
MRD-Based Therapeutic Decisions in Genetically Defined Subsets of Adolescents and Young Adult Philadelphia-Negative ALL

 , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. ALL in Adolescents and Young Adult Patients
2.1. AYA Patient Identification: Age Ranges
2.2. Ph− ALL: Diagnostic Subsets Defined by Immunophenotype and Cytogenetics/Genetics in AYAs
2.3. Treatment: Traditional Adult vs. Modern Pediatric Regimens
- Intensive chemotherapy regimens inspired to modern pediatric schedules and treatment principles are superior to historical adult-type programs, as demonstrated with very few exceptions by comparative analyses among successive Phase 2 trials and many large non-comparative trials [6,16,17]. Whenever available, retrospective comparisons with historical datasets (not shown in the table and available in study references) confirm an average improvement of outcome measures of 15–25%.
- In these modern AYA or AYA-containing adult ALL studies, the projected survival rates at 5 years (range 3–7 years), assuming “cure” for most patients who remain disease-free at ≥5 years, is 50% and greater (overall survival, OS), with age-related variations and OS rates of 60–70% and occasionally higher in younger age groups.
- Unlike OS, which reflects the cumulative survival effect of both first line and salvage therapies, relapse-free and event-free survival (RFS and EFS) depict the curative potential of upfront therapy only, in CR patients and all study patients, respectively. These figures range 55–70% (RFS) and 40–74% (EFS), once again with significantly better results in younger age groups.
- The overall chemotherapy intensity is increased in pediatric-based regimens, with regard to vincristine, corticosteroids, antimetabolites (cytarabine, methotrexate and 6-mercaptopurine), L-asparaginase and, more recently, Pegylated-asparaginase (Peg-ASP). Consequently, drug-related toxicity may be higher, requiring higher clinical skills for the management and prevention of toxic side effects.
- The improved pediatric-like protocol may consist of an unmodified or modified pediatric schedule, in the latter case adapting some treatment elements to an increasing patient age with attending risks of treatment toxicity. The issue of Peg-ASP dosing and toxicity is highly critical in patients at older age [52,53].
- The patients who achieve CR, namely about 90% of all patients (≥95% in younger age groups), are usually risk-stratified to assess the individual risk class and decide about the application of risk-specific treatments that range, for high-risk (HR) patients, from chemotherapy intensification to allogeneic hematopoietic cell transplantation (HCT) and/or experimental new agents. Most patients at standard-risk (SR) or intermediate-risk can achieve cure with a full chemotherapy regimen including maintenance as standard of care, without HCT, this lowering the incidence of non-lethal and lethal toxicities (10–15% average mortality from HCT).
- In the risk stratification process, by analogy with pediatric trials, the analysis of post-induction MRD is crucial since it has been demonstrated to be the most powerful predictive factor for marrow relapse in multivariable analysis from several studies [7,8,9,10]. Therefore, MRD is currently used together with other risk factors for the definition of risk groups and individual risk profiles.
3. Methods of MRD Assessment
3.1. Multiparameter Flow Cytometry
3.2. PCR for Fusion Genes and Transcripts
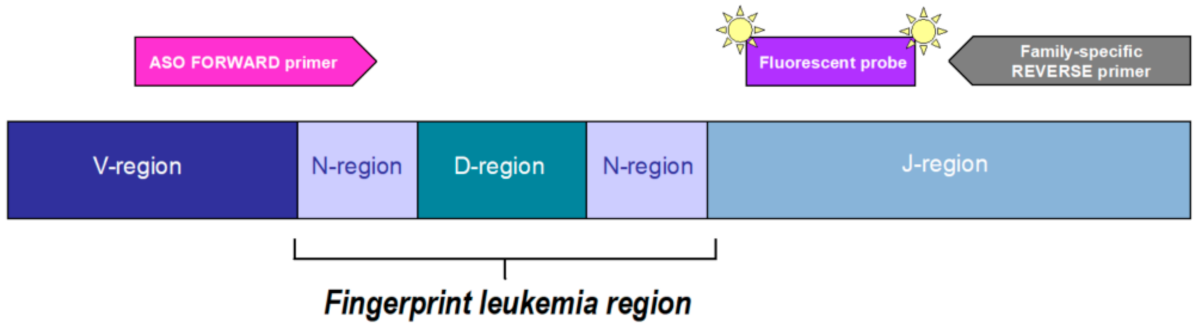
3.3. PCR for Ig and TCR Gene Rearrangements
3.4. Next Generation Sequencing
3.5. Digital Droplet PCR
4. MRD in AYA Ph− ALL
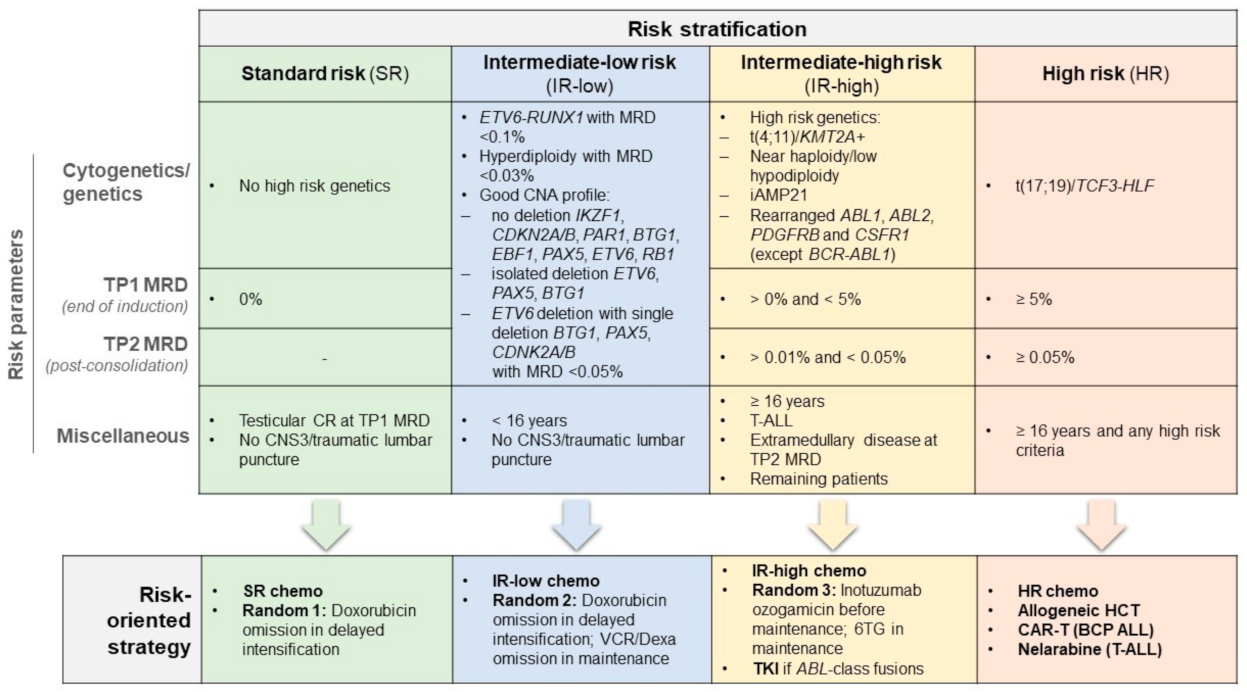
4.1. MRD Study Results for Risk Stratification

{kind=link}
{kind=link}
{kind=link}
| Trial/Study (Ref.) | Patient Age (Years), Median (Range) | MRD Analysis | Favorable MRD Response | Comparative Outcomes: Favorable MRD Cut-Offs Vs. Not | ||
|---|---|---|---|---|---|---|
| Evaluable/CR, No. (%) | Method 1 | Cut-Off 2 | No. (%) | |||
| AYA only (maximum age 40 years) | ||||||
| MRC-UKALL2003 [11] | NR (16–24) | 223/229 (97) | Mol | Negative/<0.01% d29, negative at EOC | 54 (24) | 5-y EFS 93% vs. 63–71% (p = 0.0001) 3 |
| PETHEMA ALL08 [35] | 20 (15–30) | 61/68 (90) | MFC | <0.1% w5–6, <0.05% w19–20 | 48 (77) | NR |
| GIMEMA LAL1308 [39] | NR (18–35) | 64/68 (94)/ 66/68 (97) 49/68 (72)/ 50/68 (73) | MFC Mol | <0.1% d33/78 | 37 (58)/54 (82) 28 (57)/38 (76) | 4-y OS by d33 MRD 67–75% vs. 27–41% (p = 0.002) 4 4-y RFS by d33 MRD 67–73% vs. 27–43% (p ≤ 0.025) 4 4-y OS by d78 MRD 74–77% vs. 31–39% (p ≤ 0.01) 4 4-y RFS by d78 MRD 71–72% vs. 26–34% (p ≤ 0.01) 4 |
| MDACC (aBFM/HyperCVAD) * [36] | 22 (13–39) | 93/199 (47) | MFC | <0.01% d29/d84 | 58 (62) | 5-y OS by d29 MRD 75% vs. 40% (p = 0.004) 5-y OS by d84 MRD 75% vs. 22% (p = 0.0004) |
| CALGB 10,403 [37] | 24 (17–39) | 80/237 (34) | Mol | Negative at EOI | 35 (44) | 3-y RFS 85% vs. 54% (p = 0.0006) |
| AYA and adults (maximum age > 40 years) | ||||||
| NOPHO 2008 [14] | 26 (18–45) | NR/218 | Mol | <0.1% d29/d79 | (56–64) 5 | NR |
| GRAALL 2003–2005 [26] | 31 (15–59) | 423/860 (49) | Mol | Negative/<0.01% w6 | 265 (63) | 5-y CIR 23–31% vs. 60% (p ≤ 0.01) |
| GMALL 07/03 [74] | 34 (16–65) | 1057/1857 (57) | Mol | Negative w16 | 625 (59) | 5-y OS 83% vs. 43–68% (p < 0.0001) 6 |
| MDACC [75] | 37 (15–86) | 215/394 (55) | MFC | <0.01% d24/d108 | 147 (68)/194 (90) | 3-y OS by d24 MRD 76% vs. 49–61% (p = 0.001) 7 3-y EFS by d24 MRD 65% vs.16–46% (p < 0.001) 7 |
