
The Prognostic Value of Methylation Signatures and NF2 Mutations in Atypical Meningiomas

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
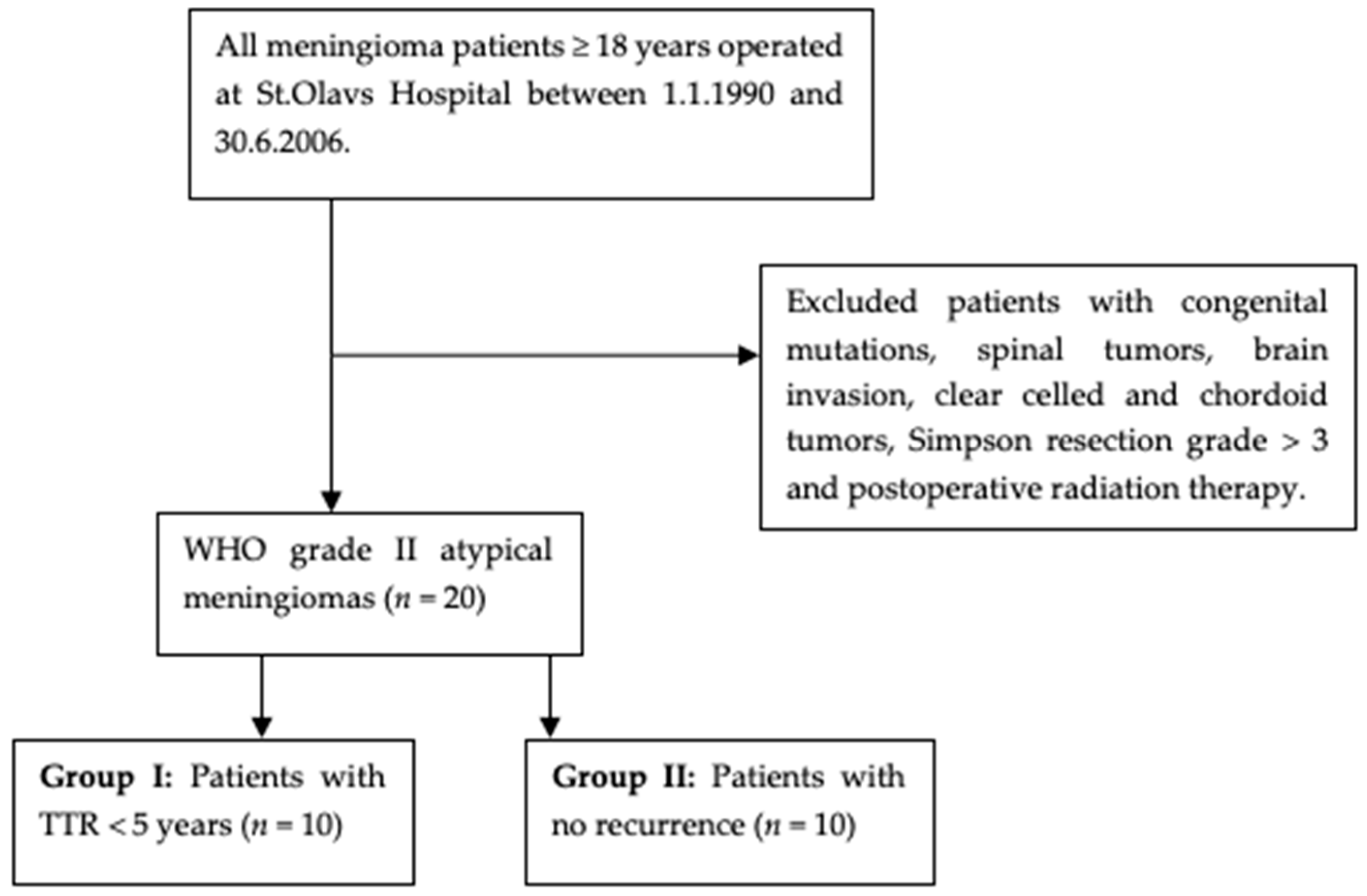
2. Materials and Methods
2.1. Material Collection
2.2. DNA Purification
2.3. Next-Generation Sequencing
2.4. Copy Number Variation Detection Based on NGS Panel Data and Illumina Infinium Methylation EPIC Bead Chip Array Analysis
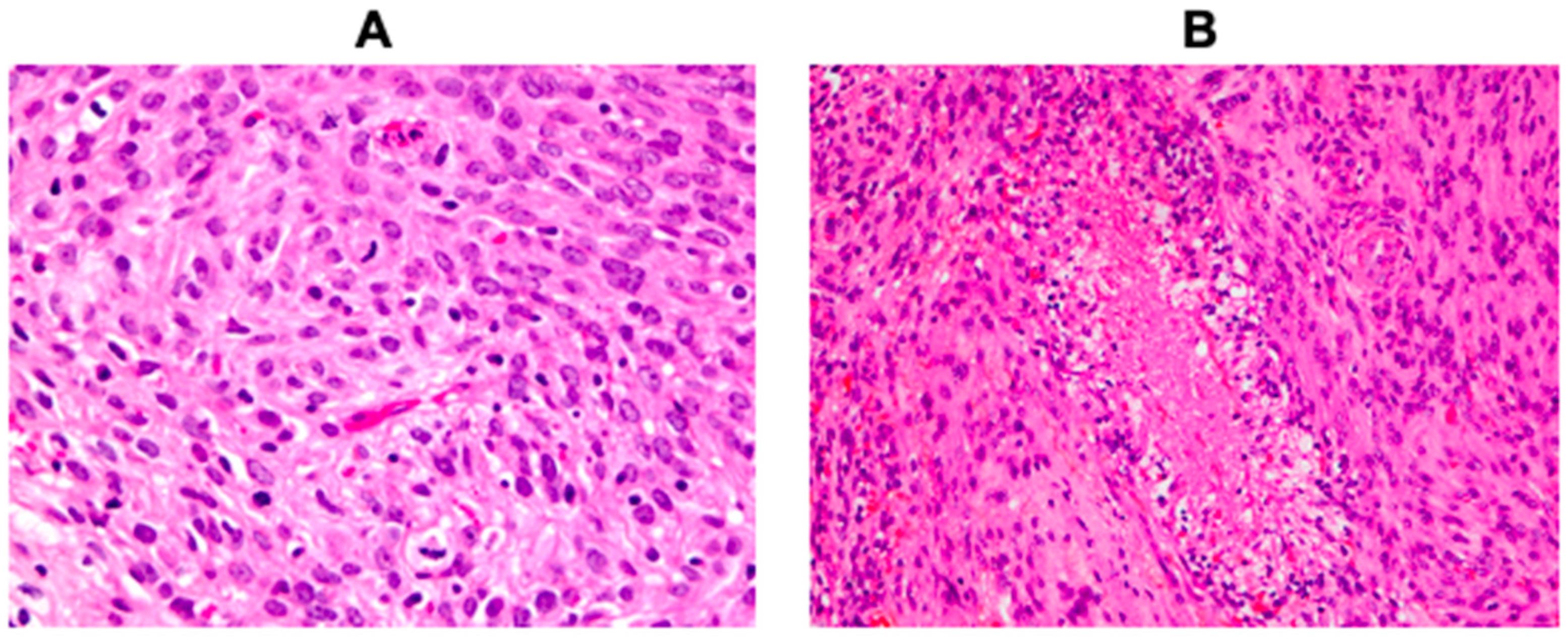
3. Results
4. Discussion
4.1. Genome-Wide DNA Methylation Analysis
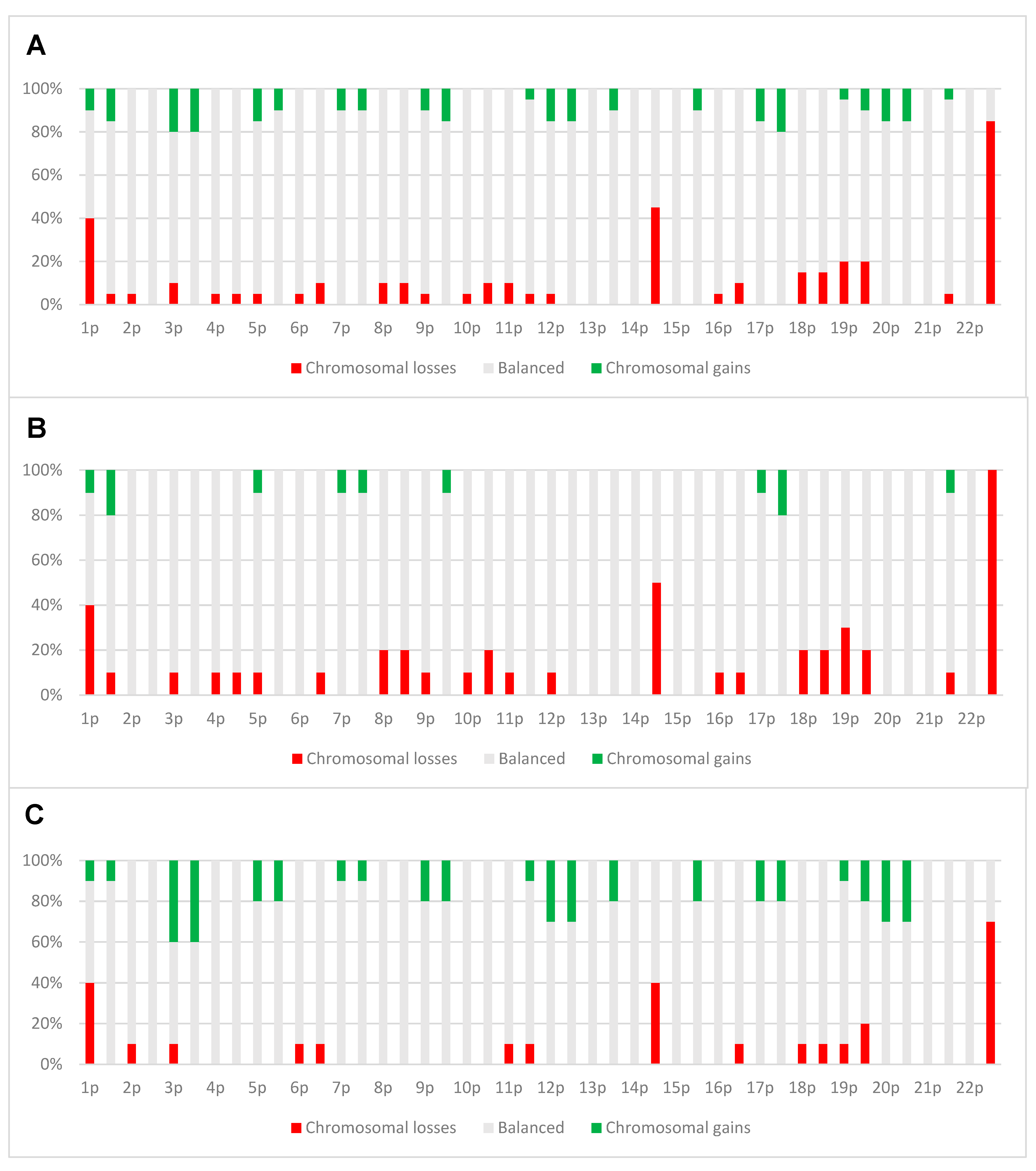
4.2. Copy Number Alterations
4.3. NF1/2 Mutations
4.4. Strengths and Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perry, A.; Gutmann, D.H.; Reifenberger, G. Molecular pathogenesis of meningiomas. J. Neuro-Oncol. 2004, 70, 183–202. [Google Scholar] [CrossRef] [PubMed]
- Mawrin, C.; Perry, A. Pathological classification and molecular genetics of meningiomas. J. Neuro-Oncol. 2010, 99, 379–391. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro-Oncology 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Perry, A.; Louis, D.N.; Budka, H.; von Deimling, A.; Sahm, F.; Rushing, E.; Mawrin, C.; Claus, E.B.; Loeffler, J.; Sadetzki, S. Meningioma. In WHO Classification of Tumours of the Central Nervous System; IARC: Lyon, France, 2016; pp. 231–245. [Google Scholar]
- Rogers, C.L.; Perry, A.; Pugh, S.; Vogelbaum, M.A.; Brachman, D.; McMillan, W.; Jenrette, J.; Barani, I.; Shrieve, D.; Sloan, A.; et al. Pathology concordance levels for meningioma classification and grading in NRG Oncology RTOG Trial 0539. Neuro-Oncology 2016, 18, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suppiah, S.; Nassiri, F.; Bi, W.L.; Dunn, I.F.; Hanemann, C.O.; Horbinski, C.M.; Hashizume, R.; James, C.D.; Mawrin, C.; Noushmehr, H.; et al. Molecular and translational advances in meningiomas. Neuro-Oncology 2019, 21, i4–i17. [Google Scholar] [CrossRef]
- Harter, P.N.; Braun, Y.; Plate, K.H. Classification of meningiomas—Advances and controversies. Chin. Clin. Oncol. 2017, 6, S2. [Google Scholar] [CrossRef]
