
Natural Trienoic Acids as Anticancer Agents: First Stereoselective Synthesis, Cell Cycle Analysis, Induction of Apoptosis, Cell Signaling and Mitochondrial Targeting Studies

 ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
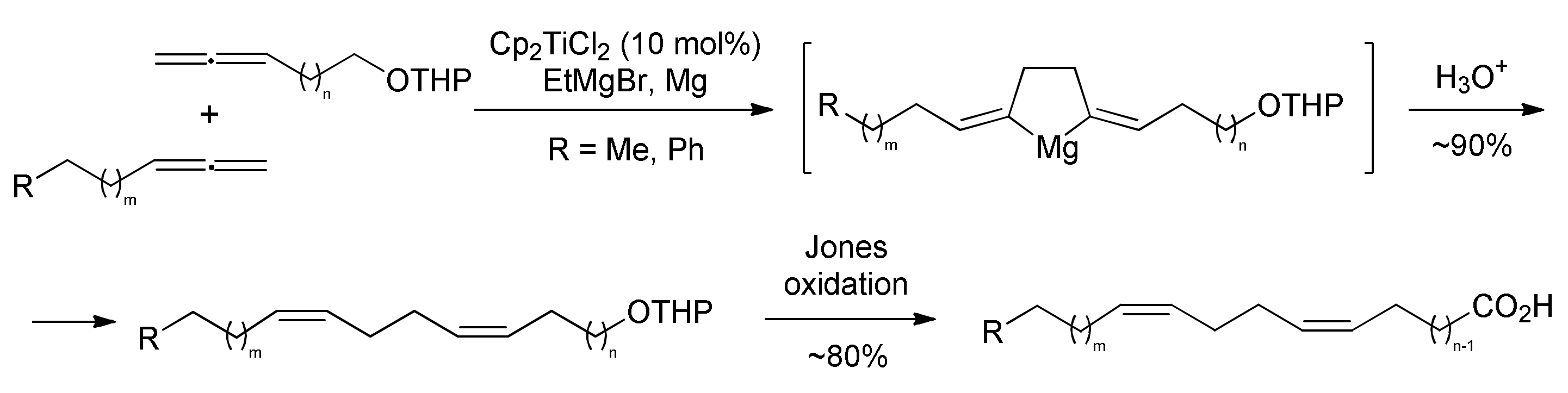
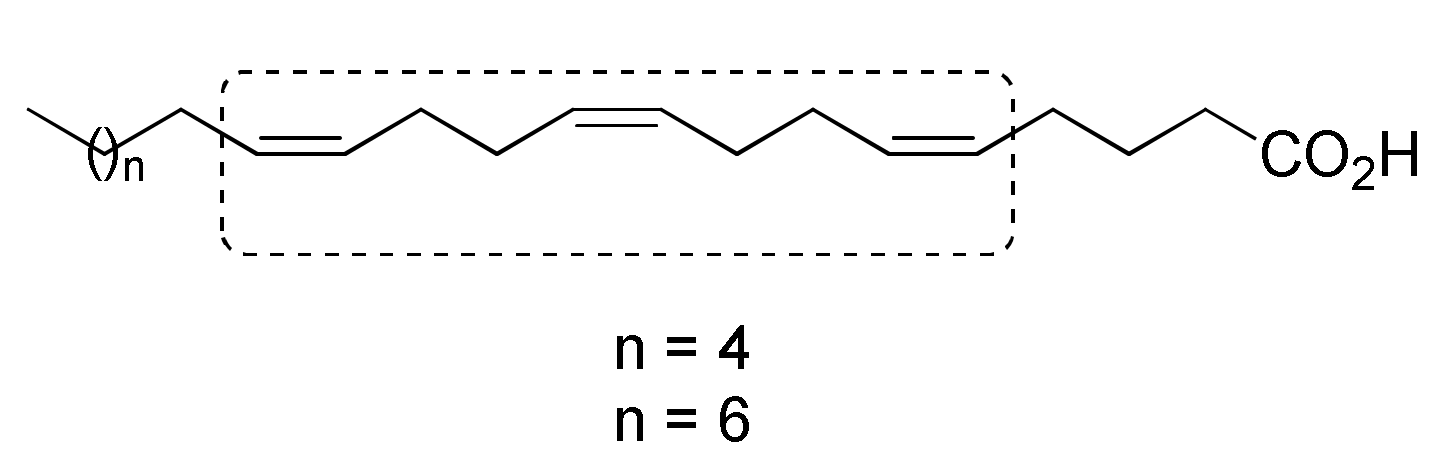
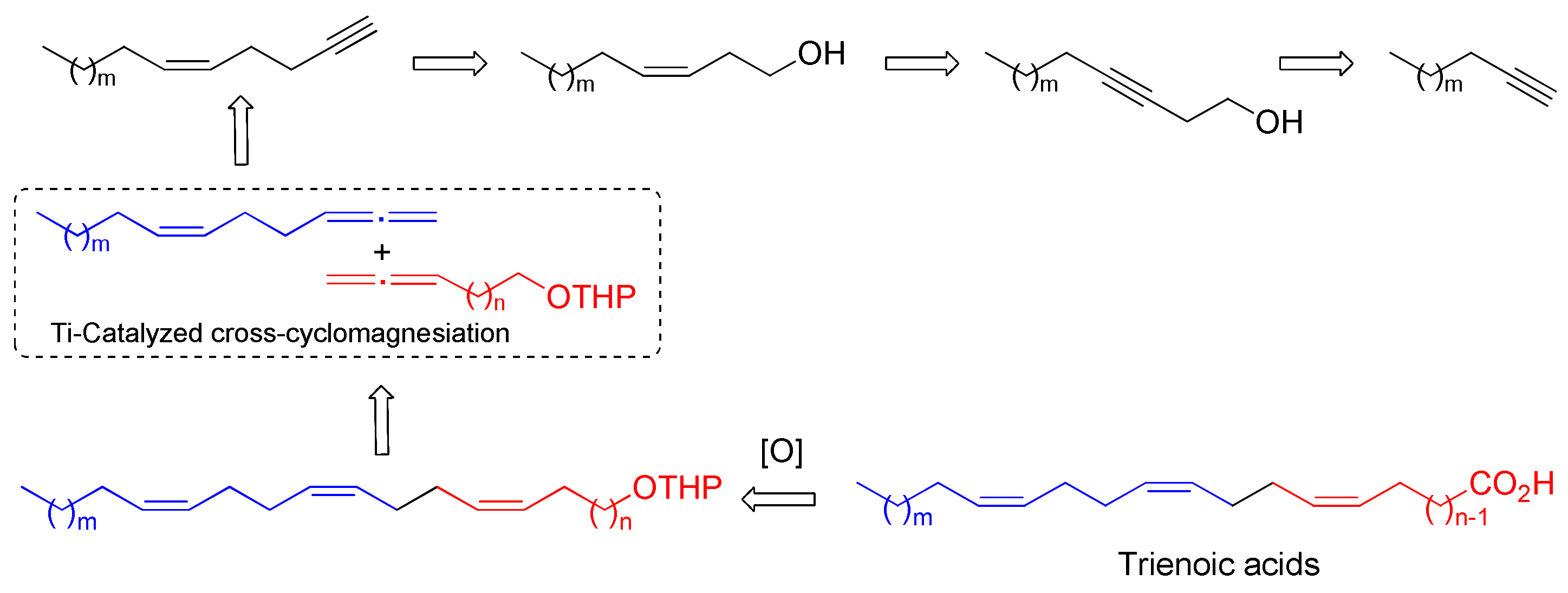
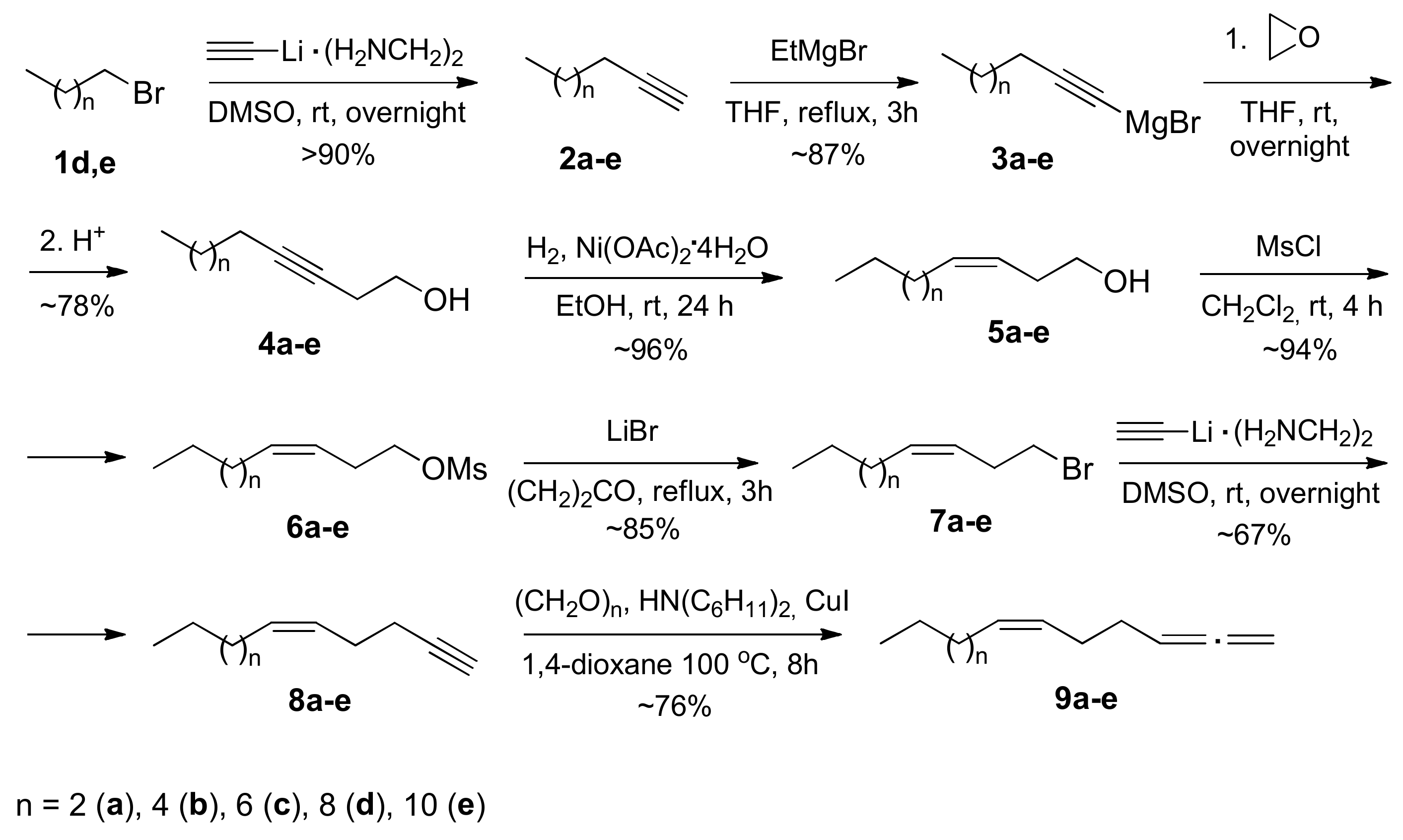
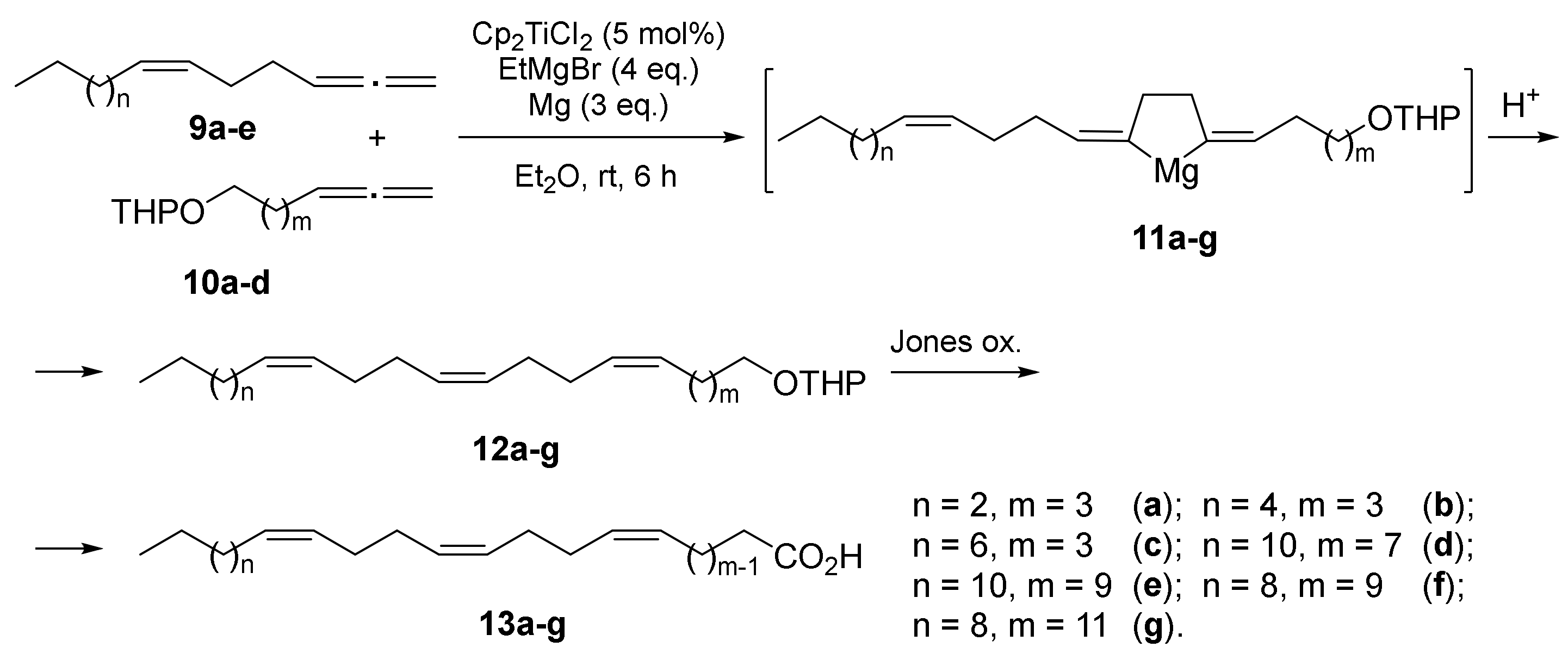
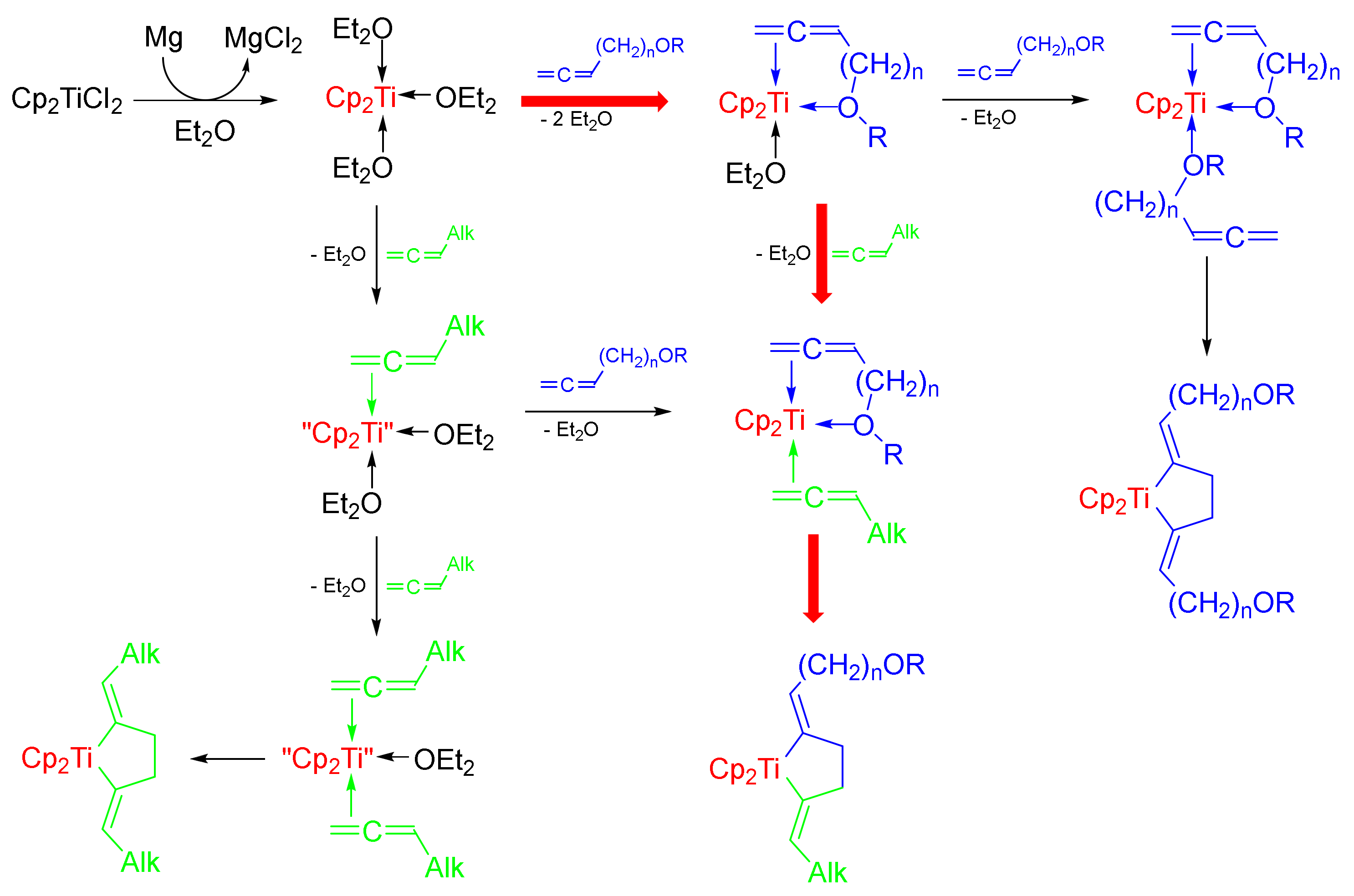
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Cytotioxic Activity In Vitro
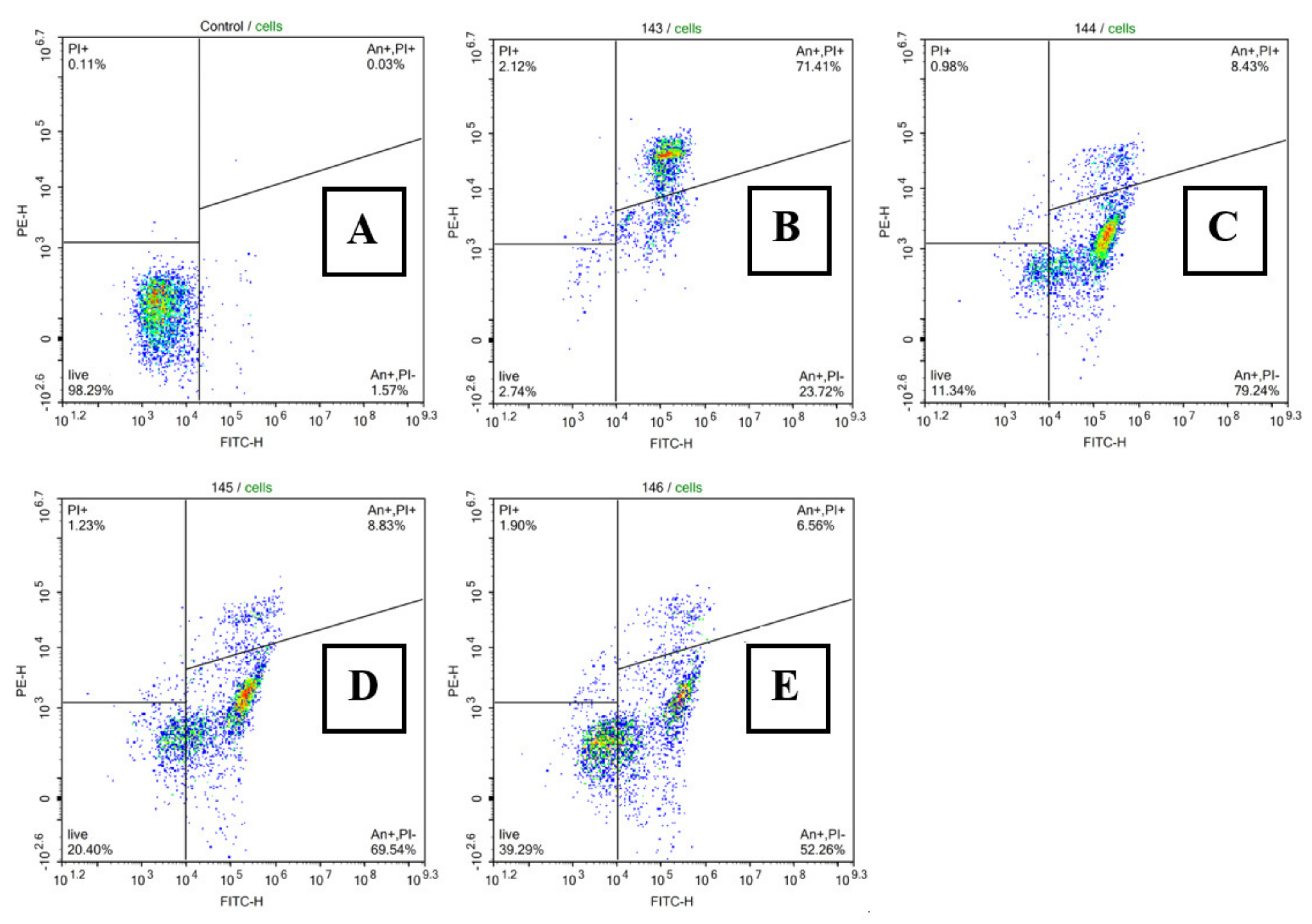
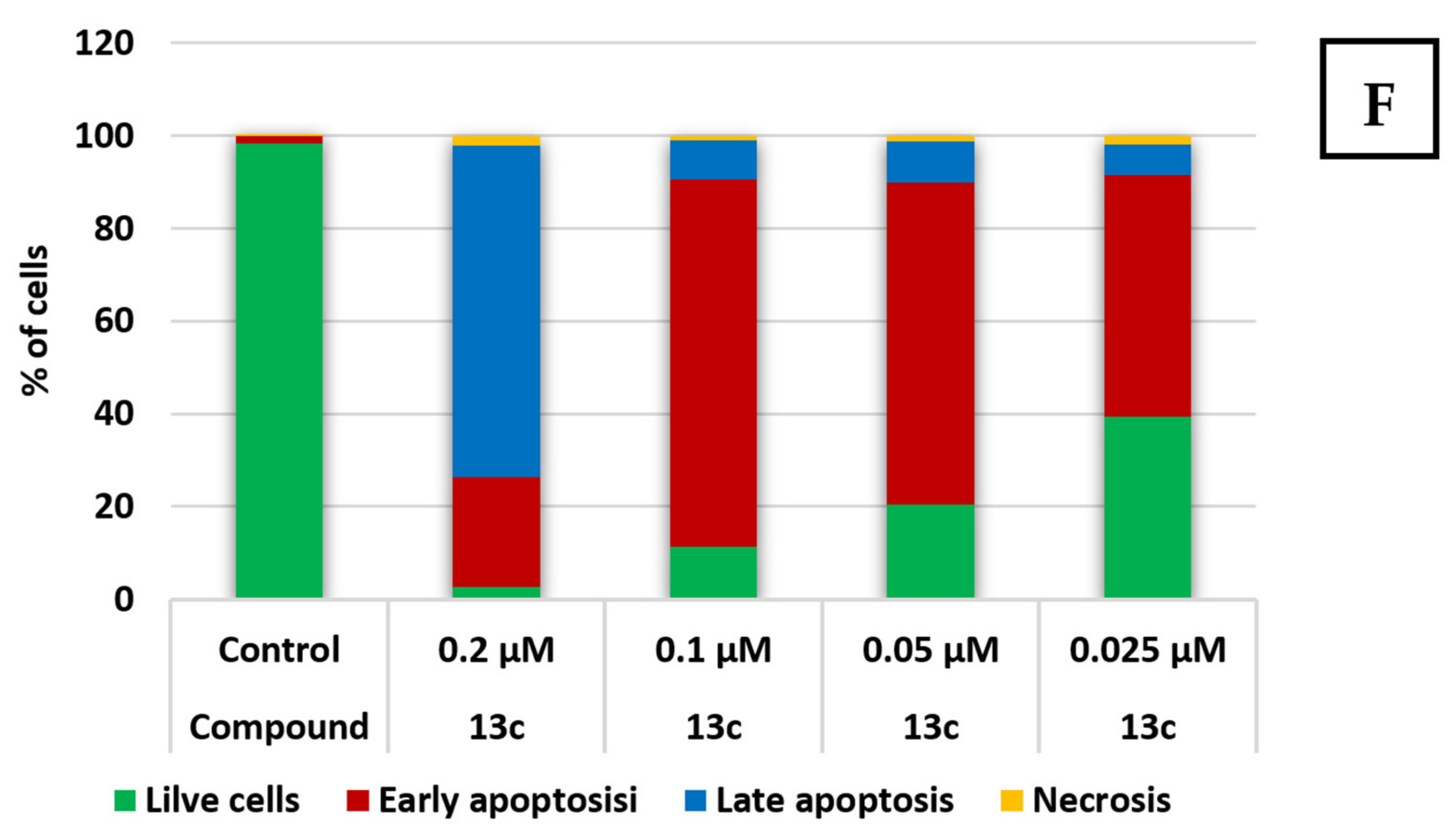
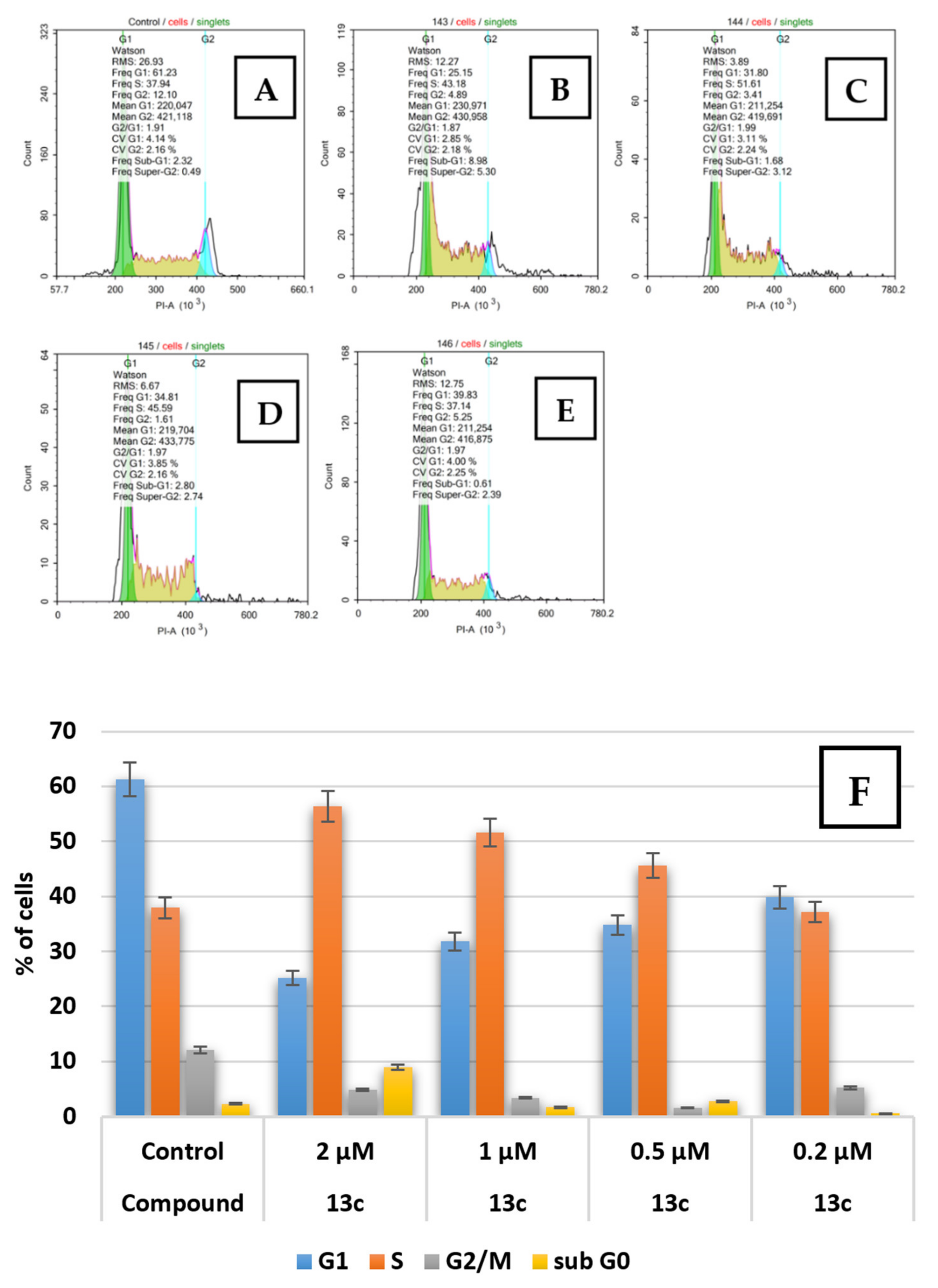
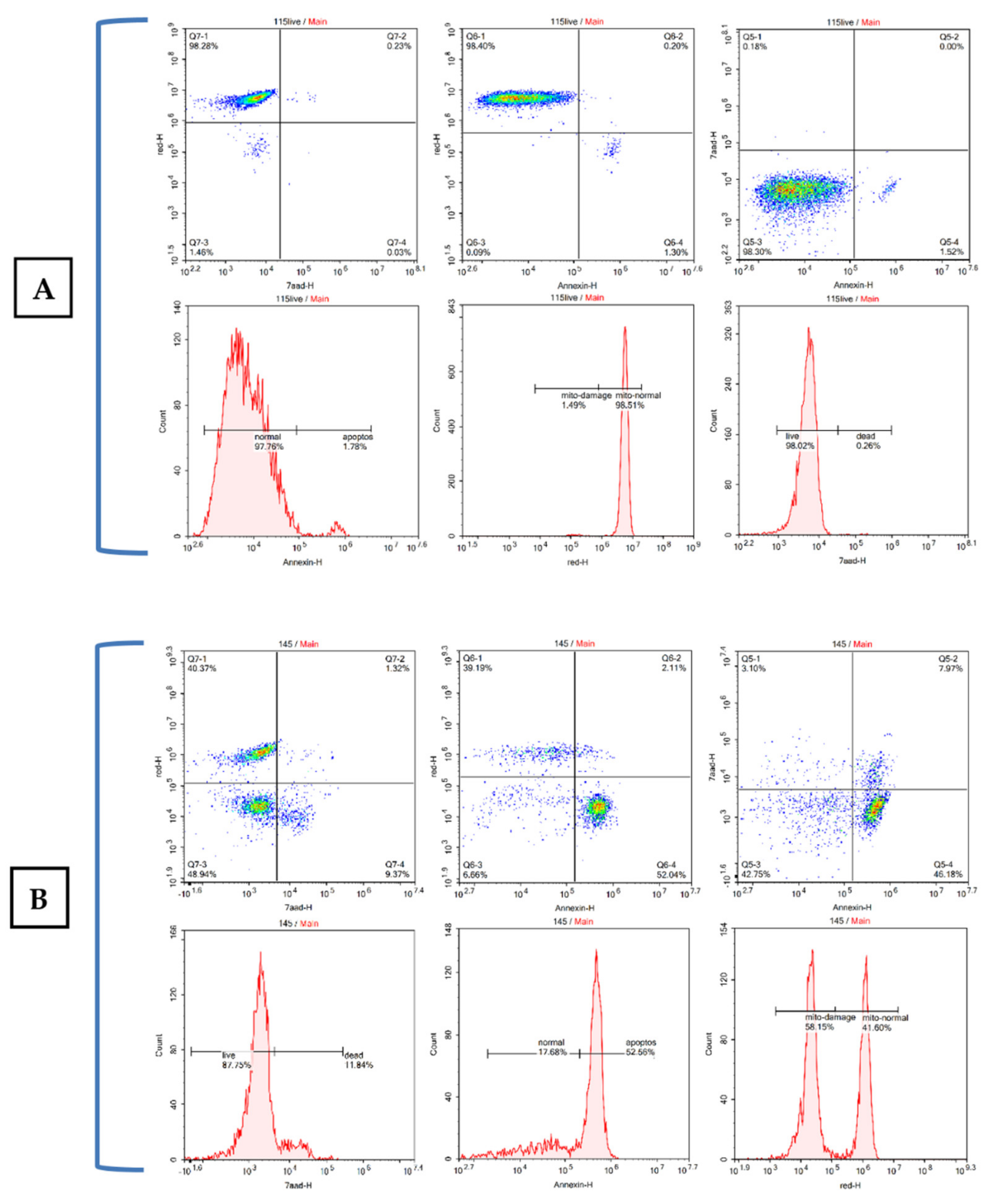
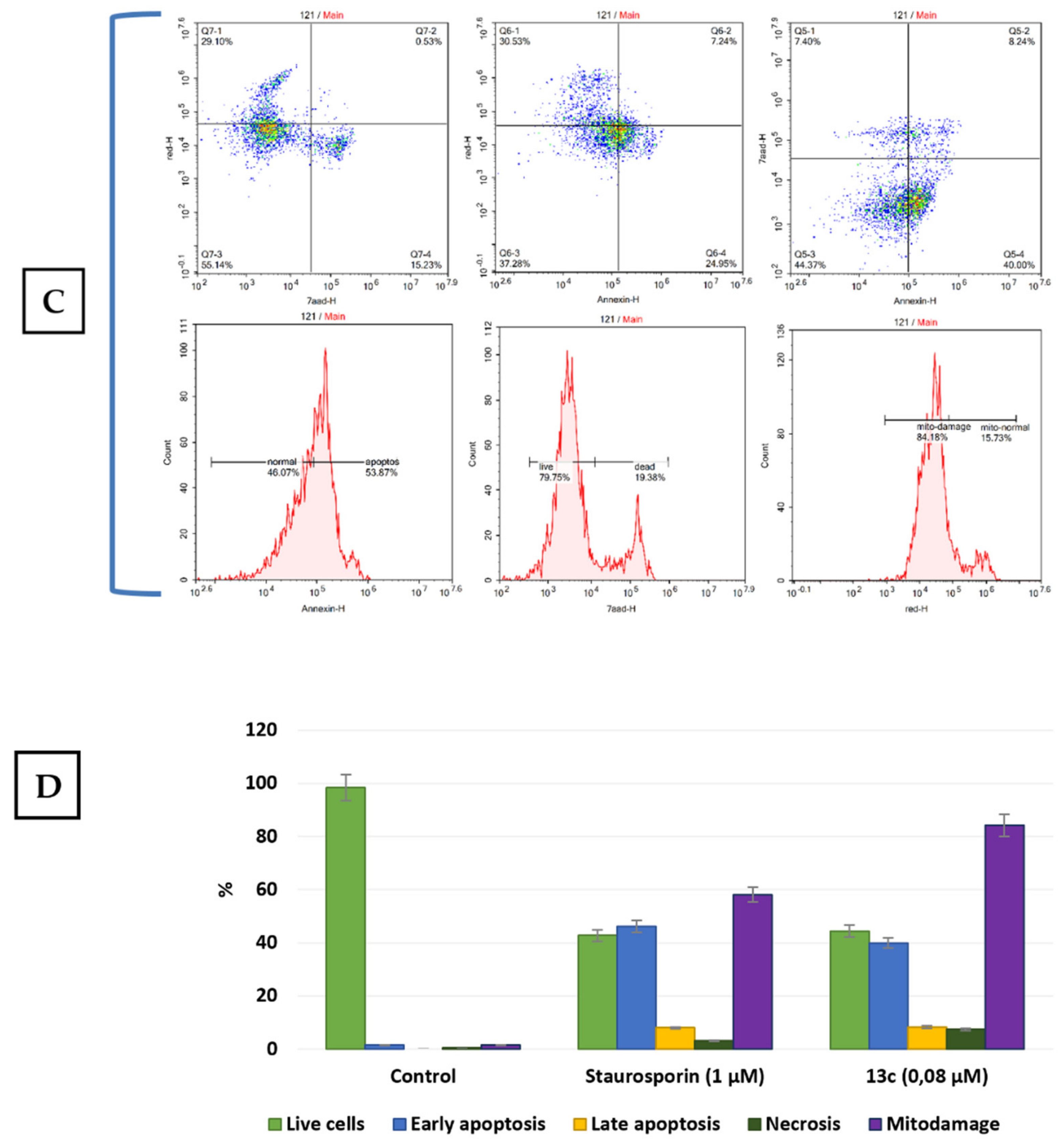
2.2.2. Apoptosis and Cell Cycle Research
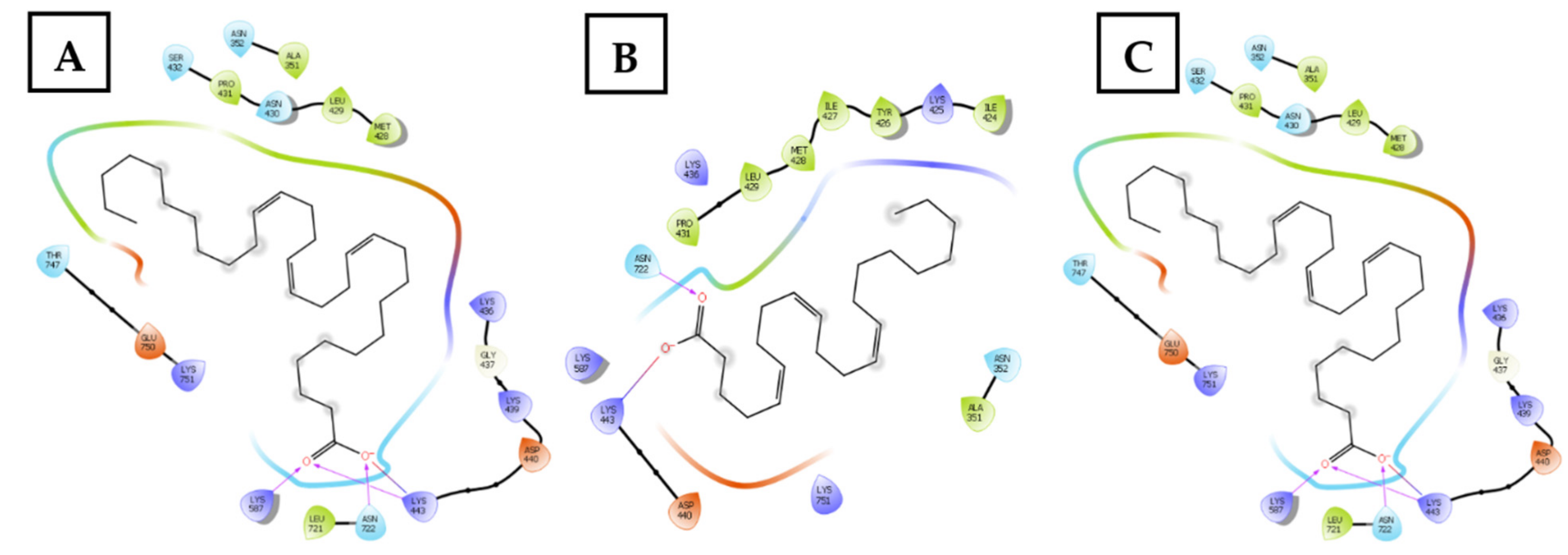
2.2.3. Topoisomerase I Inhibition Assay and Molecular Docking Studies
2.2.4. Studying the Effect of Trienoic Acids on Mitochondria
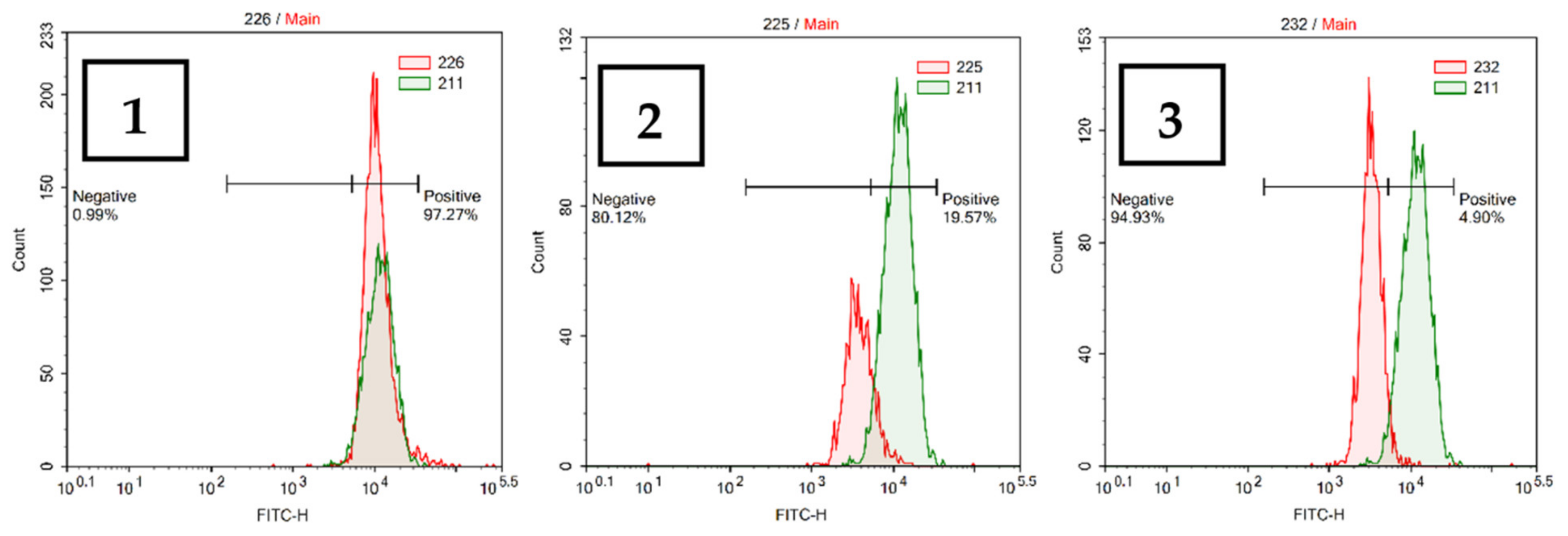
2.2.5. Cytochrome C Release from Mitochondria
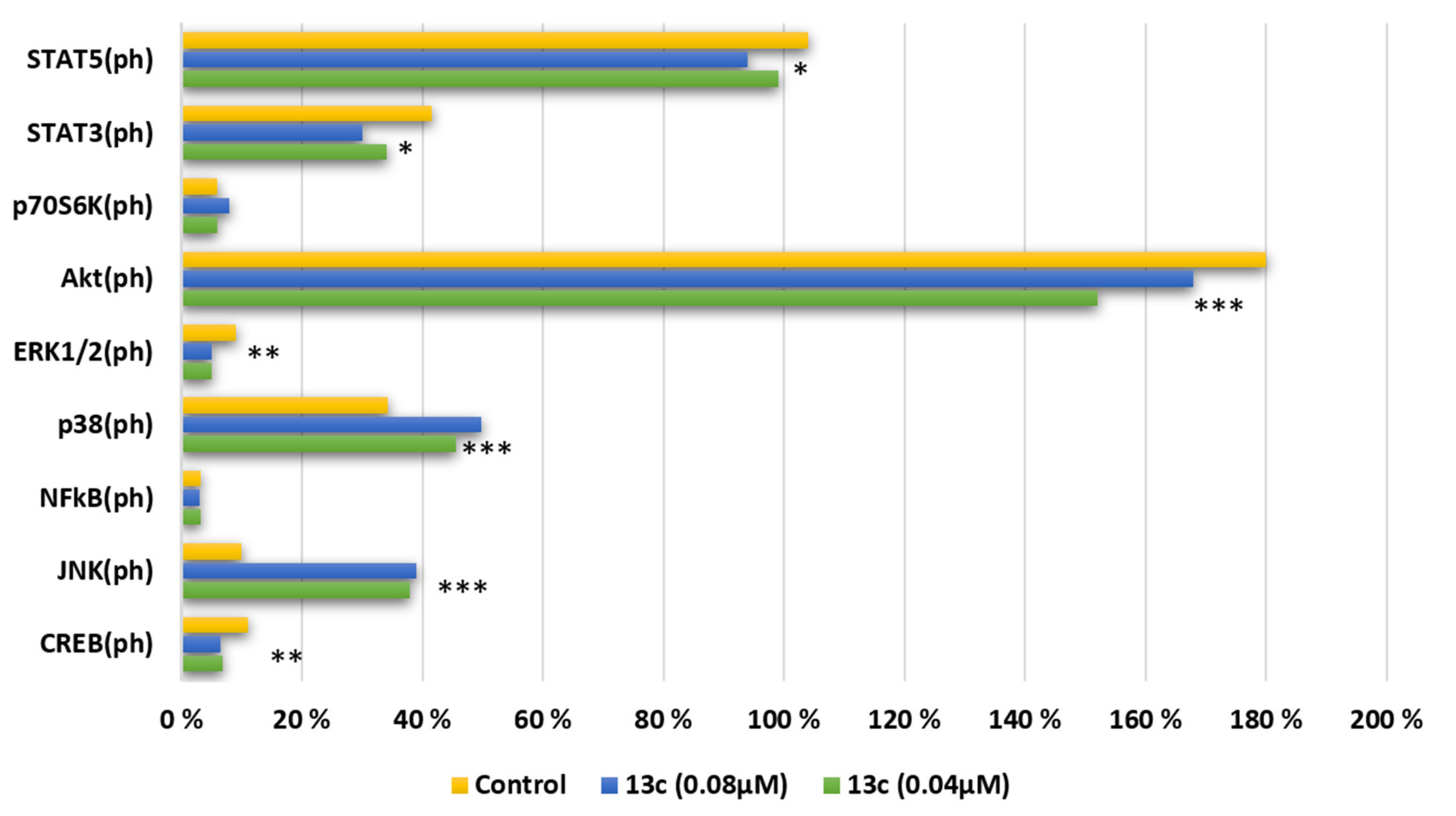
2.2.6. Major Kinases and Their Phosphorylation Status of Nine Signaling Pathways for Cell Growth and Proliferation
3. Materials and Methods
3.1. Chemistry
3.2. Cell Culturing
3.3. DNA Topoisomerase I Assay
3.4. Cytotoxicity Assay
3.5. Viability and Apoptosis
3.6. Cell Cycle Analysis
3.7. Mitochondrial Damage
3.8. Histone H2A.X Analysis
3.9. Cytochrome C Release Analysis
3.10. Multiplex Analysis of Early Apoptosis Markers
3.11. Chemical Experimental Data
3.11.1. Procedure for Preparation of Alka-3-in-1-ols (4a–e)
Oct-3-yn-1-ol (4a)
Dec-3-yn-1-ol (4b)
Dodec-3-yn-1-ol (4c)
Tetradec-3-yn-1-ol (4d)
Hexadec-3-yn-1-ol (4e)
3.11.2. Synthesis of (Z)-alk-3-en-1-ols (5)
(Z)-Oct-3-en-1-ol (5a)
(Z)-Dec-3-en-1-ol (5b)
(Z)-Dodec-3-en-1-ol (5c)
(Z)-Tetradec-3-en-1-ol (5d)
(Z)-Hexadec-3-en-1-ol (5e)
3.11.3. Synthesis of (Z)-1-Bromoalk-3-enes (7)
(Z)-1-Bromoct-3-ene (7a)
(Z)-1-Bromdec-3-ene (7b)
