
Novel Molecular Targets for Hepatocellular Carcinoma

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Expression Analysis
2.2. Survival Analysis
2.3. HCC Samples Collection and Transcriptomic Analysis
2.4. Biological Networks and Interaction Analysis
2.5. HLA Class I Epitope Prediction
2.6. BLAST Homology Search
2.7. Epitope Modeling and Molecular Docking
2.8. Peptide Synthesis and Solubilization
2.9. Peptide Binding Affinity and BFA Decay Assays
2.10. pMHC Multimer Preparation and T-Cell Staining
2.11. Statistical Analysis
3. Results
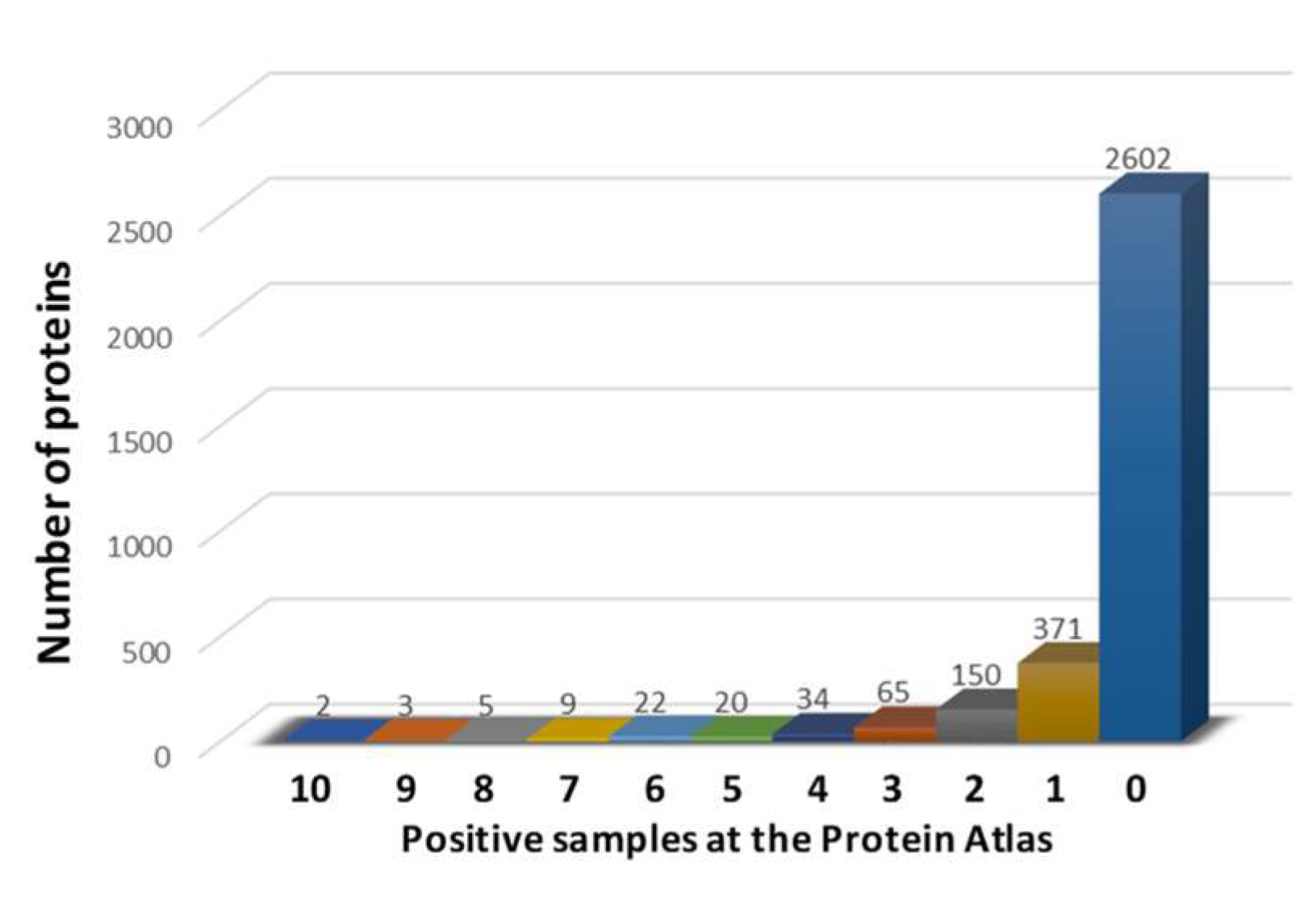
3.1. Identification of HCC-Related Proteins
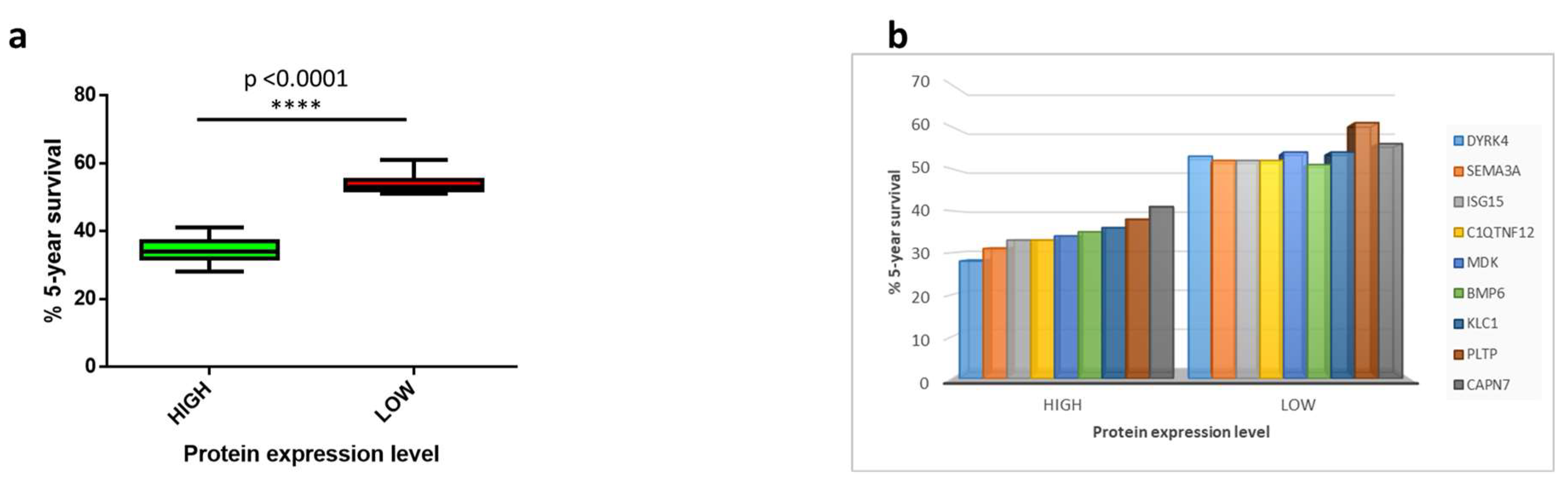
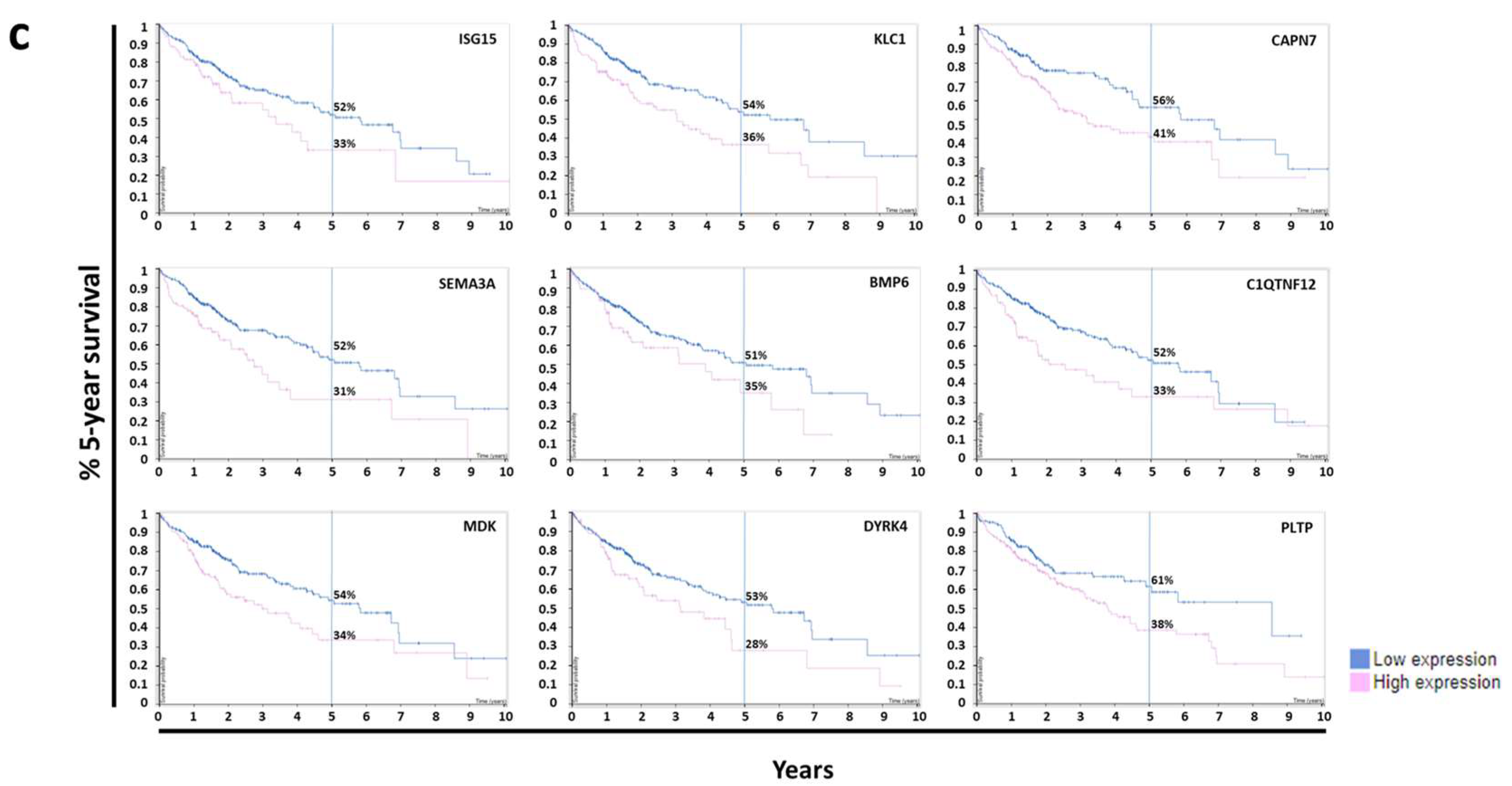
3.2. Survival Analysis
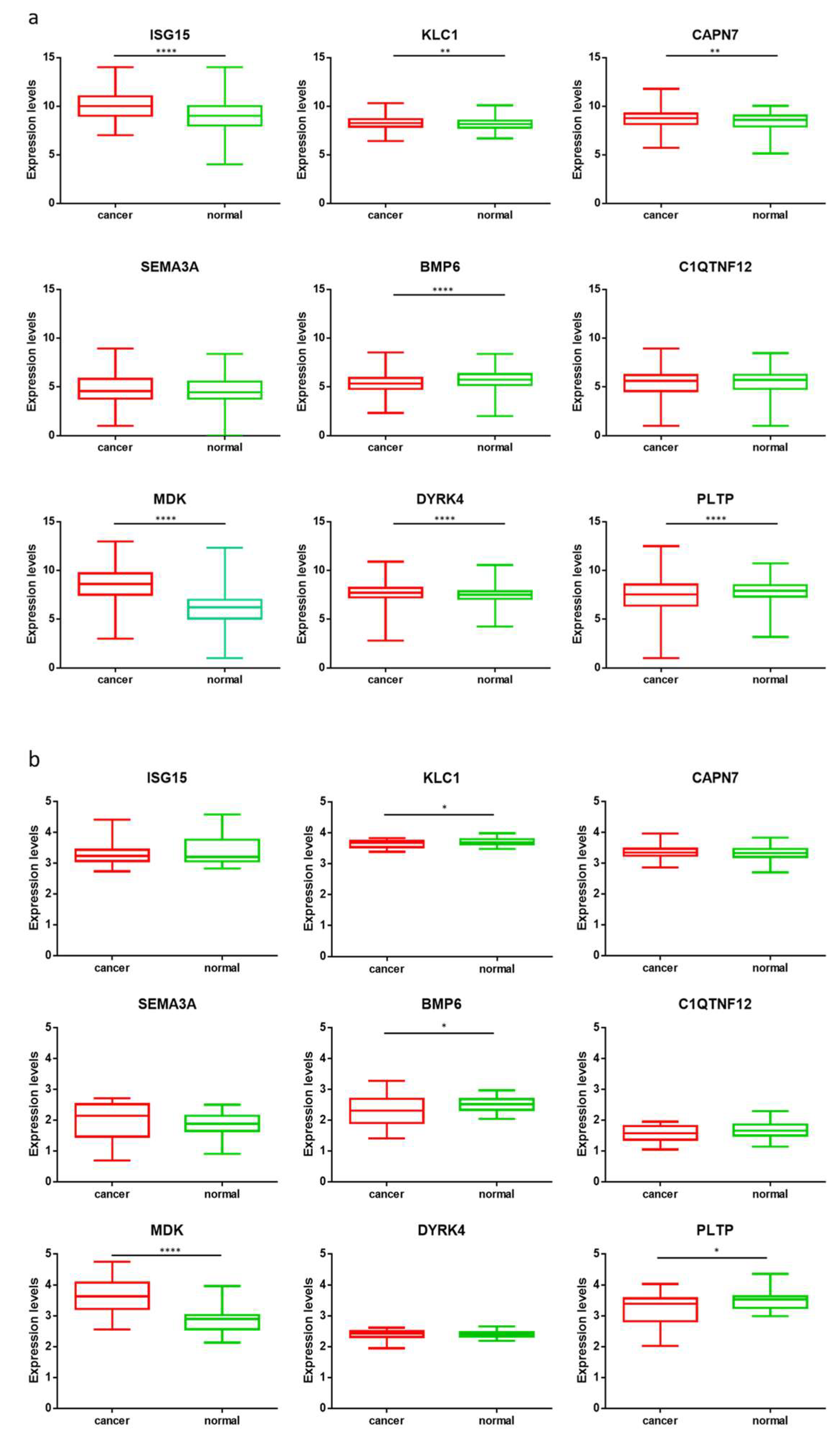
3.3. Gene Expression Analysis
3.4. Analysis of Pathways
3.5. Peptide Prediction
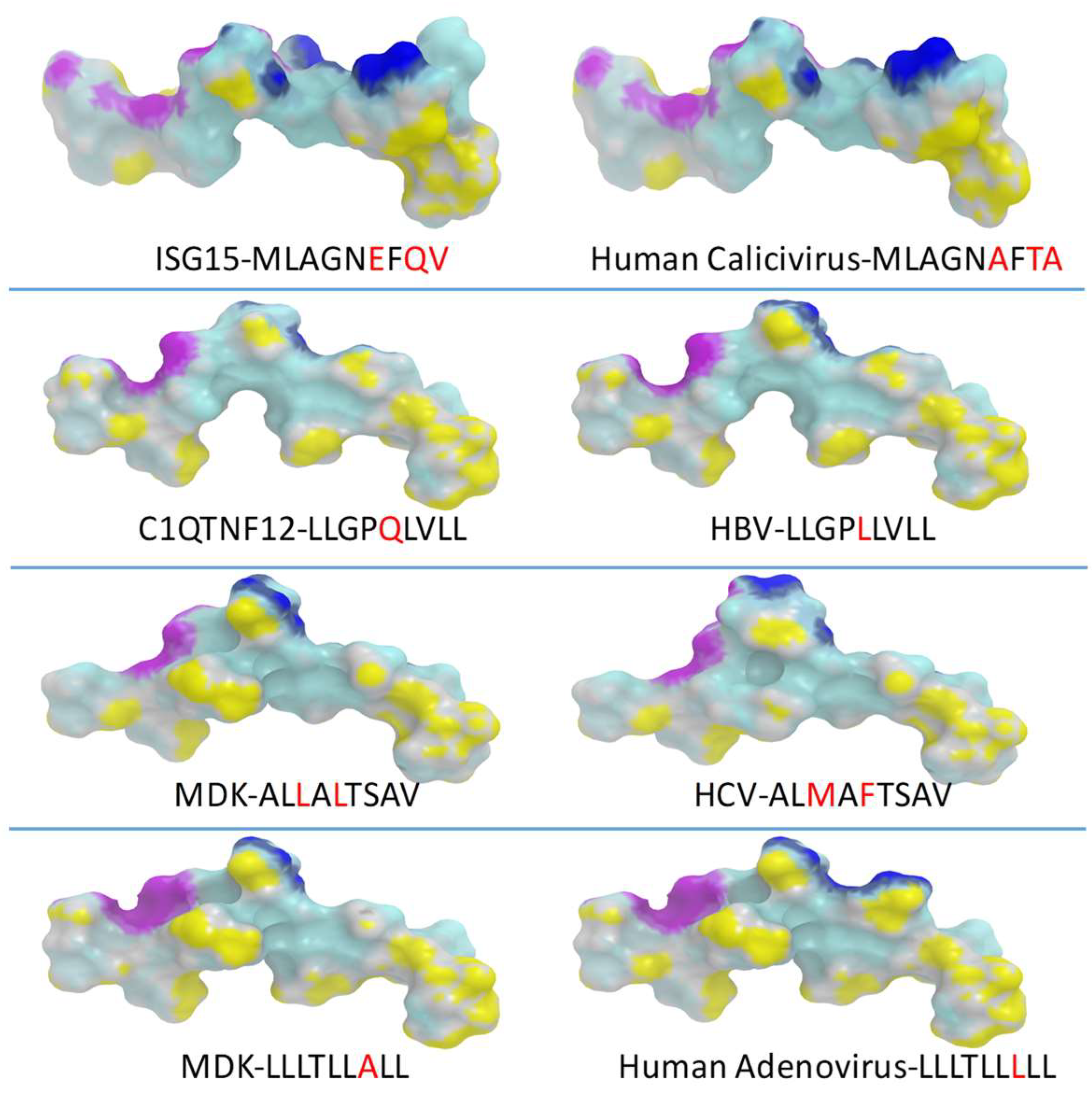
3.6. Identification of Homologous Viral Epitopes
