
Long-Term Antitumor CD8+ T Cell Immunity Induced by Endogenously Engineered Extracellular Vesicles

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Vector Synthesis
2.2. Cell Cultures and Transfection
2.3. EV Isolation
2.4. Western Blot Analysis
2.5. Mice Immunization
2.6. IFN-γ EliSpot Analysis
2.7. Intracellular Cytokine Staining (ICS)
2.8. qRT-PCR
2.9. Tumor Challenge and Re-Challenge
2.10. Statistical Analysis
3. Results
3.1. Optimization of the CD8+ T Immunogenicity Induced by Nefmut-Based DNA Vectors through Electroporation
3.2. Optimization of the HPV16-Specific CD8+ T Immunogenicity through Co-Injection of DNA Vectors Separately Expressing E6 and E7 Fused with Nefmut
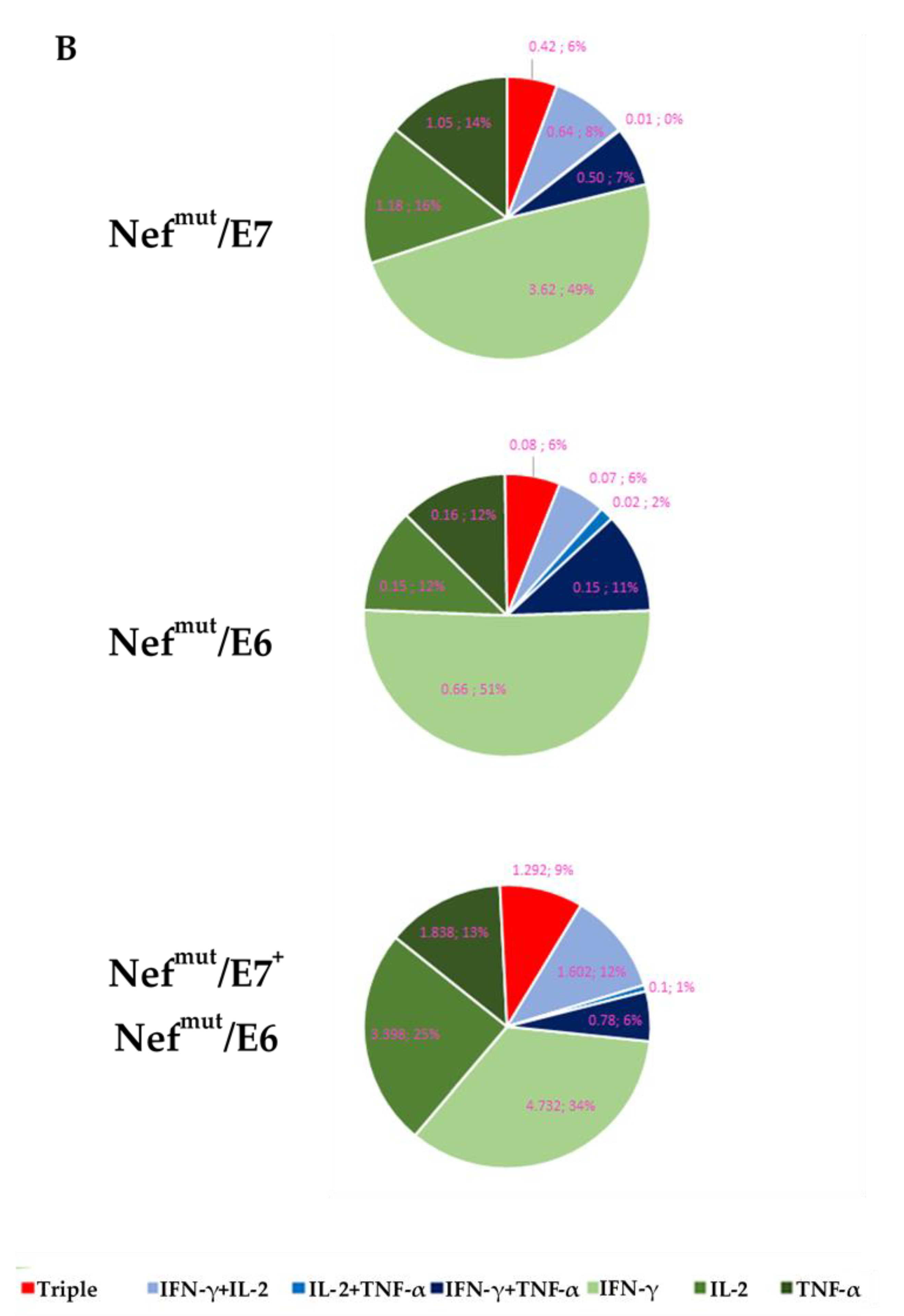
3.3. HPV16-E6 and –E7-Specific CD8+ T Cell Immune Response in Mice Immunized after Tumor Implantation
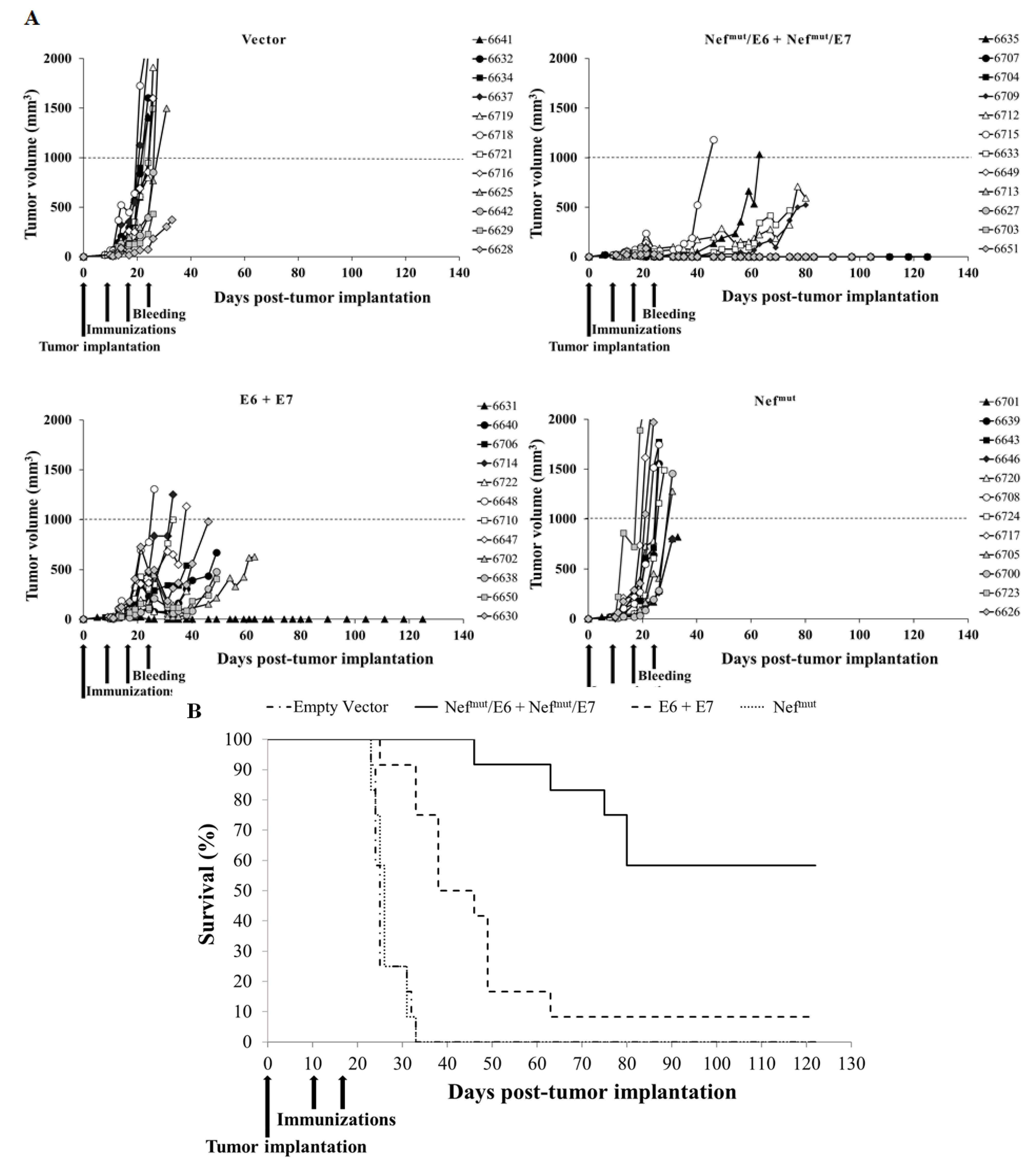
3.4. Antitumor Therapeutic Effect in Mice Co-Injected with Vectors Expressing E6- and E7-Based Fusion Proteins
3.5. Cured Mice Resisted the Tumor Re-Challenging
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Teng, F.; Fussenegger, M. Shedding Light on Extracellular Vesicle Biogenesis and Bioengineering. Adv. Sci. 2020, 8, 2003505. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma Microvesicles Transport RNA and Proteins That Promote Tumour Growth and Provide Diagnostic Biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Mathivanan, S.; Ji, H.; Simpson, R.J. Exosomes: Extracellular Organelles Important in Intercellular Communication. J. Proteom. 2010, 73, 1907–1920. [Google Scholar] [CrossRef]
- Manandhar, S.; Kothandan, V.K.; Oh, J.; Yoo, S.H.; Hwang, J.; Hwang, S.R. A Pharmaceutical Investigation into Exosomes. J. Pharm. Investig. 2018, 48, 617–626. [Google Scholar] [CrossRef]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; Del Portillo, H.A.; et al. Applying Extracellular Vesicles Based Therapeutics in Clinical Trials—An ISEV Position Paper. J. Extracell Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef]
- Bell, B.M.; Kirk, I.D.; Hiltbrunner, S.; Gabrielsson, S.; Bultema, J.J. Designer Exosomes as Next-Generation Cancer Immunotherapy. Nanomedicine 2016, 12, 163–169. [Google Scholar] [CrossRef]
