
Geographical Distribution of E-cadherin Germline Mutations in the Context of Diffuse Gastric Cancer: A Systematic Review

 , , , ,
, , , ,  , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Study Accuracy and Selection
2.2. Group Description
2.3. Data Extraction, Statistical Methods and Quality Assessment of Studies
2.4. Missense Variant Scoring
3. Results
3.1. Study Features
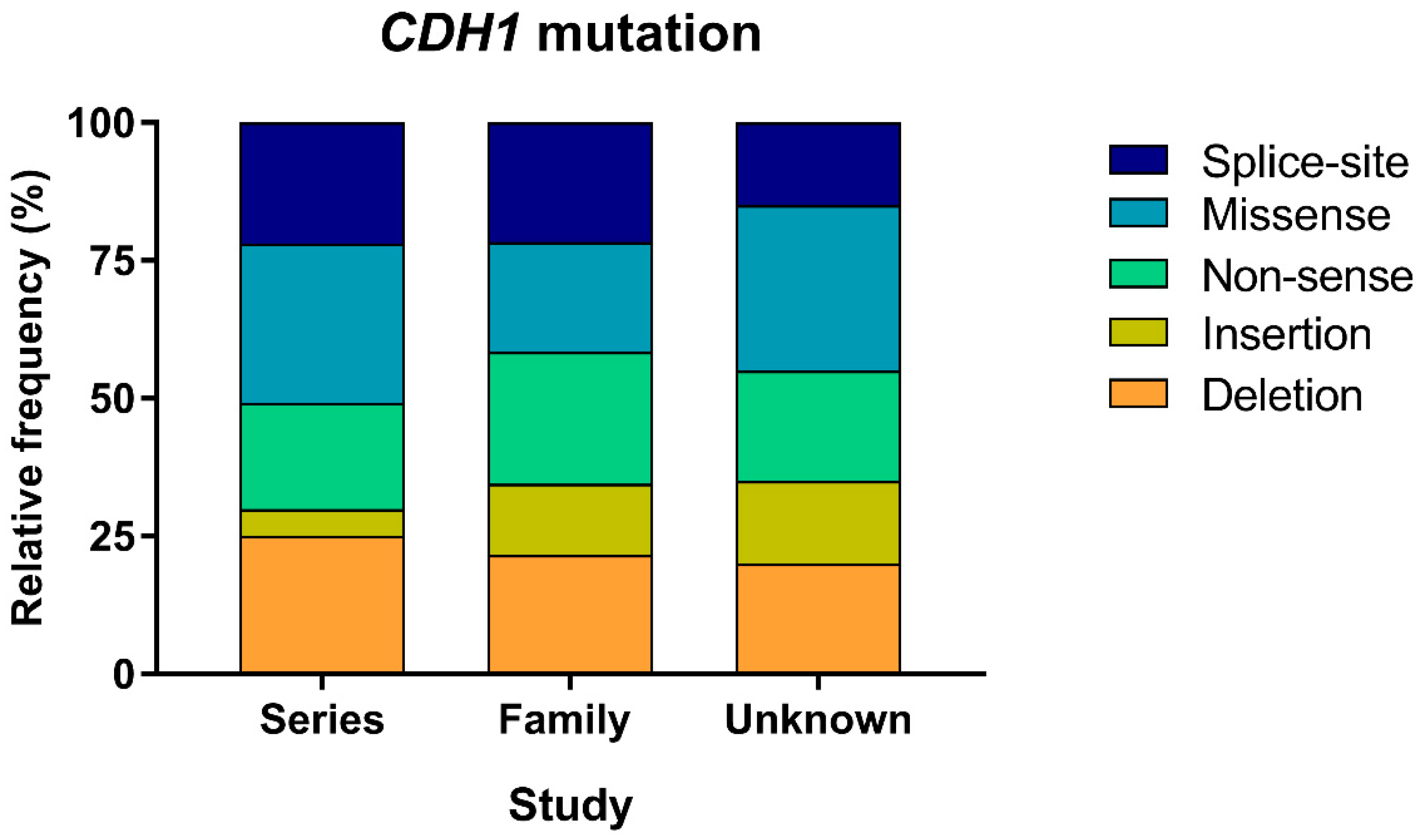
3.2. Mutation Type Frequencies
3.3. Geographical Distribution of CDH1 Mutations
3.4. Missense Variant Relevance across Geographical Regions and Study Contexts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lyons, K.; Le, L.C.; Pham, Y.T.; Borron, C.; Park, J.Y.; Tran, C.T.D.; Tran, T.V.; Tran, H.T.; Vu, K.T.; Do, C.D.; et al. Gastric cancer: Epidemiology, biology, and prevention: A mini review. Eur. J. Cancer Prev. 2019, 28, 397–412. [Google Scholar] [CrossRef]
- GBD 2017 Stomach Cancer Collaborators. The global, regional, and national burden of stomach cancer in 195 countries, 1990–2017: A systematic analysis for the Global Burden of Disease study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Global Burden of Disease Cancer Collaboration; Fitzmaurice, C.; Abate, D.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdel-Rahman, O.; Abdelalim, A.; Abdoli, A.; Abdollahpour, I.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2017: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2019, 5, 1749–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carioli, G.; Bertuccio, P.; Boffetta, P.; Levi, F.; La Vecchia, C.; Negri, E.; Malvezzi, M. European cancer mortality predictions for the year 2020 with a focus on prostate cancer. Ann. Oncol. 2020, 31, 650–658. [Google Scholar] [CrossRef]
- Saghier, A.A.; Sagar, M.; Kabanja, J.H.; Afreen, S. Gastric Cancer: Environmental risk factors, treatment and prevention. J. Carcinogene Mutagene 2013. [Google Scholar] [CrossRef] [Green Version]
- Ward, E.; Jemal, A.; Forman, D.; Ferlay, J.; Center, M.M.; Bray, F. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Nagini, S. Carcinoma of the stomach: A review of epidemiology, pathogenesis, molecular genetics and chemoprevention. World J. Gastrointest. Oncol. 2012, 4, 156–169. [Google Scholar] [CrossRef]
- Piazuelo, M.B.; Correa, P. Gastric cancer: Overview. Colomb. Med. 2013, 44, 192–201. [Google Scholar]
- Karimi, P.; Islami, F.; Anandasabapathy, S.; Freedman, N.D.; Kamangar, F. Gastric cancer: Descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol. Biomark. Prev. 2014, 23, 700–713. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Parkin, D.M.; Steliarova-Foucher, E. Estimates of cancer incidence and mortality in Europe in 2008. Eur. J. Cancer 2010, 46, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 15, 2893–2917. [Google Scholar] [CrossRef]
- Ferro, A.; Peleteiro, B.; Malvezzi, M.; Bosetti, C.; Bertuccio, P.; Levi, F.; Negri, E.; La Vecchia, C.; Lunet, N. Worldwide trends in gastric cancer mortality (1980–2011), with predictions to 2015, and incidence by subtype. Eur. J. Cancer 2014, 50, 1330–1344. [Google Scholar] [CrossRef] [Green Version]
- La Vecchia, C.; Negri, E.; Franceschi, S.; Gentile, A. Family history and the risk of stomach and colorectal cancer. Cancer 1992, 70, 50–55. [Google Scholar] [CrossRef]
- Corso, G.; Montagna, G.; Figueiredo, J.; La Vecchia, C.; Fumagalli Romario, U.; Fernandes, M.S.; Seixas, S.; Roviello, F.; Trovato, C.; Guerini-Rocco, E.; et al. Hereditary Gastric and Breast Cancer Syndromes Related to CDH1 Germline Mutation: A Multidisciplinary Clinical Review. Cancers 2020, 12, 1598. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Carvalho, J.; Marrelli, D.; Vindigni, C.; Carvalho, B.; Seruca, R.; Roviello, F.; Oliveira, C. Somatic mutations and deletions of the E-cadherin gene predict poor survival of patients with gastric cancer. J. Clin. Oncol. 2013, 31, 868–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilford, P.; Hopkins, J.; Harraway, J.; McLeod, M.; McLeod, N.; Harawira, P.; Taite, H.; Scoular, R.; Miller, A.; Reeve, A.E. E-cadherin germline mutations in familial gastric cancer. Nature 1998, 392, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Caldas, C.; Carneiro, F.; Lynch, H.T.; Yokota, J.; Wiesner, G.L.; Powell, S.M.; Lewis, F.R.; Huntsman, D.G.; Pharoah, P.D.; Jankowski, J.A.; et al. Familial gastric cancer: Overview and guidelines for management. J. Med. Genet. 1999, 36, 873–880. [Google Scholar] [PubMed]