| PETHEMA ALL-HR11 [49] | 40 (15–60) | 286/289 (99) | MFC | <0.1% w5–6 | 220 (82) | 5-y OS 59% vs. 38% (p < 0.001) |
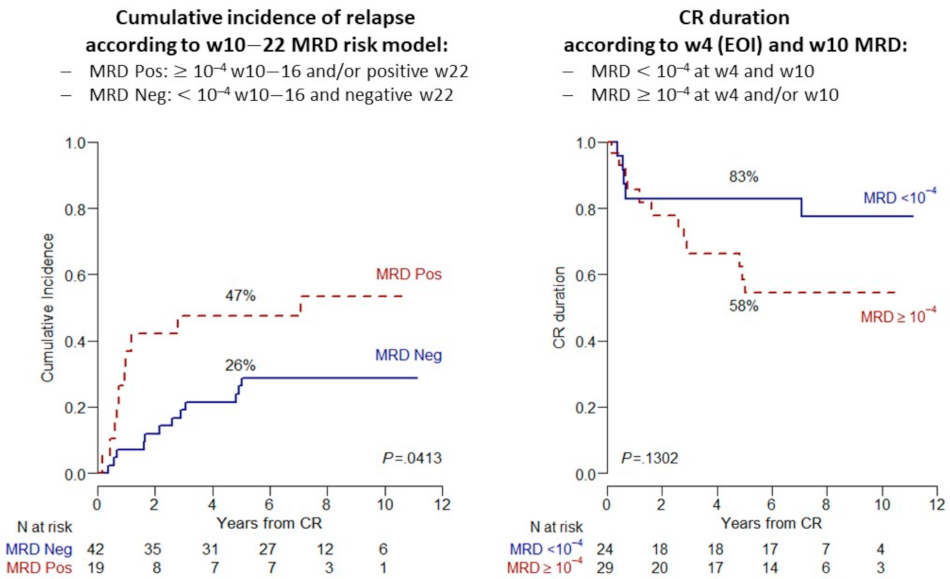
| NILG 10/07 [51] | 41 (18–65) | 109/140 (78) | Mol | <0.01% w10–16/negative w22 | 68 (62) | 5-y OS 78% vs. 34% (p < 0.0001) 5-y RFS 66% vs. 29% (p < 0.0001) |
| HOVON-100 ** [41] | 42 (18–70) | 168/297 (56) | Mol/MFC | <0.01% after consolidation 1 | 126 (75) | NR |
4.2. Terminology of MRD Response for Clinical Purposes
4.3. MRD-Related Outcomes
4.4. Risk of Relapse in MRD Responders
4.5. Allogeneic HCT for MRD Positive States
4.5.1. Allogeneic HCT Results in MRDpos AYA Ph− ALL
| Trial/Study (Ref.) | MRD+ and/or HR Patients Eligible to HCT/HR Protocol (No.) | Had Allogeneic HCT/HR Protocol (No.) | Outcomes * |
|---|---|---|---|
| AYA only (maximum age 40 years) | |||
| PETHEMA ALL08 [35] | 2 MRDpos and 20 HR to HCT or HR protocol | 5 HCT and 13 HR protocol | 4 HCT survivors (80%) and 7 HR protocol survivors (54%) |
| MRC-UKALL2003 [11] | 109 MRDpos to random study and 14 HR to HCT 1 | 64 randomized and 14 HCT | 9 HCT survivors (64%) |
| MDACC [76] | 17 MRDpos or HR to HCT | 17 HCT | 7 survivors (41%), 5 in CR (29%) |
| CALGB 10,403 [37] | 20 HR/other 2 to HCT | 20 HCT | 8 survivors (40%) |
| GIMEMA LAL1308 [40] | 21 MRDpos to HCT and 9 HR to HR protocol | 15 HCT and 12 HR protocol | 4-y OS HR 52.6% vs. SR 73.4% (p = 0.032) 4-y RFS HR 54.2% vs. SR 66.6% (p = 0.51) |
| AYA and adults (maximum age > 40 years) | |||
| NOPHO 2008 [14] | 35 MRDpos to HCT and 45 HR to HR protocol | NR | 5-y EFS 61% 3 |
| GRAALL 2003–2005 [83] | 105 HR MRDpos to HCT | 59 HCT | 3-y OS 65% vs. 40% (p = 0.001) 3-y RFS 56% vs. 22% (p = 0.002) |
| GMALL 07/03 [74] | 196 MRDpos to HCT | 121 HCT | 5-y OS 53% vs. 28% (p < 0.0001) 5-y CRD 56% vs. 9% (p < 0.0001) |
| PETHEMA ALL-HR11 [49] | 66 MRDpos and 40 HR to HCT | 62 HCT | 5-y OS 54% (as treated) |
| NILG 10/07 [51] | 41 MRDpos to HCT | 23 HCT | 5-y OS 35% vs. 14% (p = 0.02) 5-y OS RFS 43% vs. 12% (p = 0.09) |
4.5.2. Pre-Transplantation MRD Status
5. New Therapeutic Options for MRD Positive ALL
5.1. Immunotherapy for BCP ALL
5.1.1. Blinatumomab
5.1.2. Inotuzumab Ozogamicin
5.2. Chimeric Antigen Receptor-Modified T-Cell Therapy
5.3. Investigational Agents for T-ALL
5.3.1. Nelarabine for MRDpos T-ALL
5.3.2. Immunotherapy for MRDpos T-ALL
5.4. Other Experimental Approaches
5.4.1. Molecular Profiling for Precision Medicine
- Most relevant is Ph-like ALL, which is rather frequent in AYA patients and carries a higher risk of MRD persistence. Some of the associated gene abnormalities recognizable in this poor risk entity (ABL-class fusions, CLRF2 deregulation and JAK/STAT and IL7R pathway alterations, among others) are actionable by TK inhibitors, JAK inhibitors (ruxolitinib) and other similar drugs. Trials in children, AYA and adults have been incepted worldwide, sometimes with promising preliminary results [126,127,128,129]. The data from these studies may elucidate which of these new drugs or drug combinations with either chemotherapy, immunotherapy and/or other targeting agents could optimize the outcome of the distinct genetically defined Ph-like ALL subsets.
- Among the BCP ALL subsets to consider for targeted therapy is t (v;11) + ALL or ALL carrying KMT2A gene rearrangements, most frequently t (4;11) + ALL. This entity stands out for its clinical aggressiveness. In this subset, the available molecular studies point to a therapeutic use of BCL-2 inhibitors (venetoclax and navitoclax) and DOT-L1 and histone-deacetylase inhibitors; however, the relative rarity of this ALL syndrome precludes an extensive clinical evaluation of these drugs outside large collaborative clinical trials.
- There are many more candidates for targeted therapy of BCP ALL subsets or ALL in general, as extensively reviewed [122]. Most of these drugs are under investigation in early clinical trials, and it is too early to define exactly their place and/or anticipate their approval for use as standard agents for front-line therapy and/or MRDpos conditions. Worthy of mentioning are the proteasome inhibitors, again the BCL-2 inhibitors and the activators of P53-mediated apoptosis, given the frequent dysregulation of these molecular mechanisms. Likewise, the analysis of bone marrow immune cell contexture led to identify a poor risk ALL subset with PD1 + TIM3 + CD4 + bone marrow T-cells > 0.1% that might be targeted by PD1 checkpoint inhibitors [130].
- Many of the new drugs potentially active in BCP ALL could be exploited in T-ALL as well, namely inhibitors of the antiapoptotic BCL-2 family members and inhibitors of JAK/STAT, PI3K/Akt/mTOR, MAPK and Notch-1, the latter being a typical T-ALL target. While experience with Notch-1 inhibitors has been rather disappointing so far, BCL-2 inhibitors navitoclax and venetoclax induced a CR in six of 16 patients with refractory T-ALL, achieving undetectable MRD in four [129]. ETP ALL may be sensitive to the JAK-2 inhibitor ruxolitinib.
5.4.2. Drug Sensitivity Profiling for Precision Medicine
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Surveillance, Epidemiology, and End Results (SEER) Program. SEER Cancer Statistic Review (1975–2017), National Cancer Institute, DCCPS, Surveillance Research Program, Surveillance Systems. Available online: https://Seer.Cancer.Gov/Seerstat/ (accessed on 17 December 2020).