- Kakimi, K.; Karasaki, T.; Matsushita, H.; Sugie, T. Advances in Personalized Cancer Immunotherapy. Breast Cancer 2017, 24, 16–24. [Google Scholar] [CrossRef]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mpakali, A.; Stratikos, E. The Role of Antigen Processing and Presentation in Cancer and the Efficacy of Immune Checkpoint Inhibitor Immunotherapy. Cancers 2021, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Warshauer, J.T.; Bluestone, J.A. Is Autoimmunity the Achilles’ Heel of Cancer Immunotherapy? Nat. Med. 2017, 23, 540–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, M.A.; Gwin, W.R.; Mitchell, D.A. Vaccine Therapies for Cancer: Then and Now. Target Oncol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, L.; Federico, M. A Strategy of Antigen Incorporation into Exosomes: Comparing Cross-Presentation Levels of Antigens Delivered by Engineered Exosomes and by Lentiviral Virus-like Particles. Vaccine 2012, 30, 7229–7237. [Google Scholar] [CrossRef] [PubMed]
- Ferrantelli, F.; Chiozzini, C.; Manfredi, F.; Leone, P.; Federico, M. A New Concept on Anti-SARS-CoV-2 Vaccines: Strong CD8+ T-Cell Immune Response in Both Spleen and Lung Induced in Mice by Endogenously Engineered Extracellular Vesicles. Vaccines 2021, 9, 240. [Google Scholar] [CrossRef] [PubMed]
- Di Bonito, P.; Chiozzini, C.; Arenaccio, C.; Anticoli, S.; Manfredi, F.; Olivetta, E.; Ferrantelli, F.; Falcone, E.; Ruggieri, A.; Federico, M. Antitumor HPV E7-Specific CTL Activity Elicited by in Vivo Engineered Exosomes Produced through DNA Inoculation. Int. J. Nanomed. 2017, 12, 4579–4591. [Google Scholar] [CrossRef] [Green Version]
- Anticoli, S.; Manfredi, F.; Chiozzini, C.; Arenaccio, C.; Olivetta, E.; Ferrantelli, F.; Capocefalo, A.; Falcone, E.; Ruggieri, A.; Federico, M. An Exosome-Based Vaccine Platform Imparts Cytotoxic T Lymphocyte Immunity Against Viral Antigens. Biotechnol. J. 2018, 13, e1700443. [Google Scholar] [CrossRef]
- Anticoli, S.; Aricò, E.; Arenaccio, C.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Ferrantelli, F.; Lattanzi, L.; D’Urso, M.T.; Proietti, E.; et al. Engineered Exosomes Emerging from Muscle Cells Break Immune Tolerance to HER2 in Transgenic Mice and Induce Antigen-Specific CTLs upon Challenge by Human Dendritic Cells. J. Mol. Med. 2018, 96, 211–221. [Google Scholar] [CrossRef]