- Kaurah, P.; MacMillan, A.; Boyd, N.; Senz, J.; De Luca, A.; Chun, N.; Suriano, G.; Zaor, S.; Van Manen, L.; Gilpin, C.; et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007, 297, 2360–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks-Wilson, A.R.; Kaurah, P.; Suriano, G.; Leach, S.; Senz, J.; Grehan, N.; Butterfield, Y.S.; Jeyes, J.; Schinas, J.; Bacani, J.; et al. Germline E-cadherin mutations in hereditary diffuse gastric cancer: Assessment of 42 new families and review of genetic screening criteria. J. Med. Genet. 2004, 41, 508–517. [Google Scholar] [CrossRef]
- Van der Post, R.S.; Vogelaar, I.P.; Carneiro, F.; Guilford, P.; Huntsman, D.; Hoogerbrugge, N.; Caldas, C.; Schreiber, K.E.; Hardwick, R.H.; Ausems, M.G.; et al. Hereditary diffuse gastric cancer: Updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J. Med. Genet. 2015, 52, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Corso, G.; Figueiredo, J.; La Vecchia, C.; Veronesi, P.; Pravettoni, G.; Macis, D.; Karam, R.; Lo Gullo, R.; Provenzano, E.; Toesca, A.; et al. Hereditary lobular breast cancer with an emphasis on E-cadherin genetic defect. J. Med. Genet. 2018, 55, 431–441. [Google Scholar] [CrossRef]
- Hansford, S.; Kaurah, P.; Li-Chang, H.; Woo, M.; Senz, J.; Pinheiro, H.; Schrader, K.A.; Schaeffer, D.F.; Shumansky, K.; Zogopoulos, G.; et al. Hereditary Diffuse Gastric Cancer Syndrome: CDH1 Mutations and Beyond. JAMA Oncol. 2015, 1, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suriano, G.; Seixas, S.; Rocha, J.; Seruca, R. A model to infer the pathogenic significance of CDH1 germline missense variants. J. Mol. Med. 2006, 84, 1023–1031. [Google Scholar] [CrossRef]
- Lee, K.; Krempely, K.; Roberts, M.E.; Anderson, M.J.; Carneiro, F.; Chao, E.; Dixon, K.; Figueiredo, J.; Ghosh, R.; Huntsman, D.; et al. Specifications of the ACMG/AMP variant curation guidelines for the analysis of germline CDH1 sequence variants. Hum. Mutat. 2018, 39, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
- Chakravorty, S.; Hegde, M. Gene and variant annotation for mendelian disorders in the era of advanced sequencing technologies. Ann. Rev. Genom. Hum. Genet. 2017, 18, 229–256. [Google Scholar] [CrossRef]
- Corso, G.; Marrelli, D.; Pascale, V.; Vindigni, C.; Roviello, F. Frequency of CDH1 germline mutations in gastric carcinoma coming from high- and low-risk areas: Metanalysis and systematic review of the literature. BMC Cancer 2012, 12, 8. [Google Scholar] [CrossRef] [Green Version]
- Blair, V.R.; McLeod, M.; Carneiro, F.; Coit, D.G.; D’Addario, J.L.; van Dieren, J.M.; Harris, K.L.; Hoogerbrugge, N.; Oliveira, C.; van der Post, R.S.; et al. Hereditary diffuse gastric cancer: Updated clinical practice guidelines. Lancet Oncol. 2020, 21, 386–397. [Google Scholar] [CrossRef]
- Shin, S.; Kim, Y.; Lee, J.K.; Lee, K.A. Frequency and Clinical Characteristics of Unselected Korean Gastric Cancer Patients with a Germline CDH1 V832M Mutation. J. Cancer 2020, 11, 208–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindalini, R.S.C.; Cormedi, M.C.V.; Maistro, S.; Pasini, F.S.; Branas, P.C.A.A.; Dos Santos, L.; de Lima Pereira, G.F.; de Bock, G.H.; Saccaro, D.M.; Katayama, M.L.H.; et al. Frequency of CDH1 germline variants and contribution of dietary habits in early age onset gastric cancer patients in Brazil. Gastric Cancer 2019, 22, 920–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Cho, M.Y.; Kim, J.; Kim, S.N.; Oh, S.C.; Lee, K.A. Profiling cancer-associated genetic alterations and molecular classification of cancer in Korean gastric cancer patients. Oncotarget 2017, 8, 69888–69905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakkaart, C.; Ellison-Loschmann, L.; Day, R.; Sporle, A.; Koea, J.; Harawira, P.; Cheng, S.; Gray, M.; Whaanga, T.; Pearce, N.; et al. Germline CDH1 mutations are a significant contributor to the high frequency of early-onset diffuse gastric cancer cases in New Zealand Maori. Fam. Cancer 2019, 18, 83–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedaldi, G.; Pirini, F.; Tebaldi, M.; Zampiga, V.; Cangini, I.; Danesi, R.; Arcangeli, V.; Ravegnani, M.; Abou Khouzam, R.; Molinari, C.; et al. Multigene Panel Testing Increases the Number of Loci Associated with Gastric Cancer Predisposition. Cancers 2019, 11, 1340. [Google Scholar] [CrossRef] [Green Version]