- Nowosielski, M.; Galldiks, N.; Iglseder, S.; Kickingereder, P.; Von Deimling, A.; Bendszus, M.; Wick, W.; Sahm, F. Diagnostic challenges in meningioma. Neuro-Oncology 2017, 19, 1588–1598. [Google Scholar] [CrossRef]
- Budohoski, K.P.; Clerkin, J.; Millward, C.P.; O’Halloran, P.J.; Waqar, M.; Looby, S.; Young, A.M.H.; Guilfoyle, M.R.; Fitzroll, D.; Devadass, A.; et al. Predictors of early progression of surgically treated atypical meningiomas. Acta Neurochir. 2018, 160, 1813–1822. [Google Scholar] [CrossRef] [Green Version]
- Olar, A.; Goodman, L.D.; Wani, K.M.; Boehling, N.S.; Sharma, D.S.; Mody, R.R.; Gumin, J.; Claus, E.B.; Lang, F.F.; Cloughesy, T.F.; et al. A gene expression signature predicts recurrence-free survival in meningioma. Oncotarget 2018, 9, 16087–16098. [Google Scholar] [CrossRef]
- Bi, W.L.; Greenwald, N.F.; Abedalthagafi, M.; Wala, J.; Gibson, W.J.; Agarwalla, P.K.; Horowitz, P.; Schumacher, S.E.; Esaulova, E.; Mei, Y.; et al. Genomic landscape of high-grade meningiomas. NPJ Genom. Med. 2017, 2. [Google Scholar] [CrossRef]
- Galani, V.; Lampri, E.; Varouktsi, A.; Alexiou, G.; Mitselou, A.; Kyritsis, A.P. Genetic and epigenetic alterations in meningiomas. Clin. Neurol. Neurosurg. 2017, 158, 119–125. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, Y.S. Molecular characteristics of meningiomas. J. Pathol. Transl. Med. 2020, 54, 45–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirian, C.; Duun-Henriksen, A.K.; Juratli, T.; Sahm, F.; Spiegl-Kreinecker, S.; Peyre, M.; Biczok, A.; Tonn, J.-C.; Goutagny, S.; Bertero, L.; et al. Poor prognosis associated with TERT gene alterations in meningioma is independent of the WHO classification: An individual patient data meta-analysis. J. Neurol. Neurosurg. Psychiatry 2020, 91, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Fürtjes, G.; Köchling, M.; Peetz-Dienhart, S.; Wagner, A.; Heß, K.; Hasselblatt, M.; Senner, V.; Stummer, W.; Paulus, W.; Brokinkel, B. hTERT promoter methylation in meningiomas and central nervous hemangiopericytomas. J. Neuro-Oncol. 