- Di Bonito, P.; Ridolfi, B.; Columba-Cabezas, S.; Giovannelli, A.; Chiozzini, C.; Manfredi, F.; Anticoli, S.; Arenaccio, C.; Federico, M. HPV-E7 Delivered by Engineered Exosomes Elicits a Protective CD8+ T Cell-Mediated Immune Response. Viruses 2015, 7, 1079–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manfredi, F.; di Bonito, P.; Ridolfi, B.; Anticoli, S.; Arenaccio, C.; Chiozzini, C.; Baz Morelli, A.; Federico, M. The CD8+ T Cell-Mediated Immunity Induced by HPV-E6 Uploaded in Engineered Exosomes Is Improved by ISCOMATRIXTM Adjuvant. Vaccines 2016, 4, 42. [Google Scholar] [CrossRef] [Green Version]
- Chiozzini, C.; Manfredi, F.; Ferrantelli, F.; Leone, P.; Giovannelli, A.; Olivetta, E.; Federico, M. CD8+ T cell immunogenicity induced by endogenous EVs engineered by antigens fused to a truncated Nefmut EV-anchoring protein. bioRxiv 2021. [Google Scholar] [CrossRef]
- Smahel, M.; Síma, P.; Ludvíková, V.; Vonka, V. Modified HPV16 E7 Genes as DNA Vaccine against E7-Containing Oncogenic Cells. Virology 2001, 281, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Cid-Arregui, A.; Juárez, V.; zur Hausen, H. A Synthetic E7 Gene of Human Papillomavirus Type 16 That Yields Enhanced Expression of the Protein in Mammalian Cells and Is Useful for DNA Immunization Studies. J. Virol. 2003, 77, 4928–4937. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and Characterization of Exosomes from Cell Culture Supernatants and Biological Fluids. Curr. Protoc. Cell Biol. 2006. [Google Scholar] [CrossRef]
- Bauer, S.; Heeg, K.; Wagner, H.; Lipford, G.B. Identification of H-2Kb Binding and Immunogenic Peptides from Human Papilloma Virus Tumour Antigens E6 and E7. Scand. J. Immunol. 1995, 42, 317–323. [Google Scholar] [CrossRef]
- De Oliveira, L.M.F.; Morale, M.G.; Chaves, A.A.M.; Cavalher, A.M.; Lopes, A.S.; de Diniz, M.O.; Schanoski, A.S.; de Melo, R.L.; de Ferreira, L.C.S.; de Oliveira, M.L.S.; et al. Design, Immune Responses and Anti-Tumor Potential of an HPV16 E6E7 Multi-Epitope Vaccine. PLoS ONE 2015, 10, e0138686. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, M.; Holst, P.J.; Bukh, J.; Thomsen, A.R.; Christensen, J.P. Enhanced and Sustained CD8+ T Cell Responses with an Adenoviral Vector-Based Hepatitis C Virus Vaccine Encoding NS3 Linked to the MHC Class II Chaperone Protein Invariant Chain. J. Immunol. 2011, 186, 2355–2364. [Google Scholar] [CrossRef] [Green Version]