(Z)-1-Bromdodec-3-ene (7c)
(Z)-1-Bromtetradec-3-ene (7d)
(Z)-1-Bromhexadec-3-ene (7e)
3.11.4. Synthesis of Alk-5-en-1-ynes (8a–e)
(Z)-Dec-5-en-1-yne (8a)
(Z)-Dodec-5-en-1-yne (8b)
(Z)-Tetradec-5-en-1-yne (8c)
(Z)-Hexadec-5-en-1-yne (8d)
(Z)-Octadec-5-en-1-yne (8e)
3.11.5. Synthesis of (Z)-alk-1,2,6-trienes (9a–e)
(Z)-Undeca-1,2,6-triene (9a)
(Z)-Trideca-1,2,6-triene (9b)
(Z)-Pentadeca-1,2,6-triene (9c)
(Z)-Heptadeca-1,2,6-triene (9d)
(Z)-Nonadeca-1,2,6-triene (9e)
3.11.6. Cross-Cyclomagnesiation of (Z)-alk-1,2,6-trienes (9a–e) and Allene Alcohol Tetrahydropyran Ethers (10a–d) with EtMgBr in the Presence of Mg Metal and Cp2TiCl2 Catalyst (General Procedure)
2-((5Z,9Z,13Z)-Octadeca-5,9,13-trien-1-yloxy)tetrahydro-2H-pyran (12a)
2-((5Z,9Z,13Z)-Icosa-5,9,13-trien-1-yloxy)tetrahydro-2H-pyran (12b)
2-((5Z,9Z,13Z)-Docosa-5,9,13-trien-1-yloxy)tetrahydro-2H-pyran (12c)
2-((9Z,13Z,17Z)-Triaconta-9,13,17-trien-1-yloxy)tetrahydro-2H-pyran (12d)
2-((11Z,15Z,19Z)-Triaconta-11,15,19-trien-1-yloxy)tetrahydro-2H-pyran (12e)
2-((11Z,15Z,19Z)-Dotriaconta-11,15,19-trien-1-yloxy)tetrahydro-2H-pyran (12f)
2-((13Z,17Z,21Z)-Dotriaconta-13,17,21-trien-1-yloxy)tetrahydro-2H-pyran (12g)
3.11.7. General Procedures for the Preparation of 1Z,5Z,9Z-Dienoic Acids 13a–g
(5Z,9Z,13Z)-Octadeca-5,9,13-trienoic acid (13a)
(5Z,9Z,13Z)-Icosa-5,9,13-trienoic acid (13b)
(5Z,9Z,13Z)-Docosa-5,9,13-trienoic acid (13c)
(9Z,13Z,17Z)-Triaconta-9,13,17-trienoic acid (13d)
(11Z,15Z,19Z)-Triaconta-11,15,19-trienoic acid (13e)
(11Z,15Z,19Z)-Dotriaconta-11,15,19-trienoic acid (13f)
(13Z,17Z,21Z)-Dotriaconta-13,17,21-trienoic acid (13g)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase Isozymes: The Biology of Prostaglandin Synthesis and Inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkup, S.E.; Cheng, Z.; Elmes, M.; Wathes, D.C.; Abayasekara, D.R.E. Polyunsaturated fatty acids modulate prostaglandin synthesis by ovine amnion cells in vitro. Reproduction 2010, 140, 943–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uttaro, A.D. Biosynthesis of Polyunsaturated Fatty Acids in Lower Eukaryotes. IUBMB Life 2006, 58, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-G.; An, J.