2016, 130, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.M.; Hielscher, T.; Liechty, B.; Silverman, J.; Zagzag, D.; Sen, R.; Wu, P.; Golfinos, J.G.; Reuss, D.; Neidert, M.C.; et al. Loss of histone H3K27me3 identifies a subset of meningiomas with increased risk of recurrence. Acta Neuropathol. 2018, 135, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA methylation-based classification and grading system for meningioma: A multicentre, retrospective analysis. Lancet Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef] [Green Version]
- Backer-Grøndahl, T.; Moen, B.H.; Torp, S.H. The histopathological spectrum of human meningiomas. Int. J. Clin. Exp. Pathol. 2012, 5, 231–242. [Google Scholar]
- Backer-Grøndahl, T.; Moen, B.H.; Sundstrøm, S.H.; Torp, S.H. Histopathology and prognosis in human meningiomas. APMIS 2014, 122, 856–866. [Google Scholar] [CrossRef]
- Zacher, A.; Kaulich, K.; Stepanow, S.; Wolter, M.; Köhrer, K.; Felsberg, J.; Malzkorn, B.; Reifenberger, G. Molecular Diagnostics of Gliomas Using Next Generation Sequencing of a Glioma-Tailored Gene Panel. Brain Pathol. 2017, 27, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.K.; Boldt, H.B.; Sørensen, M.D.; Blach, S.; Dahlrot, R.H.; Hansen, S.; Burton, M.; Thomassen, M.; Kruse, T.; Poulsen, F.R.; et al. Targeted next-generation sequencing of adult gliomas for retrospective prognostic evaluation and up-front diagnostics. Neuropathol. Appl. Neurobiol. 2021, 47, 108–126. [Google Scholar] [CrossRef] [PubMed]
- Priesterbach-Ackley, L.P.; Boldt, H.B.; Petersen, J.K.; Bervoets, N.; Scheie, D.; Ulhøi, B.P.; Gardberg, M.; Brännström, T.; Torp, S.H.; Aronica, E.; et al. Brain tumour diagnostics using a DNA methylation-based classifier as a diagnostic support tool. Neuropathol. Appl. Neurobiol. 2020, 46, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Jones, D.T.W.; Sill, M.; Hovestadt, V.; Schrimpf, D.; Sturm, D.; Koelsche, C.; Sahm, F.; Chavez, L.; Reuss, D.E.; et al. DNA methylation-based classification of central nervous system tumours. Nature 2018, 555, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Capper, D.; Stichel, D.; Sahm, F.; Jones, D.T.W.; Schrimpf, D.; Sill, M.; Schmid, S.; Hovestadt, V.; Reuss, D.E.; Koelsche, C.; et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: The Heidelberg experience. Acta Neuropathol. 2018, 136, 181–210. [Google Scholar] [CrossRef] [Green Version]