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadié, M.; Simonetti, A.; Lutz, J.-M.; et al. Incidence of Hematologic Malignancies in Europe by Morphologic Subtype: Results of the HAEMACARE Project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef]
- Pui, C.-H.; Evans, W.E. A 50-Year Journey to Cure Childhood Acute Lymphoblastic Leukemia. Semin. Hematol. 2013, 50, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [Green Version]
- Rowe, J.M. Prognostic Factors in Adult Acute Lymphoblastic Leukaemia: Review. Br. J. Haematol. 2010. [Google Scholar] [CrossRef]
- Siegel, S.E.; Stock, W.; Johnson, R.H.; Advani, A.; Muffly, L.; Douer, D.; Reed, D.; Lewis, M.; Freyer, D.R.; Shah, B.; et al. Pediatric-Inspired Treatment Regimens for Adolescents and Young Adults With Philadelphia Chromosome–Negative Acute Lymphoblastic Leukemia: A Review. JAMA Oncol. 2018, 4, 725. [Google Scholar] [CrossRef]
- Berry, D.A.; Zhou, S.; Higley, H.; Mukundan, L.; Fu, S.; Reaman, G.H.; Wood, B.L.; Kelloff, G.J.; Jessup, J.M.; Radich, J.P. Association of Minimal Residual Disease With Clinical Outcome in Pediatric and Adult Acute Lymphoblastic Leukemia: A Meta-Analysis. JAMA Oncol. 2017, 3, e170580. [Google Scholar] [CrossRef] [PubMed]
- Bassan, R.; Intermesoli, T.; Scattolin, A.; Viero, P.; Maino, E.; Sancetta, R.; Carobolante, F.; Gianni, F.; Stefanoni, P.; Tosi, M.; et al. Minimal Residual Disease Assessment and Risk-Based Therapy in Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2017, 17, S2–S9. [Google Scholar] [CrossRef]
- Hoelzer, D.; Bassan, R.; Dombret, H.; Fielding, A.; Ribera, J.M.; Buske, C. Acute Lymphoblastic Leukaemia in Adult Patients: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2016, 27, v69–v82. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Jabbour, E.; Albitar, M.; de Lima, M.; Gore, L.; Jorgensen, J.; Logan, A.C.; Park, J.; Ravandi, F.; Shah, B.; et al. Recommendations for the Assessment and Management of Measurable Residual Disease in Adults with Acute Lymphoblastic Leukemia: A Consensus of North American Experts. Am. J. Hematol. 2019, 94, 257–265. [Google Scholar] [CrossRef]
- Hough, R.; Rowntree, C.; Goulden, N.; Mitchell, C.; Moorman, A.; Wade, R.; Vora, A. Efficacy and Toxicity of a Paediatric Protocol in Teenagers and Young Adults with Philadelphia Chromosome Negative Acute Lymphoblastic Leukaemia: Results from UKALL 2003. Br. J. Haematol. 2016, 172, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, E.C.; Devidas, M.; Chen, S.; Salzer, W.L.; Raetz, E.A.; Loh, M.L.; Mattano, L.A.; Cole, C.; Eicher, A.; Haugan, M.; et al. Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults with High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children’s Oncology Group Study AALL0232. J. Clin. Oncol. 2016, 34, 2380–2388. [Google Scholar] [CrossRef]
- Winter, S.S.; Dunsmore, K.P.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Improved Survival for Children and Young Adults with T-Lineage Acute Lymphoblastic Leukemia: Results From the Children’s Oncology Group AALL0434 Methotrexate Randomization. J. Clin. Oncol. 2018, 36, 2926–2934. [Google Scholar] [CrossRef] [PubMed]
- Toft, N.; Birgens, H.; Abrahamsson, J.; Griškevičius, L.; Hallböök, H.; Heyman, M.; Klausen, T.W.; Jónsson, Ó.; Palk, K.; Pruunsild, K.; et al. Results of NOPHO ALL2008 Treatment for Patients Aged 1–45 Years with Acute Lymphoblastic Leukemia. Leukemia 2018, 32, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Bassan, R.; Hoelzer, D. Modern Therapy of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2011, 29, 532–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carobolante, F.; Chiaretti, S.; Skert, C.; Bassan, R. Practical Guidance for the Management of Acute Lymphoblastic Leukemia in the Adolescent and Young Adult Population. Ther. Adv. Hematol. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rank, C.U.; Schmiegelow, K. Optimal Approach to the Treatment of Young Adults with Acute Lymphoblastic Leukemia in 2020. Semin. Hematol. 2020, 57, 102–114. [Google Scholar] [CrossRef]
- Roberts, K.G. Genetics and Prognosis of ALL in Children vs. Adults. Hematology 2018, 2018, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Moorman, A.V. The Clinical Relevance of Chromosomal and Genomic Abnormalities in B-Cell Precursor Acute Lymphoblastic Leukaemia. Blood Rev. 2012, 26, 123–135. [Google Scholar] [CrossRef]
- Chiaretti, S.; Vitale, A.; Cazzaniga, G.; Orlando, S.M.; Silvestri, D.; Fazi, P.; Valsecchi, M.G.; Elia, L.; Testi, A.M.; Mancini, F.; et al. Clinico-Biological Features of 5202 Patients with Acute Lymphoblastic Leukemia Enrolled in the Italian AIEOP and GIMEMA Protocols and Stratified in Age Cohorts. Haematologica 2013, 98, 1702–1710. [Google Scholar] [CrossRef]
- Lafage-Pochitaloff, M.; Baranger, L.; Hunault, M.; Cuccuini, W.; Lefebvre, C.; Bidet, A.; Tigaud, I.; Eclache, V.; Delabesse, E.; Bilhou-Nabéra, C.; et al. Impact of Cytogenetic Abnormalities in Adults with Ph-Negative B-Cell Precursor Acute Lymphoblastic Leukemia. Blood 2017, 130, 1832–1844. [Google Scholar] [CrossRef] [Green Version]
- Herold, T.; Baldus, C.D.; Gökbuget, N. Ph-like Acute Lymphoblastic Leukemia in Older Adults. N. Engl. J. Med. 2014, 371, 2235. [Google Scholar] [CrossRef]
- Hamadeh, L.; Enshaei, A.; Schwab, C.; Alonso, C.N.; Attarbaschi, A.; Barbany, G.; den Boer, M.L.; Boer, J.M.; Braun, M.; Dalla Pozza, L.; et al. Validation of the United Kingdom Copy-Number Alteration Classifier in 3239 Children with B-Cell Precursor ALL. Blood Adv. 2019, 3, 148–157. [Google Scholar] [CrossRef]
- Stanulla, M.; Dagdan, E.; Zaliova, M.; Möricke, A.; Palmi, C.; Cazzaniga, G.; Eckert, C.; te Kronnie, G.; Bourquin, J.-P.; Bornhauser, B.; et al. IKZF1plus Defines a New Minimal Residual Disease–Dependent Very-Poor Prognostic Profile in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 1240–1249. [Google Scholar] [CrossRef] [Green Version]
- Petit, A.; Trinquand, A.; Chevret, S.; Ballerini, P.; Cayuela, J.-M.; Grardel, N.; Touzart, A.; Brethon, B.; Lapillonne, H.; Schmitt, C.; et al. Oncogenetic Mutations Combined with MRD Improve Outcome Prediction in Pediatric T-Cell Acute Lymphoblastic Leukemia. Blood 2018, 131, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Beldjord, K.; Chevret, S.; Asnafi, V.; Huguet, F.; Boulland, M.-L.; Leguay, T.; Thomas, X.; Cayuela, J.-M.; Grardel, N.; Chalandon, Y.; et al. Oncogenetics and Minimal Residual Disease Are Independent Outcome Predictors in Adult Patients with Acute Lymphoblastic Leukemia. Blood 2014, 123, 3739–3749. [Google Scholar] [CrossRef] [PubMed]
- Coustan-Smith, E.; Mullighan, C.G.; Onciu, M.; Behm, F.G.; Raimondi, S.C.; Pei, D.; Cheng, C.; Su, X.; Rubnitz, J.E.; Basso, G.; et al. Early T-Cell Precursor Leukaemia: A Subtype of Very High-Risk Acute Lymphoblastic Leukaemia. Lancet Oncol. 2009, 10, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Lamb, A.V.; O’Brien, S.; Ravandi, F.; Konopleva, M.; Jabbour, E.; Zuo, Z.; Jorgensen, J.; Lin, P.; Pierce, S.; et al. Early T-Cell Precursor Acute Lymphoblastic Leukemia/Lymphoma (ETP-ALL/LBL) in Adolescents and Adults: A High-Risk Subtype. Blood 2016, 127, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.; Graux, C.; Lhermitte, L.; Lara, D.; Cluzeau, T.; Leguay, T.; Cieslak, A.; Trinquand, A.; Pastoret, C.; Belhocine, M.; et al. Early Response–Based Therapy Stratification Improves Survival in Adult Early Thymic Precursor Acute Lymphoblastic Leukemia: A Group for Research on Adult Acute Lymphoblastic Leukemia Study. J. Clin. Oncol. 2017, 35, 2683–2691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boissel, N.; Auclerc, M.-F.; Lhéritier, V.; Perel, Y.; Thomas, X.; Leblanc, T.; Rousselot, P.; Cayuela, J.-M.; Gabert, J.; Fegueux, N.; et al. Should Adolescents With Acute Lymphoblastic Leukemia Be Treated as Old Children or Young Adults? Comparison of the French FRALLE-93 and LALA-94 Trials. J. Clin. Oncol. 2003, 21, 774–780. [Google Scholar] [CrossRef]
- Stock, W.; La, M.; Sanford, B.; Bloomfield, C.D.; Vardiman, J.W.; Gaynon, P.; Larson, R.A.; Nachman, J. What Determines the Outcomes for Adolescents and Young Adults with Acute Lymphoblastic Leukemia Treated on Cooperative Group Protocols? A Comparison of Children’s Cancer Group and Cancer and Leukemia Group B Studies. Blood 2008, 112, 1646–1654. [Google Scholar] [CrossRef] [Green Version]
- For the Japan Adult Leukemia Study Group (JALSG); Hayakawa, F.; Sakura, T.; Yujiri, T.; Kondo, E.; Fujimaki, K.; Sasaki, O.; Miyatake, J.; Handa, H.; Ueda, Y.; et al. Markedly Improved Outcomes and Acceptable Toxicity in Adolescents and Young Adults with Acute Lymphoblastic Leukemia Following Treatment with a Pediatric Protocol: A Phase II Study by the Japan Adult Leukemia Study Group. Blood Cancer J. 2014, 4, e252. [Google Scholar] [CrossRef] [Green Version]