-U.; Ko, Y.-J.; Parkd, J.-B.; Oh, D.-K. Enzymatic synthesis of new hepoxilins and trioxilins from polyunsaturated fatty acids. Green Chem. 2019, 21, 3172–3181. [Google Scholar] [CrossRef]
- Endo, Y.; Tsunokake, K.; Ikeda, I. Effects of Non-Methylene-Interrupted Polyunsaturated Fatty Acid, Sciadonic (All-cis-5,11,14-eicosatrienoic Acid) on Lipid Metabolism in Rats. Biosci. Biotechnol. Biochem. 2009, 73, 577–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppenbrouwers, T.; Hogervorst, J.H.C.; Garssen, J.; Wichers, H.J.; Willemsen, L.E.M. Long Chain Polyunsaturated Fatty Acids (LCPUFAs) in the Prevention of Food Allergy. Front. Immunol. 2019, 10, 1118. [Google Scholar] [CrossRef]
- Tasdemir, D.; Topaloglu, B.; Perozzo, R.; Brun, R.; O’Neill, R.; Carballeira, N.M.; Zhang, X.; Tonge, P.J.; Lindeng, A.; Ruedi, P. Marine natural products from the Turkish sponge Agelas oroides that inhibit the enoyl reductases from Plasmodium falciparum, Mycobacterium tuberculosis and Escherichia coli. Bioorganic Med. Chem. 2007, 15, 6834–6845. [Google Scholar] [CrossRef]
- Gu, Z.; Shan, K.; Chen, H.; Chen, Y.Q. Polyunsaturated Fatty Acids and Their Role in Cancer Chemoprevention. Curr. Pharmacol. Rep. 2015, 1, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dembitsky, V.M.; Srebnik, M. Natural halogenated fatty acids:their analogues and derivatives. Prog. Lipid Res. 2002, 41, 315–367. [Google Scholar] [CrossRef]
- D’yakonov, V.A.; Dzhemileva, L.U.; Dzhemilev, U.M. Natural Compounds with bis-Methylene-Interrupted Z-Double Bonds: Plant Sources, Strategies of Total Synthesis, Biological Activity, and Perspectives. Phytochem. Rev. 2021, 20, 325–342. [Google Scholar] [CrossRef]