3.7. Peptide Modeling and Molecular Docking
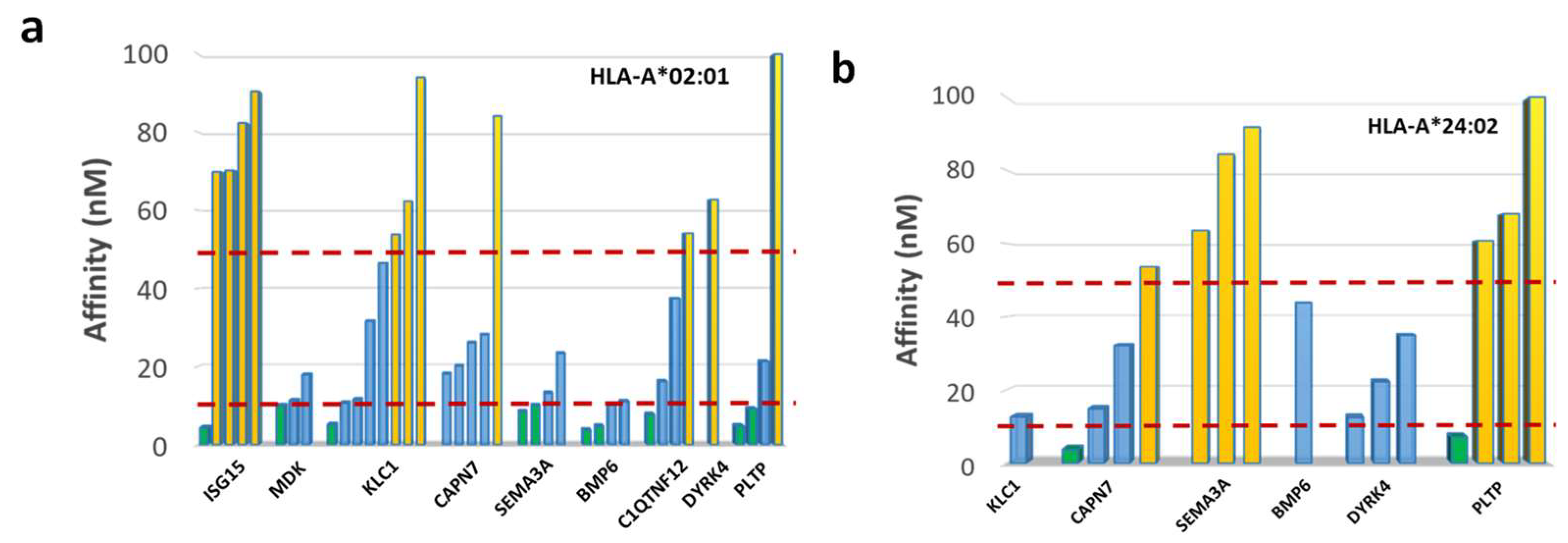
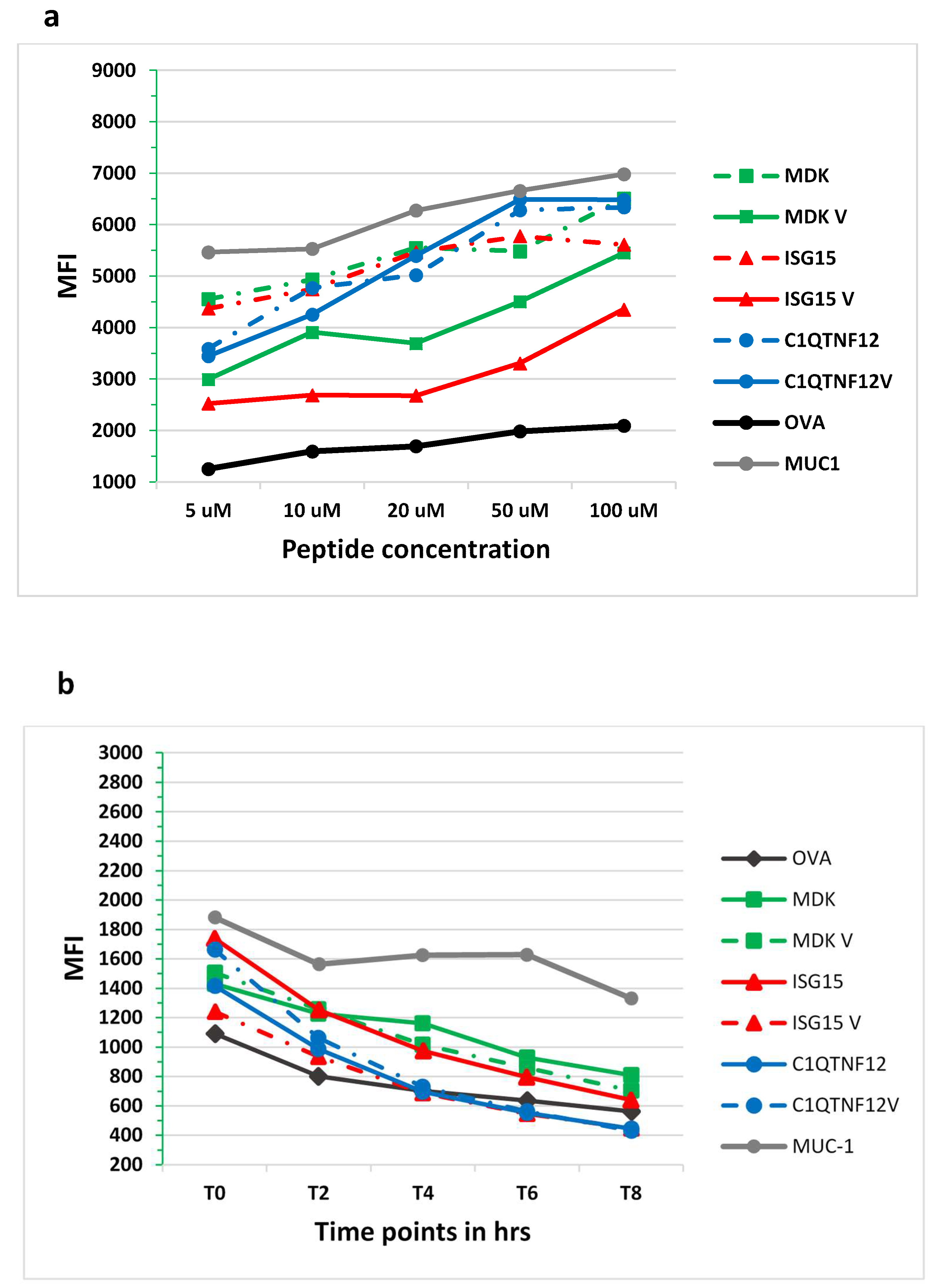
3.8. In Vitro Analysis of Binding Affinity and Stability to HLA-A*02:01 Molecule
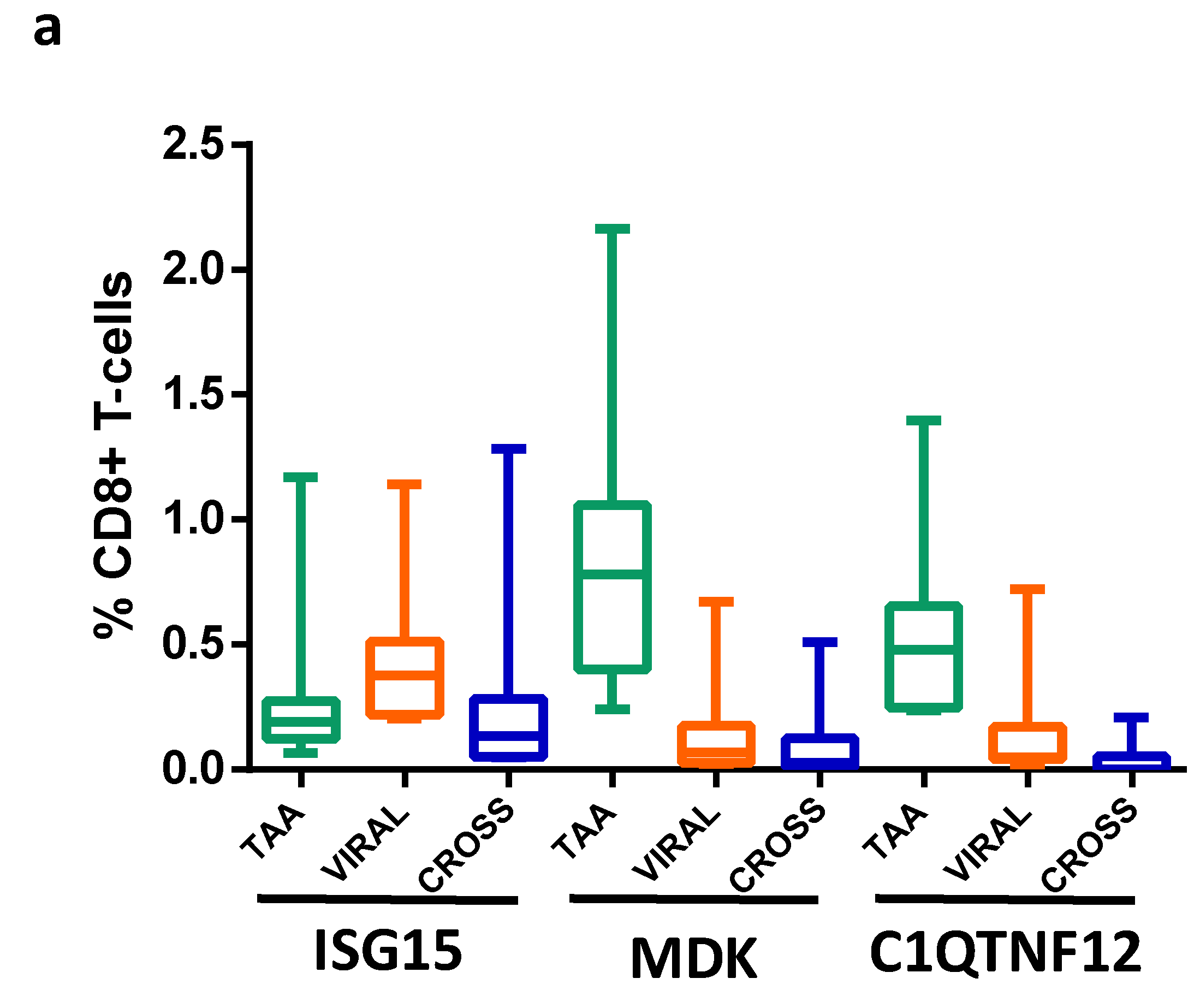
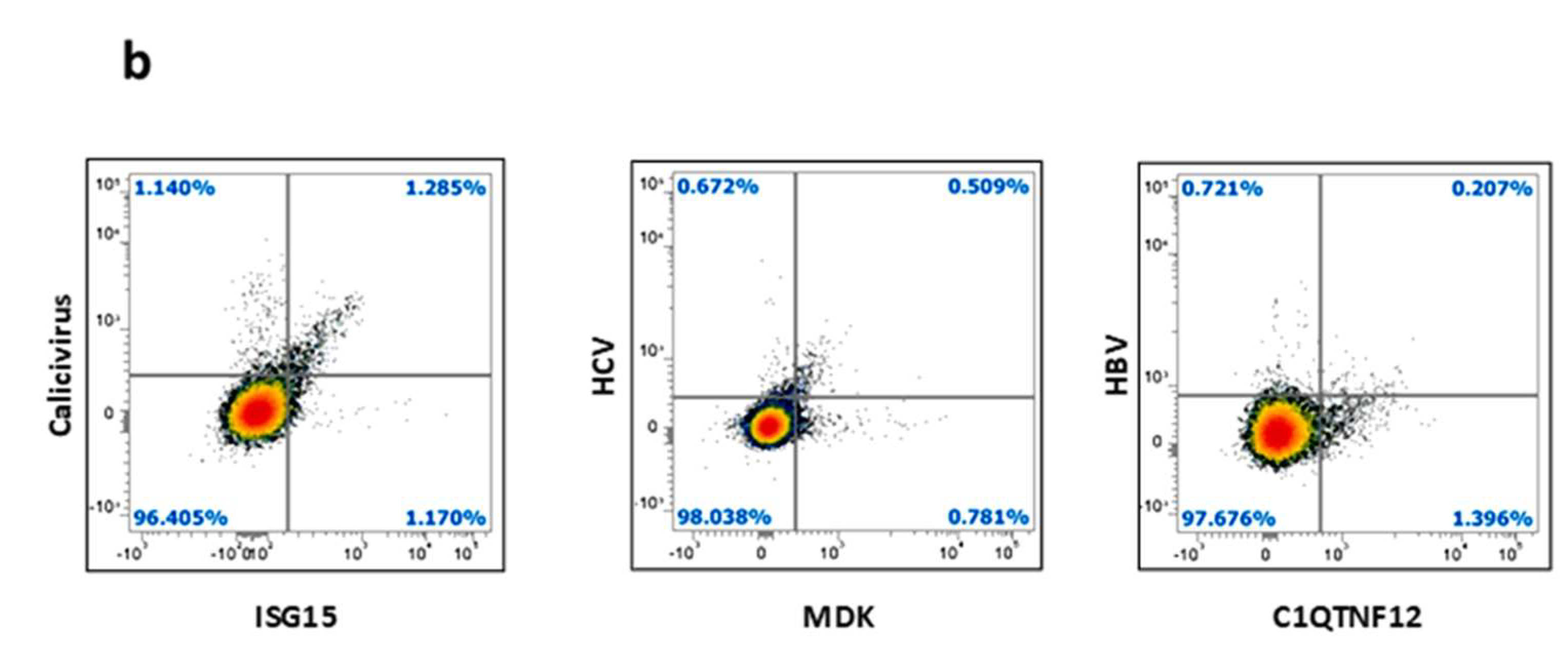
3.9. Detection of Epitope-Specific CD8+ T-Cell Clones
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, G.; Yang, S.; Yuan, J.; Threapleton, D.; Zhao, Q.; Chen, F.; Cao, H.; Jiang, T.; Li, L. Comparative efficacy of treatment strategies for hepatocellular carcinoma: Systematic review and network meta-analysis. BMJ Open 2018, 8, e021269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bangaru, S.; Marrero, J.A.; Singal, A.G. Review article: New therapeutic interventions for advanced hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2020, 51, 78–89. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.M.; Hwang, S.; Keam, B.; Yu, Y.S.; Kim, J.H.; Kim, D.