- Molinaro, V.; Pensotti, V.; Marabelli, M.; Feroce, I.; Barile, M.; Pozzi, S.; Laghi, L.; Serrano, D.; Bernard, L.; Bonanni, B.; et al. Complementary molecular approaches reveal heterogeneous CDH1 germline defects in Italian patients with hereditary diffuse gastric cancer (HDGC) syndrome. Genes Chromosomes Cancer 2014, 53, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Garziera, M.; Canzonieri, V.; Cannizzaro, R.; Geremia, S.; Caggiari, L.; De Zorzi, M.; Maiero, S.; Orzes, E.; Perin, T.; Zanussi, S.; et al. Identification and characterization of CDH1 germline variants in sporadic gastric cancer patients and in individuals at risk of gastric cancer. PLoS ONE 2013, 8, e77035. [Google Scholar] [CrossRef] [PubMed]
- Bacani, J.T.; Soares, M.; Zwingerman, R.; di Nicola, N.; Senz, J.; Riddell, R.; Huntsman, D.G.; Gallinger, S. CDH1/E-cadherin germline mutations in early-onset gastric cancer. J. Med. Genet. 2006, 43, 867–872. [Google Scholar] [CrossRef] [Green Version]
- Corso, G.; Pedrazzani, C.; Pinheiro, H.; Fernandes, E.; Marrelli, D.; Rinnovati, A.; Pascale, V.; Seruca, R.; Oliveira, C.; Roviello, F. E-cadherin genetic screening and clinico-pathologic characteristics of early onset gastric cancer. Eur. J. Cancer 2011, 47, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Senz, J.; Kaurah, P.; Pinheiro, H.; Sanges, R.; Haegert, A.; Corso, G.; Schouten, J.; Fitzgerald, R.; Vogelsang, H.; et al. Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum. Mol. Genet. 2009, 18, 1545–1555. [Google Scholar] [CrossRef]
- Suriano, G.; Yew, S.; Ferreira, P.; Senz, J.; Kaurah, P.; Ford, J.M.; Longacre, T.A.; Norton, J.A.; Chun, N.; Young, S.; et al. Characterization of a recurrent germ line mutation of the E-cadherin gene: Implications for genetic testing and clinical management. Clin. Cancer Res. 2005, 11, 5401–5409. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.H.; Deng, W.; Li, X.W.; Liu, X.F.; Wang, J.M.; Wang, L.F.; Xiao, N.; He, Q.; Wang, Y.P.; Fan, Y.M. Novel CDH1 germline mutations identified in Chinese gastric cancer patients. World J. Gastroenterol. 2013, 19, 909–916. [Google Scholar] [CrossRef]
- Richards, F.M.; McKee, S.A.; Rajpar, M.H.; Cole, T.R.; Evans, D.G.; Jankowski, J.A.; McKeown, C.; Sanders, D.S.; Maher, E.R. Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum. Mol. Genet. 1999, 8, 607–610. [Google Scholar] [CrossRef]
- Shinmura, K.; Kohno, T.; Takahashi, M.; Sasaki, A.; Ochiai, A.; Guilford, P.; Hunter, A.; Reeve, A.E.; Sugimura, H.; Yamaguchi, N.; et al. Familial gastric cancer: Clinicopathological characteristics, RER phenotype and germline p53 and E-cadherin mutations. Carcinogenesis 1999, 20, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Benusiglio, P.R.; Malka, D.; Rouleau, E.; De Pauw, A.; Buecher, B.; Noguès, C.; Fourme, E.; Colas, C.; Coulet, F.; Warcoin, M.; et al. CDH1 germline mutations and the hereditary diffuse gastric and lobular breast cancer syndrome: A multicentre study. J. Med. Genet. 2013, 50, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Shinmura, K.; Ito, H.; Kasami, M.; Sasaki, N.; Shima, H.; Ikeda, M.; Tao, H.; Goto, M.; Ozawa, T.; et al. Germline alterations in the CDH1 gene in familial gastric cancer in the Japanese population. Cancer Sci. 2011, 102, 1782–1788. [Google Scholar] [CrossRef]
- Suriano, G.; Oliveira, C.; Ferreira, P.; Machado, J.C.; Bordin, M.C.; De Wever, O.; Bruyneel, E.A.; Moguilevsky, N.; Grehan, N.; Porter, T.R.; et al. Identification of CDH1 germline missense mutations associated with functional inactivation of the E-cadherin protein in young gastric cancer probands. Hum. Mol. Genet. 2003, 12, 575–582. [Google Scholar] [CrossRef]