- Mir, L.M.; Bureau, M.F.; Gehl, J.; Rangara, R.; Rouy, D.; Caillaud, J.M.; Delaere, P.; Branellec, D.; Schwartz, B.; Scherman, D. High-efficiency gene transfer into skeletal muscle mediated by electric pulses. Proc. Natl. Acad. Sci. USA 1999, 96, 4262–4267. [Google Scholar] [CrossRef] [Green Version]
- Ascione, A.; Arenaccio, C.; Mallano, A.; Flego, M.; Gellini, M.; Andreotti, M.; Fenwick, C.; Pantaleo, G.; Vella, S.; Federico, M. Development of a Novel Human Phage Display-Derived Anti-LAG3 ScFv Antibody Targeting CD8+ T Lymphocyte Exhaustion. BMC Biotechnol. 2019, 19, 67. [Google Scholar] [CrossRef] [Green Version]
- Lu, S. Immunogenicity of DNA Vaccines in Humans: It Takes Two to Tango. Hum. Vaccin. 2008, 4, 449–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierini, S.; Perales-Linares, R.; Uribe-Herranz, M.; Pol, J.G.; Zitvogel, L.; Kroemer, G.; Facciabene, A.; Galluzzi, L. Trial Watch: DNA-Based Vaccines for Oncological Indications. Oncoimmunology 2017, 6, e1398878. [Google Scholar] [CrossRef] [PubMed]
- Schalk, J.A.C.; Mooi, F.R.; Berbers, G.A.M.; van Aerts, L.A.G.J.M.; Ovelgönne, H.; Kimman, T.G. Preclinical and Clinical Safety Studies on DNA Vaccines. Hum. Vaccin. 2006, 2, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babiuk, S.; Baca-Estrada, M.E.; Foldvari, M.; Middleton, D.M.; Rabussay, D.; Widera, G.; Babiuk, L.A. Increased gene expression and inflammatory cell infiltration caused by electroporation are both important for improving the efficacy of DNA vaccines. J. Biotechnol. 2004, 110, 1–10. [Google Scholar] [CrossRef]
- Chiarella, P.; Massi, E.; De Robertis, M.; Sibilio, A.; Parrella, P.; Fazio, V.M.; Signori, E. Electroporation of skeletal muscle induces danger signal release and antigen-presenting cell recruitment independently of DNA vaccine administration. Expert Opin. Biol. Ther. 2008, 8, 1645–1657. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Wang, L.; Chow, E.K.-H.; Lim, C.T.; Goh, B.-C. Exosomes in Cancer Nanomedicine and Immunotherapy: Prospects and Challenges. Trends Biotechnol. 2017, 35, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Roy, S.; Saha, P.; Chatterjee, N.; Bishayee, A. Trends in Research on Exosomes in Cancer Progression and Anticancer Therapy. Cancers 2021, 13, 326. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, E.H.; Kwak, G.; Chi, S.-G.; Kim, S.H.; Yang, Y. Exosomes: Cell-Derived Nanoplatforms for the Delivery of Cancer Therapeutics. Int. J. Mol. Sci. 2020, 22, 14. [Google Scholar] [CrossRef] [PubMed]