-S.; Bae, S.H.; Kim, G.-D.; Lee, J.K.; Seo, Y.B.; et al. Gene Signature for Sorafenib Susceptibility in Hepatocellular Carcinoma: Different Approach with a Predictive Biomarker. Liver Cancer 2020, 9, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. IMbrave150 Investigators. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 14, 1894–1905. [Google Scholar]
- Tagliamonte, M.; Mauriello, A.; Cavalluzzo, B.; Ragone, C.; Manolio, C.; Petrizzo, A.; Buonaguro, L. Tackling hepatocellular carcinoma with individual or combinatorial immunotherapy approaches. Cancer Lett. 2020, 473, 25–32. [Google Scholar] [CrossRef]
- Montalbano, M.; Georgiadis, J.; Masterson, A.L.; McGuire, J.T.; Prajapati, J.; Shirafkan, A.; Rastellini, C.; Cicalese, L. Biology and function of glypican-3 as a candidate for early cancerous transformation of hepatocytes in hepatocellular carcinoma (Review). Oncol. Rep. 2017, 37, 1291–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagliamonte, M.; Petrizzo, A.; Tornesello, M.L.; Ciliberto, G.; Buonaguro, F.M.; Buonaguro, L. Combinatorial immunotherapy strategies for hepatocellular carcinoma. Curr. Opin. Immunol. 2016, 39, 103–113. [Google Scholar] [CrossRef]
- Buonaguro, L.; HEPAVAC Consortium. Developments in cancer vaccines for hepatocellular carcinoma. Cancer Immunol. Immunother. 2016, 65, 93–99. [Google Scholar] [CrossRef]
- Buonaguro, L.; HEPAVAC Consortium. New vaccination strategies in liver cancer. Cytokine Growth Factor Rev. 2017, 36, 125–129. [Google Scholar] [CrossRef]
- Löffler, M.W.; Mohr, C.; Bichmann, L.; Freudenmann, L.K.; Walzer, M.; Schroeder, C.M.; Trautwein, N.; Hilke, F.J.; Zinser, R.S.; Mühlenbruch, L.; et al. Multi-omics discovery of exome-derived neoantigens in hepatocellular carcinoma. Genome Med. 2019, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Mauriello, A.; Zeuli, R.; Cavalluzzo, B.