- Kim, S.; Chung, J.W.; Jeong, T.D.; Park, Y.S.; Lee, J.H.; Ahn, J.Y.; Kim, D.H.; Choi, K.D.; Lee, W.; Song, H.J.; et al. Searching for E-cadherin gene mutations in early onset diffuse gastric cancer and hereditary diffuse gastric cancer in Korean patients. Fam. Cancer 2013, 12, 503–507. [Google Scholar] [CrossRef]
- Yoon, K.A.; Ku, J.L.; Yang, H.K.; Kim, W.H.; Park, S.Y.; Park, J.G. Germline mutations of E-cadherin gene in Korean familial gastric cancer patients. J. Hum. Genet. 1999, 44, 177–180. [Google Scholar] [CrossRef] [Green Version]
- Tsukanov, A.C.; Shelygin, I.A.; Kashnikov, V.N.; Frolov, S.A.; Liubchenko, L.N.; Shubin, V.P.; Karpukhin, A.V.; Muzaffarova, T.A.; Pospekhova, N.I. Molecular genetics study of hereditary predisposition to diffuse gastric cancer in Russian patients. Vopr. Onkol. 2013, 59, 580–584. [Google Scholar] [PubMed]
- Kim, H.C.; Wheeler, J.M.; Kim, J.C.; Ilyas, M.; Beck, N.E.; Kim, B.S.; Park, K.C.; Bodmer, W.F. The E-cadherin gene (CDH1) variants T340A and L599V in gastric and colorectal cancer patients in Korea. Gut 2000, 47, 262–267. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, X.; Fan, Y.; Ding, J.; Xu, A.; Zhou, X.; Hu, X.; Zhu, M.; Zhang, X.; Li, S.; et al. Germline mutations and polymorphic variants in MMR, E-cadherin and MYH genes associated with familial gastric cancer in Jiangsu of China. Int. J. Cancer 2006, 119, 2592–2596. [Google Scholar] [CrossRef]
- Wang, Y.; Song, J.P.; Ikeda, M.; Shinmura, K.; Yokota, J.; Sugimura, H. Ile-Leu substitution (I415L) in germline E-cadherin gene (CDH1) in Japanese familial gastric cancer. Jpn. J. Clin. Oncol. 2003, 33, 17–20. [Google Scholar] [CrossRef] [Green Version]
- Keller, G.; Vogelsang, H.; Becker, I.; Hutter, J.; Ott, K.; Candidus, S.; Grundei, T.; Becker, K.F.; Mueller, J.; Siewert, J.R.; et al. Diffuse type gastric and lobular breast carcinoma in a familial gastric cancer patient with an E-cadherin germline mutation. Am. J. Pathol. 1999, 155, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Ascaño, J.J.; Frierson, H., Jr.; Moskaluk, C.A.; Harper, J.C.; Roviello, F.; Jackson, C.E.; El-Rifai, W.; Vindigni, C.; Tosi, P.; Powell, S.M. Inactivation of the E-cadherin gene in sporadic diffuse-type gastric cancer. Mod. Pathol. 2001, 14, 942–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabuta, T.; Shinmura, K.; Tani, M.; Yamaguchi, S.; Yoshimura, K.; Katai, H.; Nakajima, T.; Mochiki, E.; Tsujinaka, T.; Takami, M.; et al. E-cadherin gene variants in gastric cancer families whose probands are diagnosed with diffuse gastric cancer. Int. J. Cancer 2002, 101, 434–441. [Google Scholar] [CrossRef] [PubMed]
- El-Husny, A.; Raiol-Moraes, M.; Amador, M.; Ribeiro-Dos-Santos, A.M.; Montagnini, A.; Barbosa, S.; Silva, A.; Assumpção, P.; Ishak, G.; Santos, S.; et al. CDH1 mutations in gastric cancer patients from northern Brazil identified by Next- Generation Sequencing (NGS). Genet. Mol. Biol. 2016, 39, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Humar, B.; Toro, T.; Graziano, F.; Müller, H.; Dobbie, Z.; Kwang-Yang, H.; Eng, C.; Hampel, H.; Gilbert, D.; Winship, I.; et al. Novel germline CDH1 mutations in hereditary diffuse gastric cancer families. Hum. Mutat. 2002, 19, 518–525. [Google Scholar] [CrossRef]
- Roviello, F.; Corso, G.; Pedrazzani, C.; Marrelli, D.; De Falco, G.; Berardi, A.; Garosi, L.; Suriano, G.; Vindigni, C.; De Stefano, A.; et al. Hereditary diffuse gastric cancer and E-cadherin: Description of the first germline mutation in an Italian family. Eur. J. Surg. Oncol. 2007, 33, 448–451. [Google Scholar] [CrossRef]
- Norero, E.; Alarcon, M.A.; Hakkaart, C.; de Mayo, T.; Mellado, C.; Garrido, M.; Aguayo, G.; Lagos, M.; Torres, J.; Calvo, A.; et al. Identification of c.1531C>T Pathogenic Variant in the CDH1 Gene as a Novel Germline Mutation of Hereditary Diffuse Gastric Cancer. Int. J. Mol. Sci. 2019, 20, 4980. [Google Scholar] [CrossRef] [Green Version]
- Katona, B.W.; Clark, D.F.; Domchek, S. CDH1 on Multigene Panel Testing: Look Before You Leap. J. Natl. Cancer Inst. 2020, 112, 330–334. [Google Scholar] [CrossRef]
- Obermair, F.; Rammer, M.; Burghofer, J.; Malli, T.; Schossig, A.; Wimmer, K.; Kranewitter, W.; Mayrbaeurl, B.; Duba, H.C.; Webersinke, G. Cleft lip/palate and hereditary diffuse gastric cancer: Report of a family harboring a CDH1 c.687 + 1G > A germline mutation and review of the literature. Fam. Cancer 2019, 18, 253–260. [Google Scholar] [CrossRef]