- Sibbons, C.M.; Irvine, N.A.; Perez-Mojica, J.E.; Calder, P.C.; Lillycrop, K.A.; Fielding, B.A.; Burdge, G.C. Polyunsaturated Fatty acid Biosynthesis involving Δ8 Desaturation and Differential DNA Methylation of FADS2 regulates Proliferation of human Peripheral Blood Mononuclear cells. Front. Immunol. 2018, 9, 432. [Google Scholar] [CrossRef] [Green Version]
- Sokola-Wysoczanska, E.; Wysoczanski, T.; Wagner, J.; Czyz, K.; Bodkowski, R.; Lochynski, S.; Patkowska-Sokola, B. Polyunsaturated Fatty Acids and Their Potential Therapeutic Role in Cardiovascular System Disorders—A Review. Nutrients 2018, 10, 1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, B.M.; Ma, D.W.L. Are all n-3 polyunsaturated fatty acids created equal. Lipids Health Dis. 2009, 8, 33. [Google Scholar] [CrossRef] [Green Version]
- Bazinet, R.P.; Laye, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. AOP 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Kawashima, H. Novel Non-methylene-Interrupted Dienoic and Trienoic Fatty Acids with a Terminal Double Bond in Ovaries of the Limpet Cellana toreuma. Lipids 2020, 55, 285–290. [Google Scholar] [CrossRef]
- Zakhartsev, M.V.; Naumenko, N.V.; Chelomin, V.P. Non-methylene-Interrupted Fatty Acids in Phospholipids of the Membranes of the Mussel Crenomytilus grayanus. Russ. J. Mar. Biol. 1998, 24, 183–186. [Google Scholar]
- Kornprobst, J.-M.; Barnathan, G. Demospongic Acids Revisited. Mar. Drugs 2010, 8, 2569–2577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carballeira, N.M.; Emiliano, A.; Guzman, A. Facile syntheses for (5Z,9Z)-5,9-hexadecadienoic acid, (5Z,9Z)-5,9-nonadecadienoic acid, and (5Z,9Z)-5,9-eicosadienoic acid through a common synthetic route. Chem. Phys. Lipids 1999, 100, 33–40. [Google Scholar] [CrossRef]
- Zhou, X.; Shang, J.; Qin, M.; Wang, J.; Jiang, B.; Yang, H.; Zhang, Y. Fractionated Antioxidant and Anti-inflammatory Kernel Oil from Torreya fargesii. Molecules 2019, 24, 3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhanga, J.; Zhanga, S.-D.; Wanga, P.; Guoa, N.; Wanga, W.; Yaoa, L.-P.; Yangb, Q.; Eferthd, T.; Jiaoa, J.; Fu, Y.-J. Pinolenic acid ameliorates oleic acid-induced lipogenesis and oxidative stress via AMPK/SIRT1 signaling pathway in HepG2 cells. Eur. J. Pharmacol. 2019, 861, 172618. [Google Scholar] [CrossRef]