- Do, H.; Dobrovic, A. Sequence Artifacts in DNA from Formalin-Fixed Tissues: Causes and Strategies for Minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludyga, N.; Grünwald, B.; Azimzadeh, O.; Englert, S.; Höfler, H.; Tapio, S.; Aubele, M. Nucleic acids from long-term preserved FFPE tissues are suitable for downstream analyses. Virchows Arch. 2012, 460, 131–140. [Google Scholar] [CrossRef]
- Louis, D.N.; Wesseling, P.; Aldape, K.; Brat, D.J.; Capper, D.; Cree, I.A.; Ederhart, C.; Figarella-Branger, D.; Fouladi, M.; Fuller, G.N.; et al. cIMPACT-NOW update 6: New entity and diagnostic principle recommendations of the cIMPACT-Utrecht meeting on future CNS tumor classification and grading. Brain Pathol. 2020, 30, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Olar, A.; Wani, K.M.; Wilson, C.D.; Zadeh, G.; Demonte, F.; Jones, D.T.W.; Pfister, S.M.; Sulman, E.P.; Aldape, K.D. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol. 2017, 133, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Nassiri, F.; Mamatjan, Y.; Suppiah, S.; Badhiwala, J.H.; Mansouri, S.; Karimi, S.; Saarela, O.; Poisson, L.M.; Gepfner-Tuma, I.; Schittenhelm, J.; et al. DNA methylation profiling to predict recurrence risk in meningioma: Development and validation of a nomogram to optimize clinical management. Neuro-Oncology 2019, 21, 901–910. [Google Scholar] [CrossRef] [Green Version]
- Scheie, D.; Kufaishi, H.H.A.; Broholm, H.; Lund, E.L.; De Stricker, K.; Melchior, L.C.; Grauslund, M. Biomarkers in tumors of the central nervous system—A review. APMIS 2019, 127, 265–287. [Google Scholar] [CrossRef] [Green Version]
- Yuzawa, S.; Nishihara, H.; Tanaka, S. Genetic landscape of meningioma. Brain Tumor Pathol. 2016, 33, 237–247. [Google Scholar] [CrossRef]
- Choy, W.; Kim, W.; Nagasawa, D.; Stramotas, S.; Yew, A.; Gopen, Q.; Parsa, A.T.; Yang, I. The molecular genetics and tumor pathogenesis of meningiomas and the future directions of meningioma treatments. Neurosurg. Focus 2011, 30, E6. [Google Scholar] [CrossRef] [PubMed]