- Nachman, J.B.; La, M.K.; Hunger, S.P.; Heerema, N.A.; Gaynon, P.S.; Hastings, C.; Mattano, L.A.; Sather, H.; Devidas, M.; Freyer, D.R.; et al. Young Adults With Acute Lymphoblastic Leukemia Have an Excellent Outcome With Chemotherapy Alone and Benefit From Intensive Postinduction Treatment: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2009, 27, 5189–5194. [Google Scholar] [CrossRef] [Green Version]
- Ribera, J.-M.; Oriol, A.; Sanz, M.-A.; Tormo, M.; Fernández-Abellán, P.; del Potro, E.; Abella, E.; Bueno, J.; Parody, R.; Bastida, P.; et al. Comparison of the Results of the Treatment of Adolescents and Young Adults with Standard-Risk Acute Lymphoblastic Leukemia With the Programa Español de Tratamiento En Hematología Pediatric-Based Protocol ALL-96. J. Clin. Oncol. 2008, 26, 1843–1849. [Google Scholar] [CrossRef] [PubMed]
- Ribera, J.-M.; Oriol, A.; Morgades, M.; Montesinos, P.; Sarrà, J.; González-Campos, J.; Brunet, S.; Tormo, M.; Fernández-Abellán, P.; Guàrdia, R.; et al. Treatment of High-Risk Philadelphia Chromosome–Negative Acute Lymphoblastic Leukemia in Adolescents and Adults According to Early Cytologic Response and Minimal Residual Disease after Consolidation Assessed by Flow Cytometry: Final Results of the PETHEMA ALL-AR-03 Trial. J. Clin. Oncol. 2014, 32, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Rytting, M.E.; Jabbour, E.J.; Jorgensen, J.L.; Ravandi, F.; Franklin, A.R.; Kadia, T.M.; Pemmaraju, N.; Daver, N.G.; Ferrajoli, A.; Garcia-Manero, G.; et al. Final Results of a Single Institution Experience with a Pediatric-Based Regimen, the Augmented Berlin-Frankfurt-Münster, in Adolescents and Young Adults with Acute Lymphoblastic Leukemia, and Comparison to the Hyper-CVAD Regimen: Hyper-CVAD versus Pediatric-Based Regimen for AYAs with ALL. Am. J. Hematol. 2016, 91, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Stock, W.; Luger, S.M.; Advani, A.S.; Yin, J.; Harvey, R.C.; Mullighan, C.G.; Willman, C.L.; Fulton, N.; Laumann, K.M.; Malnassy, G.; et al. A Pediatric Regimen for Older Adolescents and Young Adults with Acute Lymphoblastic Leukemia: Results of CALGB 10403. Blood 2019, 133, 1548–1559. [Google Scholar] [CrossRef] [Green Version]
- Cluzeau, T.; Dhédin, N.; Huguet, F.; Raffoux, E.; Maury, S.; Mannone, L.; Escoffre-Barbe, M.; Le Calloch, R.; Delannoy, A.; Turlure, P.; et al. Dose-Intensity Impacts on Survival of Adolescents and Young Adults with Acute Lymphoblastic Leukemia Trated in Adult Departments by a Pediatric Protocol (FRALLE 2000BT). B. Blood 2012, 120, 3561. [Google Scholar] [CrossRef]
- Gökbuget, N.; Beck, J.; Brandt, K.; Brüggemann, M.; Burmeister, T.; Diedrich, H.; Faul, C.; Huettmann, A.; Kondakci, M.; Kraemer, D.M.; et al. Significant Improvement of Outcome in Adolescents and Young Adults (AYAs) Aged 15–35 Years with Acute Lymphoblastic Leukemia with a Pediatric Derived Adult ALL Protocol; Results of 1529 AYAs in 2 Consecutive Trials of the German Multicenter Study Group Dor Adult ALL (GMALL). Blood 2013, 122, 839. [Google Scholar]
- Testi, A.M.; Canichella, M.; Vitale, A.; Piciocchi, A.; Guarini, A.; Starza, I.D.; Cavalli, M.; De Propris, M.S.; Messina, M.; Elia, L.; et al. Adolescent and Young Adult Acute Lymphoblastic Leukemia. Final Results of the Phase II Pediatric-like GIMEMA LAL-1308. Trial. Am. J. Hematol 2020. [Google Scholar] [CrossRef]
- Rijneveld, A.W.; van der Holt, B.; de Weerdt, O.; Biemond, B.; van de Loosdrecht, A.; Petersen, E.; Bellido, M.; Schouten, H.; der Velden, W.; Selleslag, D.; et al. Randomized Phase III Hovon-100 Study of Clofarabine Combined with Standard Treatment in Adult Patients with Newly Diagnosed ALL. HemaSphere 2019, 3, 421–422. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Stevenson, K.E.; Dahlberg, S.E.; Silverman, L.B.; Couban, S.; Supko, J.G.; Amrein, P.C.; Ballen, K.K.; Seftel, M.D.; Turner, A.R.; et al. Long-Term Outcome of a Pediatric-Inspired Regimen Used for Adults Aged 18–50 Years with Newly Diagnosed Acute Lymphoblastic Leukemia. Leukemia 2015, 29, 526–534. [Google Scholar] [CrossRef] [PubMed]
- DeAngelo, D.J.; Stevenson, K.E.; Neuberg, D.S.; Silverman, L.B.; Ballen, K.K.; Asch, J.D.; Abou Mourad, Y.; Paulson, K.; Seftel, M.D.; Avigan, D.; et al. A Multicenter Phase II Study Using a Dose Intensified Pegylated-Asparaginase Pediatric Regimen in Adults with Untreated Acute Lymphoblastic Leukemia: A DFCI ALL Consortium Trial. Blood 2015, 126, 80. [Google Scholar] [CrossRef]
- Gökbuget, N.; Baumann, A.; Beck, J.; Brüggemann, M.; Diedrich, H.; Huettmann, A.; Leimer, L.; Zewen, S.; Mohren, M.; Reichle, A.; et al. PEG-Asparaginase Intensification in Adult Acute Lymphoblastic Leukemia (ALL): Significant Improvement of Outcome with Moderate Increase of Liver Toxicity in the German Multicenter Study Group for Adult ALL (GMALL) Study 07/2003. Blood 2010, 116, 494. [Google Scholar] [CrossRef]
- Parovichnikova, E.; Troitskaya, V. Non-Intensive but Constant and Exhausting Action on the Leukemic Clone Is a Reasonable and Effective Treatment Approach in Adult Acute Lymphoblastic Leukemia: Results of the Russian Acute Lymphoblastic Leukemia (RALL) Study Group. Blood 2014, 124, 3662. [Google Scholar] [CrossRef]
- Huguet, F.; Leguay, T.; Raffoux, E.; Thomas, X.; Beldjord, K.; Delabesse, E.; Chevallier, P.; Buzyn, A.; Delannoy, A.; Chalandon, Y.; et al. Pediatric-Inspired Therapy in Adults with Philadelphia Chromosome–Negative Acute Lymphoblastic Leukemia: The GRAALL-2003 Study. J. Clin. Oncol. 2009, 27, 911–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huguet, F.; Chevret, S.; Leguay, T.; Thomas, X.; Boissel, N.; Escoffre-Barbe, M.; Chevallier, P.; Hunault, M.; Vey, N.; Bonmati, C.; et al. Intensified Therapy of Acute Lymphoblastic Leukemia in Adults: Report of the Randomized GRAALL-2005 Clinical Trial. J. Clin. Oncol. 2018, 36, 2514–2523. [Google Scholar] [CrossRef]
- Storring, J.M.; Minden, M.D.; Kao, S.; Gupta, V.; Schuh, A.C.; Schimmer, A.D.; Yee, K.W.L.; Kamel-Reid, S.; Chang, H.; Lipton, J.H.; et al. Treatment of Adults with BCR-ABL Negative Acute Lymphoblastic Leukaemia with a Modified Paediatric Regimen. Br. J. Haematol. 2009, 146, 76–85. [Google Scholar] [CrossRef]
- Ribera, J.-M.; Morgades, M.; Ciudad, J.; Montesinos, P.; Esteve, J.; Genesca, E.; Barba, P.; Ribera, J.; García Cadenas, I.; Moreno, M.J.; et al. Chemotherapy or Allogeneic Transplantation in High-Risk Philadelphia Chromosome-Negative Adult Lymphoblastic Leukemia. Blood 2020. [Google Scholar] [CrossRef]
- Sakura, T.; Hayakawa, F.; Sugiura, I.; Murayama, T.; Imai, K.; Usui, N.; Fujisawa, S.; Yamauchi, T.; Yujiri, T.; Kakihana, K.; et al. High-Dose Methotrexate Therapy Significantly Improved Survival of Adult Acute Lymphoblastic Leukemia: A Phase III Study by JALSG. Leukemia 2018, 32, 626–632. [Google Scholar] [CrossRef]
- Bassan, R.; Pavoni, C.; Intermesoli, T.; Spinelli, O.; Tosi, M.; Audisio, E.; Marmont, F.; Cattaneo, C.; Borlenghi, E.; Cortelazzo, S.; et al. Updated Risk-Oriented Strategy for Acute Lymphoblastic Leukemia in Adult Patients 18–65 Years: NILG ALL 10/07. Blood Cancer J. 2020, 10, 119. [Google Scholar] [CrossRef]
- Stock, W.; Douer, D.; DeAngelo, D.J.; Arellano, M.; Advani, A.; Damon, L.; Kovacsovics, T.; Litzow, M.; Rytting, M.; Borthakur, G.; et al. Prevention and Management of Asparaginase/Pegasparaginase-Associated Toxicities in Adults and Older Adolescents: Recommendations of an Expert Panel. Leuk. Lymphoma 2011, 52, 2237–2253. [Google Scholar] [CrossRef]
- Burke, P.W.; Hoelzer, D.; Park, J.H.; Schmiegelow, K.; Douer, D. Managing Toxicities with Asparaginase-Based Therapies in Adult ALL: Summary of an ESMO Open—Cancer Horizons Roundtable Discussion. ESMO Open 2020, 5, e000858. [Google Scholar] [CrossRef] [PubMed]
- Giebel, S.; Marks, D.I.; Boissel, N.; Baron, F.; Chiaretti, S.; Ciceri, F.; Cornelissen, J.J.; Doubek, M.; Esteve, J.; Fielding, A.; et al. Hematopoietic Stem Cell Transplantation for Adults with Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia in First Remission: A Position Statement of the European Working Group for Adult Acute Lymphoblastic Leukemia (EWALL) and the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation (EBMT). Bone Marrow Transplant. 2019, 54, 798–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- A Treatment Study Protocol of the ALLTogether Consortium for Children and Young Adults (1-45 Years of Age) With Newly Diagnosed Acute Lymphoblastic Leukaemia (ALL): A Pilot Study, ClinicalTrials.gov Identifier: NCT03911128.