- Carballeira, N.M.; Reyes, E.D.; Sostre, A.; Rodriguez, A.D.; Rodriguez, J.L.; Gonzalez, F.A. Identification of the Novel Antimicrobial Fatty Acid (5Z,9Z)-14-Methyl-5,9-pentadecadienoic Acid in Eunicea succinea. J. Nat. Prod. 1997, 60, 502–504. [Google Scholar] [CrossRef]
- Carballeira, N.M. New advances in fatty acids as antimalarial, antimycobacterial and antifungal agents. Prog. Lipid Res. 2008, 47, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Carballeira, N.M.; Betancourt, J.E.; Orellano, E.A.; Gonzalez, F.A. Total Synthesis and Biological Evaluation of (5Z,9Z)-5,9-Hexadecadienoic Acid, an Inhibitor of Human Topoisomerase I. J. Nat. Prod. 2002, 65, 1715–1718. [Google Scholar] [CrossRef]
- Nemoto, T.; Yoshino, G.; Ojika, M.; Sakagam, Y. Amphimic Acids and Related Long-chain Fatty Acids as DNA Topoisomerase I Inhibitors from an Australian Sponge, Amphimedon sp.: Isolation, Structure, Synthesis, and Biological Evaluation. Tetrahedron 1997, 53, 16699–16710. [Google Scholar] [CrossRef]
- D’yakonov, V.A.; Makarov, A.A.; Dzhemileva, L.U.; Makarova, E.K.; Khusnutdinova, E.K.; Dzhemilev, U.M. The facile synthesis of the 5Z,9Z-dienoic acids and their topoisomerase I inhibitory activity. Chem. Commun. 2013, 49, 8401–8403. [Google Scholar] [CrossRef] [Green Version]
- D’yakonov, V.A.; Dzhemileva, L.U.; Makarov, A.A.; Mulyukova, A.R.; Baev, D.S.; Khusnutdinova, E.K.; Tolstikova, T.G.; Dzhemilev, U.M. 11-Phenylundeca-5Z,9Z-dienoic Acid: Stereoselective Synthesis and Dual Topoisomerase I/IIα Inhibition. Curr. Cancer Drug Targets 2015, 15, 504–510. [Google Scholar] [CrossRef]