- Arenaccio, C.; Chiozzini, C.; Ferrantelli, F.; Leone, P.; Olivetta, E.; Federico, M. Exosomes in Therapy: Engineering, Pharmacokinetics and Future Applications. Curr. Drug Targets 2019, 20, 87–95. [Google Scholar] [CrossRef]
- Escudier, B.; Dorval, T.; Chaput, N.; André, F.; Caby, M.-P.; Novault, S.; Flament, C.; Leboulaire, C.; Borg, C.; Amigorena, S.; et al. Vaccination of Metastatic Melanoma Patients with Autologous Dendritic Cell (DC) Derived-Exosomes: Results of The first Phase I Clinical Trial. J. Transl. Med. 2005, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morse, M.A.; Garst, J.; Osada, T.; Khan, S.; Hobeika, A.; Clay, T.M.; Valente, N.; Shreeniwas, R.; Sutton, M.A.; Delcayre, A.; et al. A Phase I Study of Dexosome Immunotherapy in Patients with Advanced Non-Small Cell Lung Cancer. J. Transl. Med. 2005, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I Clinical Trial of Autologous Ascites-Derived Exosomes Combined with GM-CSF for Colorectal Cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrantelli, F.; Manfredi, F.; Chiozzini, C.; Leone, P.; Giovannelli, A.; Olivetta, E.; Federico, M. Long-Term Antitumor CD8+ T Cell Immunity Induced by Endogenously Engineered Extracellular Vesicles. Cancers 2021, 13, 2263. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13092263
Ferrantelli F, Manfredi F, Chiozzini C, Leone P, Giovannelli A, Olivetta E, Federico M. Long-Term Antitumor CD8+ T Cell Immunity Induced by Endogenously Engineered Extracellular Vesicles. Cancers. 2021; 13(9):2263. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13092263
Chicago/Turabian StyleFerrantelli, Flavia, Francesco Manfredi, Chiara Chiozzini, Patrizia Leone, Andrea Giovannelli, Eleonora Olivetta, and Maurizio Federico. 2021. "Long-Term Antitumor CD8+ T Cell Immunity Induced by Endogenously Engineered Extracellular Vesicles" Cancers 13, no. 9: 2263. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers13092263