- Pena-Couso, L.; Perea, J.; Melo, S.; Mercadillo, F.; Figueiredo, J.; Sanches, J.M.; Sánchez-Ruiz, A.; Robles, L.; Seruca, R.; Urioste, M. Clinical and functional characterization of the CDH1 germline variant c.1679C>G in three unrelated families with hereditary diffuse gastric cancer. Eur. J. Hum. Genet. 2018, 26, 1348–1353. [Google Scholar] [CrossRef] [Green Version]
- Gullo, I.; Devezas, V.; Baptista, M.; Garrido, L.; Castedo, S.; Morais, R.; Wen, X.; Rios, E.; Pinheiro, J.; Pinto-Ribeiro, I.; et al. Phenotypic heterogeneity of hereditary diffuse gastric cancer: Report of a family with early-onset disease. Gastrointest. Endosc. 2018, 87, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Caggiari, L.; Miolo, G.; Canzonieri, V.; De Zorzi, M.; Alessandrini, L.; Corona, G.; Cannizzaro, R.; Santeufemia, D.A.; Cossu, A.; Buonadonna, A.; et al. A new mutation of the CDH1 gene in a patient with an aggressive signet-ring cell carcinoma of the stomach. Cancer Biol. Ther. 2018, 19, 254–259. [Google Scholar] [CrossRef] [Green Version]
- Betés, M.; Alonso-Sierra, M.; Valentí, V.; Patiño, A. A multidisciplinary approach allows identification of a new pathogenic CDH1 germline missense mutation in a hereditary diffuse gastric cancer family. Dig. Liver Dis. 2017, 49, 825–826. [Google Scholar] [CrossRef] [PubMed]
- Yelskaya, Z.; Bacares, R.; Salo-Mullen, E.; Somar, J.; Lehrich, D.A.; Fasaye, G.A.; Coit, D.G.; Tang, L.H.; Stadler, Z.K.; Zhang, L. CDH1 Missense Variant c.1679C>G (p.T560R) Completely Disrupts Normal Splicing through Creation of a Novel 5’ Splice Site. PLoS ONE 2016, 11, e0165654. [Google Scholar] [CrossRef] [Green Version]
- López, M.; Cervera-Acedo, C.; Santibáñez, P.; Salazar, R.; Sola, J.J.; Domínguez-Garrido, E. A novel mutation in the CDH1 gene in a Spanish family with hereditary diffuse gastric cancer. Springerplus 2016, 5, 1181. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xiao, A.; Ruggeri, J.; Bacares, R.; Somar, J.; Melo, S.; Figueiredo, J.; Simões-Correia, J.; Seruca, R.; Shah, M.A. The germline CDH1 c.48 G>C substitution contributes to cancer predisposition through generation of a pro-invasive mutation. Mutat. Res. 2014, 770, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, S.; Yamada, H.; Takahashi, M.; Morohoshi, Y.; Yamaguchi, N.; Tsunoda, Y.; Hayashi, H.; Sugimura, H.; Komatsu, H. Early-onset diffuse gastric cancer associated with a de novo large genomic deletion of CDH1 gene. Gastric Cancer 2014, 17, 745–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M.; Fukagawa, T.; Nakajima, T.; Asada, K.; Sekine, S.; Yamashita, S.; Okochi-Takada, E.; Taniguchi, H.; Kushima, R.; Oda, I.; et al. Hereditary diffuse gastric cancer in a Japanese family with a large deletion involving CDH1. Gastric Cancer 2014, 17, 750–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardram, L.; Hansen, T.V.; Gerdes, A.M.; Timshel, S.; Friis-Hansen, L.; Federspiel, B. Prophylactic total gastrectomy in hereditary diffuse gastric cancer: Identification of two novel CDH1 gene mutations-a clinical observational study. Fam. Cancer 2014, 13, 231–242. [Google Scholar] [CrossRef]
- More, H.; Humar, B.; Weber, W.; Ward, R.; Christian, A.; Lintott, C.; Graziano, F.; Ruzzo, A.M.; Acosta, E.; Boman, B.; et al. Identification of seven novel germline mutations in the human E-cadherin (CDH1) gene. Hum. Mutat. 2007, 28, 203. [Google Scholar] [CrossRef]
- Kluijt, I.; Siemerink, E.J.; Ausems, M.G.; van Os, T.A.; de Jong, D.; Simões-Correia, J.; van Krieken, J.H.; Ligtenberg, M.J.; Figueiredo, J.; van Riel, E.; et al. CDH1-related hereditary diffuse gastric cancer syndrome: Clinical variations and implications for counseling. Int. J. Cancer 2012, 131, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Guilford, P.; Humar, B.; Blair, V. Hereditary diffuse gastric cancer: Translation of CDH1 germline mutations into clinical practice. Gastric Cancer 2010, 13, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dussaulx-Garin, L.; Blayau, M.; Pagenault, M.; Le Berre-Heresbach, N.; Raoul, J.L.; Campion, J.P.; David, V.; Bretagne, J.F. A new mutation of E-cadherin gene in familial gastric linitis plastica cancer with extra-digestive dissemination. Eur. J. Gastroenterol. Hepatol. 2001, 13, 711–715. [Google Scholar] [CrossRef]