- D’yakonov, V.A.; Dzhemileva, L.U.; Tuktarova, R.A.; Ishmukhametova, S.R.; Yunusbaeva, M.M.; Ramazanova, I.R.; Dzhemilev, U.M. Novel Hybrid Molecules on the Basis of Steroids and (5Z,9Z)-Tetradeca-5,9-dienoic Acid: Synthesis, Anti-Cancer Studies and Human Topoisomerase I Inhibitory Activity. In Anticancer Agents Med. Chem.; 2017; Volume 17, pp. 1126–1135. [Google Scholar]
- D’yakonov, V.A.; Dzhemileva, L.U.; Makarov, A.A.; Mulyukova, A.R.; Baev, D.S.; Khusnutdinova, E.K.; Tolstikova, T.G.; Dzhemilev, U.M. nZ,(n+4)Z-Dienoic fatty acid: A new method for the synthesis and inhibitory action on topoisomerase I and II α. Med. Chem. Res. 2016, 25, 30–39. [Google Scholar] [CrossRef]
- Carballeira, N.M.; Medina, J.R. New∆5,9fatty acids in the phospholipids of the sea anemone Stoichactis helianthus. J. Nat. Prod. 1994, 57, 1688–1695. [Google Scholar] [CrossRef]
- Castelli, S.; Campagna, A.; Vassallo, O.; Tesauro, C.; Fiorani, P.; Tagliatesta, P.; Oteri, F.; Falconi, M.; Majumder, H.K.; Desideri, A. Conjugated eicosapentaenoic acid inhibits human topoisomerase IB with a mechanism different from camptothecin. Arch. Biochem. Biophys. 2009, 486, 103–110. [Google Scholar] [CrossRef]
- Makarov, A.A.; Dzhemileva, L.U.; Salimova, A.R.; Makarova, E.K.; Ramazanov, I.R.; D’yakonov, V.A.; Dzhemilev, U.M. New Synthetic Derivatives of Natural 5Z,9Z-Dienoic Acids: Stereoselective Synthesis and Study of the Antitumor Activity. Bioorg. Chem. 2020, 104, 104303. [Google Scholar] [CrossRef]
- Aronis, A.; Melendez, J.A.; Golan, O.; Shilo, S.; Dicter, N.; Tirosh, O. Potentiation of Fasmediated apoptosis by attenuated production of mitochondria-derived reactive oxygen species. Cell Death Differ. 2003, 10, 335–344. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- Marchetti, P.; Castedo, M.; Susin, S.A.; Zamzami, N.; Hirsch, T.; Macho, A.; Haeffner, A.; Hirsch, F.; Geuskens, M.; Kroemer, G. Mitochondrial permeability transition is a central coordinating event of apoptosis. J. Exp. Med. 1996, 184, 1155–1160. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, X. Cytochrome c-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef]