- O’Connor, D.; Enshaei, A.; Bartram, J.; Hancock, J.; Harrison, C.J.; Hough, R.; Samarasinghe, S.; Schwab, C.; Vora, A.; Wade, R.; et al. Genotype-Specific Minimal Residual Disease Interpretation Improves Stratification in Pediatric Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Moorman, A.; Kirkwood, A.; Enshaei, A.; Clifton-Hadley, L.; Lawrie, E.; Marks, D.I.; McMillan, A.; Menne, T.; Patrick, P.; Wrench, B.; et al. Clinical Efficacy of a Novel Validated Prognostic Index for Trial Design in Adult Acute Lymphoblastic Leukemia. HemaSphere 2019, 3, 748. [Google Scholar] [CrossRef]
- Mignon, L.; DelRocco, N.; Borowitz, M.J.; Rabin, K.R.; Zweidler-Mckay, P.A.; Maloney, K.; Mattano, L.A.; Larsen, E.C.; Angiolillo, A.; Schore, R.; et al. Enhanced Risk Stratification of 21,178 Children, Adolescents, and Young Adults with Acute Lymphoblastic Leukemia (ALL) Incorporating White Blood Count (WBC), Age, and Minimal Residual Disease (MRD) at Day 8 and 29 As Continuous Variables: A Children’s Oncology Group (COG) Report. Blood 2020, 136 (Suppl. S1), 39–40. [Google Scholar]
- Van Dongen, J.J.M.; Lhermitte, L.; Böttcher, S.; Almeida, J.; van der Velden, V.H.J.; Flores-Montero, J.; Rawstron, A.; Asnafi, V.; Lécrevisse, Q.; Lucio, P.; et al. EuroFlow Antibody Panels for Standardized N-Dimensional Flow Cytometric Immunophenotyping of Normal, Reactive and Malignant Leukocytes. Leukemia 2012, 26, 1908–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theunissen, P.; Mejstrikova, E.; Sedek, L.; van der Sluijs-Gelling, A.J.; Gaipa, G.; Bartels, M.; Sobral da Costa, E.; Kotrová, M.; Novakova, M.; Sonneveld, E.; et al. Standardized Flow Cytometry for Highly Sensitive MRD Measurements in B-Cell Acute Lymphoblastic Leukemia. Blood 2017, 129, 347–357. [Google Scholar] [CrossRef]
- Van Dongen, J.; Macintyre, E.; Gabert, J.; Delabesse, E.; Rossi, V.; Saglio, G.; Gottardi, E.; Rambaldi, A.; Dotti, G.; Griesinger, F.; et al. Standardized RT-PCR Analysis of Fusion Gene Transcripts from Chromosome Aberrations in Acute Leukemia for Detection of Minimal Residual Disease: Report of the BIOMED-1 Concerted Action: Investigation of Minimal Residual Disease in Acute Leukemia. Leukemia 1999, 13, 1901–1928. [Google Scholar] [CrossRef]
- Gabert, J.; Beillard, E.; van der Velden, V.H.J.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cavé, H.; et al. Standardization and Quality Control Studies of ‘Real-Time’ Quantitative Reverse Transcriptase Polymerase Chain Reaction of Fusion Gene Transcripts for Residual Disease Detection in Leukemia—A Europe Against Cancer Program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef]
- Langerak, A.W.; Groenen, P.J.T.A.; Brüggemann, M.; Beldjord, K.; Bellan, C.; Bonello, L.; Boone, E.; Carter, G.I.; Catherwood, M.; Davi, F.; et al. EuroClonality/BIOMED-2 Guidelines for Interpretation and Reporting of Ig/TCR Clonality Testing in Suspected Lymphoproliferations. Leukemia 2012, 26, 2159–2171. [Google Scholar] [CrossRef]
- Van der Velden, V.H.J.; Cazzaniga, G.; Schrauder, A.; Hancock, J.; Bader, P.; Panzer-Grumayer, E.R.; Flohr, T.; Sutton, R.; Cave, H.; Madsen, H.O.; et al. Analysis of Minimal Residual Disease by Ig/TCR Gene Rearrangements: Guidelines for Interpretation of Real-Time Quantitative PCR Data. Leukemia 2007, 21, 604–611. [Google Scholar] [CrossRef] [Green Version]
- Knecht, H.; Reigl, T.; Kotrová, M.; Appelt, F.; Stewart, P.; Bystry, V.; Krejci, A.; Grioni, A.; Pal, K.; Stranska, K.; et al. Quality Control and Quantification in IG/TR next-Generation Sequencing Marker Identification: Protocols and Bioinformatic Functionalities by EuroClonality-NGS. Leukemia 2019, 33, 2254–2265. [Google Scholar] [CrossRef] [Green Version]
- Scheijen, B.; Meijers, R.W.J.; Rijntjes, J.; van der Klift, M.Y.; Möbs, M.; Steinhilber, J.; Reigl, T.; van den Brand, M.; Kotrová, M.; Ritter, J.-M.; et al. Next-Generation Sequencing of Immunoglobulin Gene Rearrangements for Clonality Assessment: A Technical Feasibility Study by EuroClonality-NGS. Leukemia 2019, 33, 2227–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüggemann, M.; Kotrová, M.; Knecht, H.; Bartram, J.; Boudjogrha, M.; Bystry, V.; Fazio, G.; Froňková, E.; Giraud, M.; Grioni, A.; et al. Standardized Next-Generation Sequencing of Immunoglobulin and T-Cell Receptor Gene Recombinations for MRD Marker Identification in Acute Lymphoblastic Leukaemia; a EuroClonality-NGS Validation Study. Leukemia 2019, 33, 2241–2253. [Google Scholar] [CrossRef] [Green Version]
- Wren, D.; Walker, B.A.; Brüggemann, M.; Catherwood, M.A.; Pott, C.; Stamatopoulos, K.; Langerak, A.W.; Gonzalez, D. Comprehensive Translocation and Clonality Detection in Lymphoproliferative Disorders by Next-Generation Sequencing. Haematologica 2017, 102, e57–e60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavagna, R.; Guinea Montalvo, M.L.; Tosi, M.; Paris, M.; Pavoni, C.; Intermesoli, T.; Bassan, R.; Mosca, A.; Rambaldi, A.; Spinelli, O. Capture-Based Next-Generation Sequencing Improves the Identification of Immunoglobulin/T-Cell Receptor Clonal Markers and Gene Mutations in Adult Acute Lymphoblastic Leukemia Patients Lacking Molecular Probes. Cancers 2020, 12, 1505. [Google Scholar] [CrossRef] [PubMed]
- Bystry, V.; Reigl, T.; Krejci, A.; Demko, M.; Hanakova, B.; Grioni, A.; Knecht, H.; Schlitt, M.; Dreger, P.; Sellner, L.; et al. ARResT/Interrogate: An Interactive Immunoprofiler for IG/TR NGS Data. Bioinformatics 2016, btw634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duez, M.; Giraud, M.; Herbert, R.; Rocher, T.; Salson, M.; Thonier, F. Vidjil: A Web Platform for Analysis of High-Throughput Repertoire Sequencing. PLoS ONE 2016, 11, e0166126. [Google Scholar] [CrossRef]
- Faham, M.; Zheng, J.; Moorhead, M.; Carlton, V.E.H.; Stow, P.; Coustan-Smith, E.; Pui, C.-H.; Campana, D. Deep-Sequencing Approach for Minimal Residual Disease Detection in Acute Lymphoblastic Leukemia. Blood 2012, 120, 8. [Google Scholar]
- Della Starza, I.; De Novi, L.A.; Santoro, A.; Salemi, D.; Tam, W.; Cavalli, M.; Menale, L.; Soscia, R.; Apicella, V.; Ilari, C.; et al. Digital Droplet PCR and Next-Generation Sequencing Refine Minimal Residual Disease Monitoring in Acute Lymphoblastic Leukemia. Leuk. Lymphoma 2019, 60, 2838–2840. [Google Scholar] [CrossRef]
- Gökbuget, N.; Brüggemann, M.; Beck, J.; Faul, C.; Koenecke, C.; Horst, H.-A.; Kondakci, M.; Kraemer, D.M.; Lutz, C.; Spriewald, B.; et al. Evaluation of Minimal Residual Disease (MRD) and MRD-Based Treatment Decisions in Ph/BCR-ABL Negative Adult Acute Lymphoblastic Leukemia (ALL): Experience from the German Multicenter Study Group for Adult ALL (GMALL). Blood 2017, 130, 139. [Google Scholar]