- Keller, G.; Vogelsang, H.; Becker, I.; Plaschke, S.; Ott, K.; Suriano, G.; Mateus, A.R.; Seruca, R.; Biedermann, K.; Huntsman, D.; et al. Germline mutations of the E-cadherin(CDH1) and TP53 genes, rather than of RUNX3 and HPP1, contribute to genetic predisposition in German gastric cancer patients. J. Med. Genet. 2004, 41, e89. [Google Scholar] [CrossRef] [Green Version]
- Frebourg, T.; Oliveira, C.; Hochain, P.; Karam, R.; Manouvrier, S.; Graziadio, C.; Vekemans, M.; Hartmann, A.; Baert-Desurmont, S.; Alexandre, C.; et al. Cleft lip/palate and CDH1/E-cadherin mutations in families with hereditary diffuse gastric cancer. J. Med. Genet. 2006, 43, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Norton, J.A.; Ham, C.M.; Van Dam, J.; Jeffrey, R.B.; Longacre, T.A.; Huntsman, D.G.; Chun, N.; Kurian, A.W.; Ford, J.M. CDH1 truncating mutations in the E-cadherin gene: An indication for total gastrectomy to treat hereditary diffuse gastric cancer. Ann. Surg. 2007, 245, 873–879. [Google Scholar] [CrossRef]
- Rogers, W.M.; Dobo, E.; Norton, J.A.; Van Dam, J.; Jeffrey, R.B.; Huntsman, D.G.; Kingham, K.; Chun, N.; Ford, J.M.; Longacre, T.A. Risk-reducing total gastrectomy for germline mutations in E-cadherin (CDH1): Pathologic findings with clinical implications. Am. J. Surg. Pathol. 2008, 32, 799–809. [Google Scholar] [CrossRef]
- Guilford, P.J.; Hopkins, J.B.; Grady, W.M.; Markowitz, S.D.; Willis, J.; Lynch, H.; Rajput, A.; Wiesner, G.L.; Lindor, N.M.; Burgart, L.J.; et al. E-cadherin germline mutations define an inherited cancer syndrome dominated by diffuse gastric cancer. Hum. Mutat. 1999, 14, 249–255. [Google Scholar] [CrossRef]
- Mayrbaeurl, B.; Keller, G.; Schauer, W.; Burgstaller, S.; Czompo, M.; Hoebling, W.; Knoflach, P.; Duba, H.C.; Hoefler, H.; Thaler, J. Germline mutation of the E-cadherin gene in three sibling cases with advanced gastric cancer: Clinical consequences for the other family members. Eur. J. Gastroenterol. Hepatol. 2010, 22, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Wan, Y.L.; Wang, Z.J.; Zhao, B.; Zhu, J.; Huang, Y.T. Germline E-cadherin gene mutation screening in familial gastric cancer kindreds. Zhonghua Wai. Ke. Za. Zhi. 2004, 42, 914–917. [Google Scholar]
- Rodriguez-Sanjuan, J.C.; Fontalba, A.; Mayorga, M.; Bordin, M.C.; Hyland, S.J.; Trugeda, S.; Garcia, R.A.; Gomez-Fleitas, M.; Fernandez, F.; Caldas, C.; et al. A novel mutation in the E-cadherin gene in the first family with hereditary diffuse gastric cancer reported in Spain. Eur. J. Surg. Oncol. 2006, 32, 1110–1113. [Google Scholar] [CrossRef]
- Shah, M.A.; Salo-Mullen, E.; Stadler, Z.; Ruggeri, J.M.; Mirander, M.; Pristyazhnyuk, Y.; Zhang, L. De novo CDH1 mutation in a family presenting with early-onset diffuse gastric cancer. Clin. Genet. 2012, 82, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Van Domselaar, F.; Correa, D.; Vaccaro, C.; Redal, M.; Van Domselaar, R.; Huntsman, D.; Kaurah, P.; Senz, J.; Lynch, H. Hereditary diffuse gastric cancer (HDGC): Presentation of a family with a new mutation of the CDH1 gene. Acta Gastroenterol. Latinoam. 2007, 37, 158–163. [Google Scholar]
- Ghaffari, S.R.; Rafati, M.; Sabokbar, T.; Dastan, J. A novel truncating mutation in the E-cadherin gene in the first Iranian family with hereditary diffuse gastric cancer. Eur. J. Surg. Oncol. 2010, 36, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, C.; Bordin, M.C.; Grehan, N.; Huntsman, D.; Suriano, G.; Machado, J.C.; Kiviluoto, T.; Aaltonen, L.; Jackson, C.E.; Seruca, R.; et al. Screening E-cadherin in gastric cancer families reveals germline mutations only in hereditary diffuse gastric cancer kindred. Hum. Mutat. 2002, 19, 510–517. [Google Scholar] [CrossRef]
- Caron, O.; Schielke, A.; Svrcek, M.; Fléjou, J.F.; Garzon, J.; Olschwang, S.; Sézeur, A. Usefulness of prophylactic gastrectomy in a novel large hereditary diffuse gastric cancer (HDGC) family. Am. J. Gastroenterol. 2008, 103, 2160–2161. [Google Scholar] [CrossRef]