- Zamzami, N.; Kroemer, G. The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2001, 2, 67–71. [Google Scholar] [CrossRef]
- Kagan, V.E.; Borisenko, G.G.; Tyurina, Y.Y.; Tyurin, V.A.; Jiang, J.; Potapovich, A.I.; Kini, V.; Amoscato, A.A.; Fujii, Y. Oxidative lipidomics of apoptosis: Redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic. Biol. Med. 2004, 37, 1963–1985. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Xu, P.Z.; Gottlob, K.; Chen, M.L.; Sokol, K.; Shiyanova, T.; Roninson, I.; Weng, W.; Suzuki, R.; Tobe, K.; et al. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001, 15, 2203–2208. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.R.; Toker, A. Akt isoform-specific signaling in breast cancer: Uncovering an anti-migratory role for palladin. Cell Adhes. Migr. 2011, 5, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, 11189. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef] [PubMed]
- Dzhemileva, L.U.; D’yakonov, V.A.; Islamov, I.I.; Yunusbaeva, M.M.; Dzhemilev, U.M. New 1Z,5Z-diene macrodiolides: Catalytic synthesis, anticancer activity, induction of mitochondrial apoptosis, and effect on the cell cycle. Bioorg. Chem. 2020, 99, 103832. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Weng, Y.; Hongand, W.-X.; Zhang, Q. Efficient Synthesis of Unsaturated 1-Monoacyl Glycerols for in meso Crystallization of Membrane Proteins. Synlett 2011, 6, 809–812. [Google Scholar]
- Gruiec, R.; Noiretand, N.; Patin, H. Useful direct conversion of tetrahydropyranyl ethers of fatty alcohols into fatty acids. J. Am. Oil Chem. Soc. 1995, 72, 1083–1085. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acids | Jurkat, IC50, µM | K562, IC50, µM | U937, IC50, µM | HL60, IC50, µM | HEK293, IC50, µM | HeLa, IC50, µM | Fibroblasts, IC50, µM |
|---|---|---|---|---|---|---|---|
| 13a | 0.178 ± 0.022 | 0.211 ± 0.021 | 0.165 ± 0.019 | 0.159 ± 0.014 | 0.617 ± 0.054 | 0.574 ± 0.052 | 1.119 ± 0.104 |