- Yilmaz, M.; Kantarjian, H.; Wang, X.; Khoury, J.D.; Ravandi, F.; Jorgensen, J.; Short, N.J.; Loghavi, S.; Cortes, J.; Garcia-Manero, G.; et al. The Early Achievement of Measurable Residual Disease Negativity in the Treatment of Adults with Philadelphia-negative B-cell Acute Lymphoblastic Leukemia Is a Strong Predictor for Survival. Am. J. Hematol. 2020, 95, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Gökbuget, N.; Dombret, H.; Giebel, S.; Bruggemann, M.; Doubek, M.; Foà, R.; Hoelzer, D.; Kim, C.; Martinelli, G.; Parovichnikova, E.; et al. Minimal Residual Disease Level Predicts Outcome in Adults with Ph-Negative B-Precursor Acute Lymphoblastic Leukemia. Hematology 2019, 24, 337–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFilipp, Z.; Advani, A.S.; Bachanova, V.; Cassaday, R.D.; Deangelo, D.J.; Kebriaei, P.; Rowe, J.M.; Seftel, M.D.; Stock, W.; Tallman, M.S.; et al. Hematopoietic Cell Transplantation in the Treatment of Adult Acute Lymphoblastic Leukemia: Updated 2019 Evidence-Based Review from the American Society for Transplantation and Cellular Therapy. Biol. Blood Marrow Transplant. 2019, 25, 2113–2123. [Google Scholar] [CrossRef]
- Bassan, R.; Spinelli, O. Minimal Residual Disease Monitoring in Adult ALL to Determine Therapy. Curr. Hematol. Malig. Rep. 2015, 10, 86–95. [Google Scholar] [CrossRef]
- Seftel, M.D.; Neuberg, D.; Zhang, M.-J.; Wang, H.-L.; Ballen, K.K.; Bergeron, J.; Couban, S.; Freytes, C.O.; Hamadani, M.; Kharfan-Dabaja, M.A.; et al. Pediatric-Inspired Therapy Compared to Allografting for Philadelphia Chromosome-Negative Adult ALL in First Complete Remission: Chemotherapy versus Allogeneic HCT in ALL. Am. J. Hematol. 2016, 91, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Hangai, M.; Urayama, K.Y.; Tanaka, J.; Kato, K.; Nishiwaki, S.; Koh, K.; Noguchi, M.; Kato, K.; Yoshida, N.; Sato, M.; et al. Allogeneic Stem Cell Transplantation for Acute Lymphoblastic Leukemia in Adolescents and Young Adults. Biol. Blood Marrow Transplant. 2019, 25, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Wood, W.A.; Lee, S.J.; Brazauskas, R.; Wang, Z.; Aljurf, M.D.; Ballen, K.K.; Buchbinder, D.K.; Dehn, J.; Freytes, C.O.; Lazarus, H.M.; et al. Survival Improvements in Adolescents and Young Adults after Myeloablative Allogeneic Transplantation for Acute Lymphoblastic Leukemia. Biol. Blood Marrow Transplant. 2014, 20, 829–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muffly, L.; Li, Q.; Alvarez, E.; Kahn, J.; Winestone, L.; Cress, R.; Penn, D.C.; Keegan, T.H.M. Hematopoietic Cell Transplantation in Young Adult Acute Lymphoblastic Leukemia: A United States Population-Level Analysis. J. Adolesc. Young Adult Oncol. 2019, 8, 254–261. [Google Scholar] [CrossRef]
- Dhédin, N.; Huynh, A.; Maury, S.; Tabrizi, R.; Beldjord, K.; Asnafi, V.; Thomas, X.; Chevallier, P.; Nguyen, S.; Coiteux, V.; et al. Role of Allogeneic Stem Cell Transplantation in Adult Patients with Ph-Negative Acute Lymphoblastic Leukemia. Blood 2015, 125, 2486–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Z.; Gu, X.; Mao, W.; Yin, L.; Yang, L.; Zhang, Z.; Liu, K.; Wang, L.; Huang, Y. Influence of Pre-Transplant Minimal Residual Disease on Prognosis after Allo-SCT for Patients with Acute Lymphoblastic Leukemia: Systematic Review and Meta-Analysis. BMC Cancer 2018, 18, 755. [Google Scholar] [CrossRef]
- Lovisa, F.; Zecca, M.; Rossi, B.; Campeggio, M.; Magrin, E.; Giarin, E.; Buldini, B.; Songia, S.; Cazzaniga, G.; Mina, T.; et al. Pre- and Post-Transplant Minimal Residual Disease Predicts Relapse Occurrence in Children with Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2018, 180, 680–693. [Google Scholar] [CrossRef]
- Pavlů, J.; Labopin, M.; Niittyvuopio, R.; Socié, G.; Yakoub-Agha, I.; Wu, D.; Remenyi, P.; Passweg, J.; Beelen, D.W.; Aljurf, M.; et al. Measurable Residual Disease at Myeloablative Allogeneic Transplantation in Adults with Acute Lymphoblastic Leukemia: A Retrospective Registry Study on 2780 Patients from the Acute Leukemia Working Party of the EBMT. J. Hematol. Oncol.J Hematol. Oncol. 2019, 12, 108. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Liu, H.; Liu, Y.; Ma, X.; Qiu, H.; Fu, C.; Tang, X.; Han, Y.; Chen, S.; Wu, D.; et al. Gene Mutations and Pretransplant Minimal Residual Disease Predict Risk of Relapse in Adult Patients after Allogeneic Hematopoietic Stem-Cell Transplantation for T Cell Acute Lymphoblastic Leukemia. Leuk. Lymphoma 2019, 60, 2744–2753. [Google Scholar] [CrossRef] [PubMed]
- Bader, P.; Salzmann-Manrique, E.; Balduzzi, A.; Dalle, J.-H.; Woolfrey, A.E.; Bar, M.; Verneris, M.R.; Borowitz, M.J.; Shah, N.N.; Gossai, N.; et al. More Precisely Defining Risk Peri-HCT in Pediatric ALL: Pre- vs. Post-MRD Measures, Serial Positivity, and Risk Modeling. Blood Adv. 2019, 3, 3393–3405. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Liu, Y.; Xu, L.; Wang, Y.; Zhang, X.; Chen, H.; Chen, Y.; Han, W.; Sun, Y.; Yan, C.; et al. Minimal Residual Disease Status Determined by Multiparametric Flow Cytometry Pretransplantation Predicts the Outcome of Patients with ALL Receiving Unmanipulated Haploidentical Allografts. Am. J. Hematol. 2019, 94, 512–521. [Google Scholar] [CrossRef]
- Wang, X.; Fan, Q.; Xu, L.; Wang, Y.; Zhang, X.; Chen, H.; Chen, Y.; Wang, F.; Han, W.; Sun, Y.; et al. The Quantification of Minimal Residual Disease Pre- and Post-Unmanipulated Haploidentical Allograft by Multiparameter Flow Cytometry in Pediatric Acute Lymphoblastic Leukemia. Cytom. B Clin. Cytom. 2020, 98, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Kaito, S.; Najima, Y.; Harada, K.; Fukuda, T.; Noguchi, Y.; Ikegame, K.; Tanaka, M.; Ozawa, Y.; Yoshida, S.; Sawa, M.; et al. Allogeneic Hematopoietic Stem Cell Transplantation for Adult Patients with B-Cell Acute Lymphoblastic Leukemia Harboring t(1;19)(Q23;P13.3); Comparison with Normal Karyotype. Bone Marrow Transplant 2020, 55, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Topp, M.S.; Gökbuget, N.; Zugmaier, G.; Degenhard, E.; Goebeler, M.-E.; Klinger, M.; Neumann, S.A.; Horst, H.A.; Raff, T.; Viardot, A.; et al. Long-Term Follow-up of Hematologic Relapse-Free Survival in a Phase 2 Study of Blinatumomab in Patients with MRD in B-Lineage ALL. Blood 2012, 120, 5185–5187. [Google Scholar] [CrossRef] [Green Version]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.-A.; et al. Blinatumomab for Minimal Residual Disease in Adults with B-Cell Precursor Acute Lymphoblastic Leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, X.; Rijneveld, A.W.; Fracchiolla, N.; Salek, C.; Spyridonidis, A.; Chiaretti, S.; Alam, N.; Pezzani Grueter, I.; Mohammad, A.; Kormany, W.N.; et al. Real-World Effectiveness and Safety of Blinatumumab in Europe: 3-Years Results in Relapsed or Refractory Philadelphia Chromosome-Negative BCP-ALL Patients Including Those with Late First Relapse. In Proceedings of the EBMT 47th Virtual Annual Meeting, Atlanta, GA, USA, 14–17 March 2021. [Google Scholar]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.E.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Bernhardt, M.B.; et al. Effect of Postreinduction Therapy Consolidation with Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults with First Relapse of B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Jabbour, E.; Gökbuget, N.; Advani, A.; Stelljes, M.; Stock, W.; Liedtke, M.; Martinelli, G.; O’Brien, S.; Wang, T.; Laird, A.D.; et al. Impact of Minimal Residual Disease Status in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia Treated with Inotuzumab Ozogamicin in the Phase III INO-VATE Trial. Leuk. Res. 2020, 88, 106283. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia in Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Curran, K.J.; Margossian, S.P.; Kernan, N.A.; Silverman, L.B.; Williams, D.A.; Shukla, N.; Kobos, R.; Forlenza, C.J.; Steinherz, P.; Prockop, S.; et al. Toxicity and Response after CD19-Specific CAR T-Cell Therapy in Pediatric/Young Adult Relapsed/Refractory B-ALL. Blood 2019, 134, 2361–2368. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Hay, K.A.; Gauthier, J.; Hirayama, A.V.; Voutsinas, J.M.; Wu, Q.; Li, D.; Gooley, T.A.; Cherian, S.; Chen, X.; Pender, B.S.; et al. Factors Associated with Durable EFS in Adult B-Cell ALL Patients Achieving MRD-Negative CR after CD19 CAR T-Cell Therapy. Blood 2019, 133, 1652–1663. [Google Scholar] [CrossRef] [Green Version]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T Cell Expansion and Prolonged Persistence in Pediatric Patients with ALL Treated with a Low-Affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Magnani, C.F.; Gaipa, G.; Lussana, F.; Belotti, D.; Gritti, G.; Napolitano, S.; Matera, G.; Cabiati, B.; Buracchi, C.; Borleri, G.; et al. Sleeping Beauty–Engineered CAR T Cells Achieve Antileukemic Activity without Severe Toxicities. J. Clin. Investig. 2020, 130, 6021–6033. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, X.; Yang, J.; Zhang, G.; Li, J.; Song, L.; Su, Y.; Shi, Y.; Zhang, M.; He, J.; et al. Efficacy and Safety of Anti-CD19 CAR T-Cell Therapy in 110 Patients with B-Cell Acute Lymphoblastic Leukemia with High-Risk Features. Blood Adv. 2020, 4, 2325–2338. [Google Scholar] [CrossRef] [PubMed]
- Dunsmore, K.P.; Winter, S.S.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Children’s Oncology Group AALL0434: A Phase III Randomized Clinical Trial Testing Nelarabine in Newly Diagnosed T-Cell Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2020, 38, 3282–3293. [Google Scholar] [CrossRef]
- Abaza, Y.; Kantarjian, H.M.; Faderl, S.; Jabbour, E.; Jain, N.; Thomas, D.; Kadia, T.; Borthakur, G.; Khoury, J.D.; Burger, J.; et al. Hyper-CVAD plus Nelarabine in Newly Diagnosed Adult T-Cell Acute Lymphoblastic Leukemia and T-Lymphoblastic Lymphoma. Am. J. Hematol. 2018, 93, 91–99. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, K.M. The Challenge to Further Improvements in Survival of Patients with T-ALL: Current Treatments and New Insights from Disease Pathogenesis. Semin. Hematol. 2020, 57, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.L.; Winter, S.S.; Dunsmore, K.P.; Devidas, M.; Chen, S.; Asselin, B.L.; Esiashvili, N.; Loh, M.L.; Winick, N.J.; Carroll, W.L.; et al. T-Lymphoblastic Leukemia (T-ALL) Shows Excellent Outcome, Lack of Significance of the Early Thymic Precursor (ETP) Immunophenotype, and Validation of the Prognostic Value of End-Induction Minimal Residual Disease (MRD) in Children’s Oncology Group (COG) Study AALL0434. Blood 2014, 124, 21. [Google Scholar]
- Ofran, Y.; Ringelstein-Harlev, S.; Slouzkey, I.; Zuckerman, T.; Yehudai-Ofir, D.; Henig, I.; Beyar-Katz, O.; Hayun, M.; Frisch, A. Daratumumab for Eradication of Minimal Residual Disease in High-Risk Advanced Relapse of T-Cell/CD19/CD22-Negative Acute Lymphoblastic Leukemia. Leukemia 2020, 34, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, F.; Winterberg, D.; Lenk, L.; Buchmann, S.; Cario, G.; Schrappe, M.; Peipp, M.; Richter-Pechanska, P.; Kulozik, A.E.; Lentes, J.; et al. Daratumumab Eradicates Minimal Residual Disease in a Preclinical Model of Pediatric T-Cell Acute Lymphoblastic Leukemia. Blood 2019, 134, 713–716. [Google Scholar] [CrossRef]
- Cerrano, M.; Castella, B.; Lia, G.; Olivi, M.; Faraci, D.G.; Butera, S.; Martella, F.; Scaldaferri, M.; Cattel, F.; Boccadoro, M.; et al. Immunomodulatory and Clinical Effects of Daratumumab in T-cell Acute Lymphoblastic Leukaemia. Br. J. Haematol. 2020, 191. [Google Scholar] [CrossRef]
- Hixon, J.A.; Andrews, C.; Kashi, L.; Kohnhorst, C.L.; Senkevitch, E.; Czarra, K.; Barata, J.T.; Li, W.; Schneider, J.P.; Walsh, S.T.R.; et al. New Anti-IL-7Rα Monoclonal Antibodies Show Efficacy against T Cell Acute Lymphoblastic Leukemia in Pre-Clinical Models. Leukemia 2020, 34, 35–49. [Google Scholar] [CrossRef]
- Cooper, M.L.; Choi, J.; Staser, K.; Ritchey, J.K.; Devenport, J.M.; Eckardt, K.; Rettig, M.P.; Wang, B.; Eissenberg, L.G.; Ghobadi, A.; et al. An “off-the-Shelf” Fratricide-Resistant CAR-T for the Treatment of T Cell Hematologic Malignancies. Leukemia 2018, 32, 1970–1983. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, S.; Gao, L.; Yuan, Z.; Wu, K.; Liu, L.; Luo, L.; Liu, Y.; Zhang, C.; Liu, J.; et al. Abstract CT052: Clinical Safety and Efficacy Study of TruUCAR™ GC027: The First-in-Human, Universal CAR-T Therapy for Adult Relapsed/Refractory T-Cell Acute Lymphoblastic Leukemia (r/r T-ALL). Cancer Res. 2020, 80, 16. [Google Scholar]
- Bongiovanni, D.; Saccomani, V.; Piovan, E. Aberrant Signaling Pathways in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2017, 18, 1904. [Google Scholar] [CrossRef] [Green Version]
- García-Peydró, M.; Fuentes, P.; Mosquera, M.; García-León, M.J.; Alcain, J.; Rodríguez, A.; García de Miguel, P.; Menéndez, P.; Weijer, K.; Spits, H.; et al. The NOTCH1/CD44 Axis Drives Pathogenesis in a T Cell Acute Lymphoblastic Leukemia Model. J. Clin. Investig. 2018, 128, 2802–2818. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yan, J.; Phyu, T.; Fan, S.; Chung, T.-H.; Mustafa, N.; Lin, B.; Wang, L.; Eichhorn, P.J.A.; Goh, B.-C.; et al. MELK Mediates the Stability of EZH2 through Site-Specific Phosphorylation in Extranodal Natural Killer/T-Cell Lymphoma. Blood 2019, 134, 2046–2058. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-Driven Subtypes of B-Progenitor Acute Lymphoblastic Leukemia. Nat. Genet. 2019, 51, 296–307. [Google Scholar] [CrossRef]
- Autry, R.J.; Paugh, S.W.; Carter, R.; Shi, L.; Liu, J.; Ferguson, D.C.; Lau, C.E.; Bonten, E.J.; Yang, W.; McCorkle, J.R.; et al. Integrative Genomic Analyses Reveal Mechanisms of Glucocorticoid Resistance in Acute Lymphoblastic Leukemia. Nat. Cancer 2020, 1, 329–344. [Google Scholar] [CrossRef]
- Waanders, E.; Gu, Z.; Dobson, S.M.; Antić, Ž.; Crawford, J.C.; Ma, X.; Edmonson, M.N.; Payne-Turner, D.; van de Vorst, M.; Jongmans, M.C.J.; et al. Mutational Landscape and Patterns of Clonal Evolution in Relapsed Pediatric Acute Lymphoblastic Leukemia. Blood Cancer Discov. 2020, 1, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Bassan, R.; Bourquin, J.-P.; DeAngelo, D.J.; Chiaretti, S. New Approaches to the Management of Adult Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2018, 36, 3504–3519. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.G. How Advanced Are We in Targeting Novel Subtypes of ALL? Best Pract. Res. Clin. Haematol. 2019, 32, 101095. [Google Scholar] [CrossRef]
- Follini, E.; Marchesini, M.; Roti, G. Strategies to Overcome Resistance Mechanisms in T-Cell Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2019, 20, 3021. [Google Scholar] [CrossRef] [Green Version]