- Birzu, C.; Peyre, M.; Sahm, F. Molecular alterations in meningioma: Prognostic and therapeutic perspectives. Curr. Opin. Oncol. 2020, 32, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Pećina-Šlaus, N.; Kafka, A.; Lechpammer, M. Molecular Genetics of Intracranial Meningiomas with Emphasis on Canonical Wnt Signalling. Cancers 2016, 8, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Rashed, M.; Foshay, K.; Abedalthagafi, M. Recent Advances in Meningioma Immunogenetics. Front. Oncol. 2020, 9, 1472. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Case | Sex | Age | Localization | SRG | TTR in Months | Classified as Meningioma | Subclassification (Cut-Off 0.9) | Subclassification (Cut-Off 0.6) |
|---|---|---|---|---|---|---|---|---|
| Group I | ||||||||
| 1 | F | 71 | Convexity | 3 | 6.1 | Match | No match | Intermediate-A |
| 2 | F | 75 | Convexity | 2 | 0.7 | Match | No match | Intermediate-A |
| 3 | F | 72 | Basal | 2 | 20.5 | Match | No match | Intermediate-A |
| 4 | M | 65 | Falcine | 2 | 56.5 | Match | No match | No Match |
| 5 | F | 72 | Convexity | 2 | 0.1 * | No match | No match | Benign-1 |
| 6 | F | 49 | Convexity | 2 | 42.0 | Match | Benign-1 | |
| 7 | M | 47 | Fossa posterior and tentorial | 3 | 5.6 | Match | No match | Intermediate-A |
| 8 | M | 86 | Convexity | 1 | 18.9 | Match | No match | Benign-1 |
| 9 | F | 36 | Convexity | 1 | 17.6 | Match | No match | Intermediate-A |
| 10 | F | 54 | Convexity | 1 | 15.0 | Match | No match | Malignant |
| Group II | ||||||||
| 11 | F | 42 | Convexity | 2 | - | Match | No match | No match |
| 12 | F | 60 | Convexity | 1 | - | Match | No match | Benign-2 |
| 13 | F | 67 | Convexity | 2 | - | No match | No match | Benign-2 |
| 14 | F | 65 | Convexity | 1 | - | Match | No match | Benign-1 |
| 15 | F | 66 | Convexity | 2 | - | Match | No match | No match |
| 16 | F | 46 | Convexity | 1 | - | Match | Benign-1 | |
| 17 | M | 83 | Convexity | 2 | - | Match | Intermediate-A | |
| 18 | F | 80 | Basal | 2 | - | Match | No match | Intermediate-A |