| 13b | 0.147 ± 0.015 | 0.172 ± 0.016 | 0.139 ± 0.013 | 0.127 ± 0.011 | 0.523 ± 0.049 | 0.489 ± 0.041 | 0.937 ± 0.086 |
| 13c | 0.105 ± 0.009 | 0.136 ± 0.012 | 0.098 ± 0.010 | 0.086 ± 0.008 | 0.451 ± 0.039 | 0.311 ± 0.027 | 0.754 ± 0.069 |
| 13d | 0.448 ± 0.043 | 0.479 ± 0.046 | 0.456 ± 0.041 | 0.409 ± 0.037 | 1.297 ± 0.114 | 1.029 ± 0.091 | 1.738 ± 0.154 |
| 13e | 0.421 ± 0.038 | 0.467 ± 0.043 | 0.394 ± 0.037 | 0.387 ± 0.039 | 1.082 ± 0.093 | 0.983 ± 0.086 | 1.597 ± 0.139 |
| 13f | 0.454 ± 0.046 | 0.471 ± 0.051 | 0.461 ± 0.043 | 0.404 ± 0.040 | 1.211 ± 0.106 | 1.087 ± 0.094 | 1.914 ± 0.176 |
| 13g | 0.122 ± 0.011 | 0.154 ± 0.014 | 0.117 ± 0.010 | 0.104 ± 0.009 | 0.487 ± 0.046 | 0.354 ± 0.032 | 0.739 ± 0.068 |
| Acids | 13a | 13b | 13c | 13d | 13e | 13f | 13g |
|---|---|---|---|---|---|---|---|
| hTopI inhibition, µM | 0.2 | 0.2 | 0.1 | 0.8 | 0.6 | 0.8 | 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’yakonov, V.A.; Makarov, A.A.; Dzhemileva, L.U.; Ramazanov, I.R.; Makarova, E.K.; Dzhemilev, U.M. Natural Trienoic Acids as Anticancer Agents: First Stereoselective Synthesis, Cell Cycle Analysis, Induction of Apoptosis, Cell Signaling and Mitochondrial Targeting Studies. Cancers 2021, 13, 1808. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13081808
D’yakonov VA, Makarov AA, Dzhemileva LU, Ramazanov IR, Makarova EK, Dzhemilev UM. Natural Trienoic Acids as Anticancer Agents: First Stereoselective Synthesis, Cell Cycle Analysis, Induction of Apoptosis, Cell Signaling and Mitochondrial Targeting Studies. Cancers. 2021; 13(8):1808. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13081808
Chicago/Turabian StyleD’yakonov, Vladimir A., Alexey A. Makarov, Lilya U. Dzhemileva, Ilfir R. Ramazanov, Elina Kh. Makarova, and Usein M. Dzhemilev. 2021. "Natural Trienoic Acids as Anticancer Agents: First Stereoselective Synthesis, Cell Cycle Analysis, Induction of Apoptosis, Cell Signaling and Mitochondrial Targeting Studies" Cancers 13, no. 8: 1808. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13081808