- Pui, C.-H. Precision Medicine in Acute Lymphoblastic Leukemia. Front. Med. 2020, 14, 689–700. [Google Scholar] [CrossRef]
- Chiaretti, S.; Messina, M.; Foà, R. BCR/ABL1 -like Acute Lymphoblastic Leukemia: How to Diagnose and Treat?: Open Questions in BCR/ABL1-Like ALL. Cancer 2019, 125, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Tanasi, I.; Ba, I.; Sirvent, N.; Braun, T.; Cuccuini, W.; Ballerini, P.; Duployez, N.; Tanguy-Schmidt, A.; Tamburini, J.; Maury, S.; et al. Efficacy of Tyrosine Kinase Inhibitors in Ph-like Acute Lymphoblastic Leukemia Harboring ABL-Class Rearrangements. Blood 2019, 134, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.C.; Tasian, S.K. Clinical Diagnostics and Treatment Strategies for Philadelphia Chromosome-like Acute Lymphoblastic Leukemia. Blood Adv. 2020, 4, 218–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorman, A.V.; Schwab, C.; Winterman, E.; Hancock, J.; Castleton, A.; Cummins, M.; Gibson, B.; Goulden, N.; Kearns, P.; James, B.; et al. Adjuvant Tyrosine Kinase Inhibitor Therapy Improves Outcome for Children and Adolescents with Acute Lymphoblastic Leukaemia Who Have an ABL-class Fusion. Br. J. Haematol. 2020, 191, 844–851. [Google Scholar] [CrossRef]
- Hohtari, H.; Brück, O.; Blom, S.; Turkki, R.; Sinisalo, M.; Kovanen, P.E.; Kallioniemi, O.; Pellinen, T.; Porkka, K.; Mustjoki, S. Immune Cell Constitution in Bone Marrow Microenvironment Predicts Outcome in Adult ALL. Leukemia 2019, 33, 1570–1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiaretti, S.; Messina, M.; Grammatico, S.; Piciocchi, A.; Fedullo, A.L.; Di Giacomo, F.; Peragine, N.; Gianfelici, V.; Lauretti, A.; Bareja, R.; et al. Rapid Identification of BCR/ABL1 -like Acute Lymphoblastic Leukaemia Patients Using a Predictive Statistical Model Based on Quantitative Real Time-Polymerase Chain Reaction: Clinical, Prognostic and Therapeutic Implications. Br. J. Haematol. 2018, 181, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Moujalled, D.M.; Hanna, D.T.; Hediyeh-zadeh, S.; Pomilio, G.; Brown, L.; Litalien, V.; Bartolo, R.; Fleming, S.; Chanrion, M.; Banquet, S.; et al. Cotargeting BCL-2 and MCL-1 in High-Risk B-ALL. Blood Adv. 2020, 4, 2762–2767. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, D.; Gielen, O.; Jacobs, K.; Boeckx, N.; De Keersmaecker, K.; Maertens, J.; Uyttebroeck, A.; Segers, H.; Cools, J. Ruxolitinib Synergizes With Dexamethasone for the Treatment of T-Cell Acute Lymphoblastic Leukemia. HemaSphere 2019, 3, e310. [Google Scholar] [CrossRef]
- La Starza, R.; Cambò, B.; Pierini, A.; Bornhauser, B.; Montanaro, A.; Bourquin, J.-P.; Mecucci, C.; Roti, G. Venetoclax and Bortezomib in Relapsed/Refractory Early T-Cell Precursor Acute Lymphoblastic Leukemia. JCO Precis. Oncol. 2019, 1–6. [Google Scholar] [CrossRef]
- Pinkel, D. The Ninth Annual David Karnofsky Lecture. Treatment of Acute Lymphocytic Leukemia. Cancer 1979, 43, 1128–1137. [Google Scholar] [CrossRef] [PubMed]



| ALL Subset | Prognostic Category | Genetic/Cytogenetic Abnormality | Children (<15 Years) | AYA (15–40 Years) | Older Adults (>40 Years) |
|---|---|---|---|---|---|
| Ph− | Favorable | High hyperdiploidy | 20–25% | 5% | <5% |
| t(12;21)/ETV6-RUNX1 | 25% | <5% | 1% | ||
| Intermediate | Normal Karyotype | 10% | - | - | |
| t(1;19)/TCF3-PBX1 | 5% | <5% | 1% | ||
| Unfavorable | Low hypodiploidy, | 1% | 5% | >10% | |
| t(v;11)/KMT2A+ | 6% | 4% | 15% | ||
| Ph−like | 10–15% | 25–30% | 20% | ||
| Ph+ | Unfavorable * | t(9;22)/BCR-ABL1 | 2% | 6% | 25% |
| Age Groups and Trials (Ref.) | Patient Age (Years), Median (Range) | No. of Patients | Outcome Estimates (y, Years) | OS (%) | RFS (%) | EFS (%) |
|---|---|---|---|---|---|---|
| Maximum age ≤ 25 years | ||||||
| FRALLE-93 1 [30] | 15.9 (15–20) | 77 | 5-y | 78 | 72 | 67 |
| CCG 1882/191 1 [31] | 16 (16–20) | 197 | 7-y | 67 | - | 63 |
| JALSG ALL202-U [32] | 19 (15–24) | 139 | 5-y | 73 | 67 | - |
| CCG 19,61 1 [33] | (16–21) | 262 | 5-y | 78 | - | 72 |
| MRC UKALL 2003 [11] | (16–24) | 229 | 5-y | 76 | - | 72 |
| Maximum age ≤ 40 years | ||||||
| PETHEMA ALL-96 [34] | 20 (15–30) | 81 | 6-y | 69 | - | 61 |
| PETHEMA ALLRE08 [35] | 20 (15–30) | 66 | 5-y | 74 | - | - |
| MDACC (augmented BFM) * [36] | 22 (13–39) | 106 | 5-y | 60 | - | - |
| CALGB 10,403 [37] | 24 (17–39) | 295 | 3-y | 73 | 66 | 59 |
| FRALLE 2000-BT [38] | (15–29) | 89 | 5-y | 66 | - | 61 |
| GMALL 07/03 [39] | (15–35) | 887 | 5-y | 65 | 61 | - |
| GIMEMA LAL1308 [40] | (18–35) | 76 | 4-y | 60 | 60 | - |
| HOVON-100 1 [41] | (18–40) | 159 | 5-y | 60–56 2 | 58 | 61–64 2 |
| Maximum age ≤ 55 years | ||||||
| NOPHO ALL2008 [14] | 26 (18–45) | 221 | 5-y | 78 | - | - |
| DFCI 01–175 1 [42] | 28 (18–50) | 92 | 4-y | 67 | 69 | 69 |
| DFCI 06–254 1 [43] | 32 (18–50) | 89 | 3-y | 75 | 73 | 73 |
| GMALL 07/03 [44] | 35 (15–55) | 1226 | 3-y | 60–67 3 | - | - |
| Maximum age > 55 years | ||||||
| RALL 2009 [45] | 30 (15–60) | 250 | 4-y | 66 | 69 | - |
| GRAALL-2003 [46] | 31 (15–60) | 225 | 3.5-y | 60 | 59 | 55 |
| GRAALL-2005 [47] | 36 (18–59) | 787 | 5-y | 59 | - | 52 |
| Toronto (DFCI 91–01) [48] | 37 (18–60) | 85 | 5-y | 63 | 71 | - |
| PETHEMA ALL-HR-11 [49] | 40 (15–60) | 348 | 5-y | 49 | - | 40 |
| JALSG ALL 202-O [50] | 40 (24–65) | 115 | 5-y | 64 | 58 | - |
| NILG 10/07 4 [51] | 41 (18–65) | 161 | 5-y | 52 | 53 | 46 |
| Study Group | MRD | Genetics | WBC | Miscellaneous |
|---|---|---|---|---|
| RALL | + | KMT2A+, t(1;19) | - | Age > 30 |
| GMALL | + | KMT2A+ | >30 (B) | Late CR, proB, early/mature-T |
| HOVON | + | adverse | >30 (B), >100 (T) | Late CR |
| PALG | + | KMT2A+ | >30 (B), >100 (T) | CNS+ |
| FALL | + | Abn11q, hypodiploid | >100 | Late CR, D15 BM blasts > 25% |
| GIMEMA | + | adverse | >100 | Early/mature-T |
| UKALL | + | adverse | High counts | - |
| SVALL | + | KMT2A+, hypodiploidy | - | EOI BM blasts > 5% |
| CELL | + | - | - | - |
| PETHEMA | + | - | - | - |
| GRAALL | + | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tosi, M.; Spinelli, O.; Leoncin, M.; Cavagna, R.; Pavoni, C.; Lussana, F.; Intermesoli, T.; Frison, L.; Perali, G.; Carobolante, F.; et al. MRD-Based Therapeutic Decisions in Genetically Defined Subsets of Adolescents and Young Adult Philadelphia-Negative ALL. Cancers 2021, 13, 2108. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13092108
Tosi M, Spinelli O, Leoncin M, Cavagna R, Pavoni C, Lussana F, Intermesoli T, Frison L, Perali G, Carobolante F, et al. MRD-Based Therapeutic Decisions in Genetically Defined Subsets of Adolescents and Young Adult Philadelphia-Negative ALL. Cancers. 2021; 13(9):2108. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13092108
Chicago/Turabian StyleTosi, Manuela, Orietta Spinelli, Matteo Leoncin, Roberta Cavagna, Chiara Pavoni, Federico Lussana, Tamara Intermesoli, Luca Frison, Giulia Perali, Francesca Carobolante, and et al. 2021. "MRD-Based Therapeutic Decisions in Genetically Defined Subsets of Adolescents and Young Adult Philadelphia-Negative ALL" Cancers 13, no. 9: 2108. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13092108