- Black, M.D.; Kaneshiro, R.; Lai, J.I.; Shimizu, D.M. Hereditary diffuse gastric cancer associated with E-cadherin germline mutation: A case report. Hawaii J. Med. Public Health 2014, 73, 204–207. [Google Scholar] [PubMed]
- Oliveira, C.; Ferreira, P.; Nabais, S.; Campos, L.; Ferreira, A.; Cirnes, L.; Alves, C.C.; Veiga, I.; Fragoso, M.; Regateiro, F.; et al. E-Cadherin (CDH1) and p53 rather than SMAD4 and Caspase-10 germline mutations contribute to genetic predisposition in Portuguese gastric cancer patients. Eur. J. Cancer 2004, 40, 1897–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, H.T.; Kaurah, P.; Wirtzfeld, D.; Rubinstein, W.S.; Weissman, S.; Lynch, J.F.; Grady, W.; Wiyrick, S.; Senz, J.; Huntsman, D.G. Hereditary diffuse gastric cancer: Diagnosis, genetic counseling, and prophylactic total gastrectomy. Cancer 2008, 112, 2655–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões-Correia, J.; Figueiredo, J.; Lopes, R.; Stricher, F.; Oliveira, C.; Serrano, L.; Seruca, R. E-cadherin destabilization accounts for the pathogenicity of missense mutations in hereditary diffuse gastric cancer. PLoS ONE 2012, 7, e33783. [Google Scholar] [CrossRef] [Green Version]
- Charlton, A.; Blair, V.; Shaw, D.; Parry, S.; Guilford, P.; Martin, I.G. Hereditary diffuse gastric cancer: Predominance of multiple foci of signet ring cell carcinoma in distal stomach and transitional zone. Gut 2004, 53, 814–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santucci, C.; Carioli, G.; Bertuccio, P.; Malvezzi, M.; Pastorino, U.; Boffetta, P.; Negri, E.; Bosetti, C.; La Vecchia, C. Progress in cancer mortality, incidence, and survival: A global overview. Eur. J. Cancer Prev. 2020, 29, 367–381. [Google Scholar] [CrossRef]
- Hemminki, K.; Sundquist, J.; Ji, J. Familial risk for gastric carcinoma: An updated study from Sweden. Br. J. Cancer 2007, 96, 1272–1277. [Google Scholar] [CrossRef]
- Kawasaki, K.; Kanemitsu, K.; Yasuda, T.; Kamigaki, T.; Kuroda, D.; Kuroda, Y. Family history of cancer in Japanese gastric cancer patients. Gastric Cancer 2007, 10, 173–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xicola, R.M.; Li, S.; Rodriguez, N.; Reinecke, P.; Karam, R.; Speare, V.; Blanck, M.H.; LaDuca, H.; Llor, X. Clinical features and cancer risk in families with pathogenic CDH1 variants irrespective of clinical criteria. J. Med. Genet. 2019, 56, 838–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.E.; Ranola, J.M.; Marshall, M.; Susswein, L.; Graceffo, S.; Bohenert, K.; Tsai, G.; Klein, R.; Hruska, K.; Shirts, B. Comparison of CDH1 Penetrance Estimates in Clinically Ascertained Families vs. Families Ascertained for Multiple Gastric Cancers. JAMA Oncol. 2019, 5, 1325–1331. [Google Scholar] [CrossRef]
- Palli, D.; Russo, A.; Ottini, L.; Masala, G.; Saieva, C.; Amorosi, A.; Cama, A.; D’Amico, C.; Falchetti, M.; Palmirotta, R.; et al. Red meat, family history, and increased risk of gastric cancer with microsatellite instability. Cancer Res. 2001, 61, 5415–5419. [Google Scholar]
- Gonzalez, C.A.; Riboli, E. Diet and cancer prevention: Where we are, where we are going. Nutr. Cancer 2006, 56, 225–231. [Google Scholar] [CrossRef]
- Corso, G.; Pedrazzani, C.; Marrelli, D.; Pinto, E.; Roviello, F. Familial gastric cancer and Li-Fraumeni syndrome. Eur. J. Cancer Care 2010, 19, 377–781. [Google Scholar] [CrossRef]
- Melo, S.; Figueiredo, J.; Fernandes, M.S.; Gonçalves, M.; Morais-de-Sá, E.; Sanches, J.M.; Seruca, R. Predicting the Functional Impact of CDH1 Missense Mutations in Hereditary Diffuse Gastric Cancer. Int. J. Mol. Sci. 2017, 12, 2687. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, J.; Söderberg, O.; Simões-Correia, J.; Grannas, K.; Suriano, G.; Seruca, R. The importance of E-cadherin binding partners to evaluate the pathogenicity of E-cadherin missense mutations associated to HDGC. Eur. J. Hum. Genet. 2013, 21, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueiredo, J.; Melo, S.; Carneiro, P.; Moreira, A.M.; Fernandes, M.S.; Ribeiro, A.S.; Guilford, P.; Paredes, J.; Seruca, R. Clinical spectrum and pleiotropic nature of CDH1 germline mutations. J. Med. Genet. 2019, 56, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Type | Series Study | Family Study | Unknown Study | Total | p-Value |