| 19 | M | 47 | Convexity | 1 | - | Match | Intermediate-B | |
| 20 | M | 38 | Convexity | 1 | - | Match | No match | Benign-2 |
| Case | NF1/NF2 Status | NF1/NF2 Mutation; DNA (Protein) | 22q Loss |
|---|---|---|---|
| Group I | |||
| 1 | No mutations identified | + | |
| 2 | NF2 mutation 2 | c.1048delG, p.(Glu350AsnfsTer14) | + |
| 3 | NF1 mutation 1 | c.8301_8302delGAinsTT, p.(Gln2767_Ser2768delinsHisCys) | + |
| 4 | No mutations identified | + | |
| 5 | NF2 mutation 2 | c.432C > G, p.(Tyr144Ter) | + |
| 6 | NF2 mutation 2 | c.114 + 3A > C, p.(?) # | + |
| 7 | NF2 mutation 2 | c.837_838delAA, p.(Lys279AsnfsTer14) | + |
| 8 | NF2 mutation 2 | c.467_473delGTGTTCA, p.(Ser156ThrfsTer16) | + |
| 9 | NF2 mutation 2 | c.297delA, p.(Lys99AsnfsTer24) | + |
| 10 | NF2 mutation 2 | c.1093G > T, p.(Glu365Ter) | + |
| Group II | |||
| 11 | NF1 mutation 1 | c.3336C > A, p.(Asn1112Lys) | + |
| 12 | No mutations identified | ||
| 13 | No mutations identified | ||
| 14 | NF2 mutation 2 | c.543_544delGGinsTT, p.(Glu182Ter) | + |
| 15 | NF2 mutation 2 | c.1575-6C > A, p.(?)¤ | + |
| 16 | No mutations identified | + | |
| 17 | NF2 mutation 2 | c.448-1G > C, p.(?) | + |
| 18 | No mutations identified | + | |
| 19 | NF2 mutation 2 | c.1396C > T, p.(Arg466Ter) | + |
| 20 | No mutations identified | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meta, R.; Boldt, H.B.; Kristensen, B.W.; Sahm, F.; Sjursen, W.; Torp, S.H. The Prognostic Value of Methylation Signatures and NF2 Mutations in Atypical Meningiomas. Cancers 2021, 13, 1262. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061262
Meta R, Boldt HB, Kristensen BW, Sahm F, Sjursen W, Torp SH. The Prognostic Value of Methylation Signatures and NF2 Mutations in Atypical Meningiomas. Cancers. 2021; 13(6):1262. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061262
Chicago/Turabian StyleMeta, Rahmina, Henning B. Boldt, Bjarne W. Kristensen, Felix Sahm, Wenche Sjursen, and Sverre H. Torp. 2021. "The Prognostic Value of Methylation Signatures and NF2 Mutations in Atypical Meningiomas" Cancers 13, no. 6: 1262. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061262