13, no. 5: 1145. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13051145