|---|---|---|---|---|---|
| Deletion | 46 (24.6%) | 77 (21.6%) | 4 (20.0%) | 127 (22.6%) | 0.05 |
| Insertion | 9 (4.8%) | 46 (12.9%) | 3 (15.0%) | 58 (10.3%) | - |
| Non-sense | 36 (19.3%) | 85 (23.9%) | 4 (20.0%) | 125 (22.2%) | - |
| Missense | 54 (28.9%) | 71 (19.9%) | 6 (30.0%) | 131 (23.3%) | - |
| Splice-site | 41 (21.9%) | 77 (21.6) | 3 (15.0%) | 121 (21.5%) | - |
| Imbalance | 1 (0.5%) | 0 (0.0%) | 0 (0.0%) | 1 (0.2%) | - |
| Total | 187 (33.2%) | 356 (63.2%) | 20 (3.6%) | 563 | - |
| HGVS | Protein Change | ClinVar Classification | In Vitro | In Silico | Total + | Total − | + Frequency% | Variant Relevance |
|---|---|---|---|---|---|---|---|---|
| 2T>C | M1T | +++ | 0 | ++/− | 5 | 1 | 83 | Yes |
| 3G>C | M1I | +++ A | 0 | ++/− | 5 | 1 | 83 | Yes |
| 3G>A | M1I | +++ | 0 | ++/− | 5 | 1 | 83 | Yes |
| 48G>C | Q16H | 0 | +++ | ++/− | 5 | 1 | 83 | Yes |
| 79C>T | P27S | +/− | 0 | +/−− | 2 | 3 | 40 | No |
| 185G>T | G62V | +/− | +++ | ++/− | 6 | 2 | 75 | Yes |
| 286A>G | I96V | +/−− | 0 | +/−− | 2 | 4 | 33 | No |
| 313T>A | S105T | +/− | 0 | +/−− | 2 | 3 | 40 | No |
| 353C>G | T118R | +/− | +++ | −−− | 4 | 4 | 50 | Unknown |
| 387G>T | Q129H | +/−− | 0 | −−− | 1 | 5 | 17 | No |
| 554A>T | E185V | +/− | --- | ++/− | 3 | 5 | 38 | No |
| 604G>A | V202I | −−− | 0 | +/−− | 1 | 5 | 17 | No |
| 641T>C | L214P | +++ A | +++ | +++ | 9 | 0 | 100 | Yes |
| 695C>G | S232C | +/− | --- | ++/− | 3 | 5 | 38 | No |
| 715G>A | G239R | ++/− | +++ | ++/− | 7 | 2 | 78 | Yes |
| 731A>G | D244G | +/− | 0 | ++/− | 3 | 2 | 60 | Yes |
| 820G>A | G274S | +/−− | --- | +/−− | 2 | 7 | 22 | No |
| 892G>A | A298T | −−− | +++ | ++/− | 5 | 4 | 56 | Yes |
| 977T>A | I326N | 0 | +++ | +++ | 6 | 0 | 100 | Yes |
| 1018A>G | T340A | −−− | +++ | −−− | 3 | 6 | 33 | No |
| 1118C>T | P373L | +/−− | +++ | +++ | 7 | 2 | 78 | Yes |
| 1225T>C | W409R | +/−− | +++ | +++ | 7 | 2 | 78 | Yes |
| 1243A>C | I415L | +/− | +/- | +/−− | 3 | 4 | 43 | No |
| 1285C>T | P429S | +/− | +++ | ++/− | 6 | 2 | 75 | Yes |
| 1409C>T | T470I | −−− | +++ | +++ | 6 | 3 | 67 | Yes |
| 1460T>C | V487G | +++ A | 0 | +/−− | 4 | 2 | 67 | Yes |
| 1676G>A | S559N | +/− | 0 | +/−− | 2 | 3 | 40 | No |
| 1679C>G | T560R | +++ | +++ | ++/− | 8 | 1 | 89 | Yes |
| 1748T>G | L583R | +++ A | +++ | ++/− | 8 | 1 | 89 | Yes |
| 1774G>A | A592T | −−− | −−− | ++/− | 2 | 7 | 22 | No |
| 1806C>A | F602L | 0 | 0 | +/−− | 1 | 2 | 33 | No |
| 1849G>A | A617T | −−− | −−− | +/−− | 1 | 8 | 11 | No |
| 1888C>G | L630V | −−− | 0 | ++/− | 2 | 4 | 33 | No |
| 1901C>T | A634V | ++/− | +++ | +/−− | 6 | 3 | 67 | Yes |
| 2195G>A | R732Q | ++/− | +++ | ++/− | 7 | 2 | 78 | Yes |
| 2245C>T | R749W | +/− | +++ | +++ | 7 | 1 | 88 | Yes |
| 2248G>A | D750N | +/− | +/− | +++ | 5 | 2 | 71 | Yes |
| 2315T>A | L772Q | 0 | 0 | +++ | 3 | 0 | 100 | Yes |
| 2343A>T | E781D | +/− | +++ | +/−− | 5 | 3 | 63 | Yes |
| 2396C>G | P799R | +/− | +++ | +++ | 7 | 1 | 88 | Yes |
| 2413G>A | D805N | −−− | +++ | +++ | 6 | 3 | 67 | Yes |
| 2494G>A | V832M | −−− | +++ | +++ | 6 | 3 | 67 | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Corso, G.; Corso, F.; Bellerba, F.; Carneiro, P.; Seixas, S.; Cioffi, A.; La Vecchia, C.; Magnoni, F.; Bonanni, B.; Veronesi, P.; et al. Geographical Distribution of E-cadherin Germline Mutations in the Context of Diffuse Gastric Cancer: A Systematic Review. Cancers 2021, 13, 1269. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061269
Corso G, Corso F, Bellerba F, Carneiro P, Seixas S, Cioffi A, La Vecchia C, Magnoni F, Bonanni B, Veronesi P, et al. Geographical Distribution of E-cadherin Germline Mutations in the Context of Diffuse Gastric Cancer: A Systematic Review. Cancers. 2021; 13(6):1269. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061269
Chicago/Turabian StyleCorso, Giovanni, Federica Corso, Federica Bellerba, Patrícia Carneiro, Susana Seixas, Antonio Cioffi, Carlo La Vecchia, Francesca Magnoni, Bernardo Bonanni, Paolo Veronesi, and et al. 2021. "Geographical Distribution of E-cadherin Germline Mutations in the Context of Diffuse Gastric Cancer: A Systematic Review" Cancers 13, no. 6: 1269. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13061269