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M.; Ceccarelli, M.; Tagliamonte, M.; Buonaguro, L. High Somatic Mutation and Neoantigen Burden Do Not Correlate with Decreased Progression-Free Survival in HCC Patients not Undergoing Immunotherapy. Cancers 2019, 11, 1824. [Google Scholar] [CrossRef] [Green Version]
- Petrizzo, A.; Caruso, F.P.; Tagliamonte, M.; Tornesello, M.L.; Ceccarelli, M.; Costa, V.; Aprile, M.; Esposito, R.; Ciliberto, G.; Buonaguro, F.M.; et al. Identification and Validation of HCC-specific Gene Transcriptional Signature for Tumor Antigen Discovery. Sci. Rep. 2016, 6, 29258. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.-L.; Ye, Q.-H.; Qin, L.-X.; Budhu, A.; Forgues, M.; Chen, Y.; Liu, Y.-K.; Sun, H.-C.; Wang, L.; Lu, H.-Z.; et al. Gene Expression Profiling Reveals Potential Biomarkers of Human Hepatocellular Carcinoma. Clin. Cancer Res. 2007, 13, 1133–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, A.; Lukk, M.; Kutter, C.; Davies, S.; Alexander, G.; Odom, D.T. Global gene expression profiling reveals SPINK1 as a potential hepatocellular carcinoma marker. PLoS ONE 2013, 8, e59459. [Google Scholar] [CrossRef] [Green Version]
- Shirota, Y.; Kaneko, S.; Honda, M.; Kawai, H.F.; Kobayashi, K. Identification of differentially expressed genes in hepatocellular carcinoma with cDNA microarrays. Hepatology 2001, 33, 832–840. [Google Scholar] [CrossRef]
- Xia, Q.; Li, Z.; Zheng, J.; Zhang, X.; Di, Y.; Ding, J.; Yu, D.; Yan, L.; Shen, L.; Yan, D.; et al. Identification of novel biomarkers for hepatocellular carcinoma using transcriptome analysis. J. Cell Physiol. 2019, 234, 4851–4863. [Google Scholar] [CrossRef] [PubMed]
- Ocker, M. Biomarkers for hepatocellular carcinoma: What’s new on the horizon? World J. Gastroenterol. 2018, 24, 3974–3979. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Oksvold, P.; Fagerberg, L.; Lundberg, E.; Jonasson, K.; Forsberg, M.; Zwahlen, M.; Kampf, C.; Wester, K.; Hober, S.; et al. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010, 28, 1248–1250. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.; Fenoy, E.; Harndahl, M.; Kristensen, A.B.; Nielsen, I.K.; Nielsen, M.; Buus, S. Pan-Specific Prediction of Peptide–MHC Class I Complex Stability, a Correlate of T Cell Immunogenicity. J. Immunol. 2016, 197, 1517–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garboczi, D.N.; Ghosh, P.; Utz, U.; Fan, Q.R.; Biddison, W.E.; Wiley, D.C. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nat. Cell Biol. 1996, 384, 134–141. [Google Scholar] [CrossRef]
- Saini, S.K.; Tamhane, T.; Anjanappa, R.; Saikia, A.; Ramskov, S.; Donia, M.; Svane, I.M.; Jakobsen, S.N.; Garcia-Alai, M.; Zacharias, M.; et al. Empty peptide-receptive MHC class I molecules for efficient detection of antigen-specific T cells. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Petrizzo, A.; Tagliamonte, M.; Mauriello, A.; Costa, V.; Aprile, M.; Esposito, R.; Caporale, A.; Luciano, A.; Arra, C.; Tornesello, M.L.; et al. Unique true predicted neoantigens (TPNAs) correlates with anti-tumor immune control in HCC patients. J. Transl. Med. 2018, 16, 286. [Google Scholar] [CrossRef]
- Ragone, C.; Manolio, C.; Cavalluzzo, B.; Mauriello, A.; Tornesello, M.L.; Buonaguro, F.M.; Castiglione, F.; Vitagliano, L.; Iaccarino, E.; Ruvo, M.; et al. Identification and validation of viral antigens sharing sequence and structural homology with tumor-associated antigens (TAAs). J. Immunother. Cancer 2021, 9, e002694. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Cerullo, V. Pathogens: Our Allies against Cancer? Mol. Ther. 2021, 29, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Hall, A.J.; Robinson, A.E.; Verhoef, L.; Premkumar, P.; Parashar, U.D.; Koopmans, M.; Lopman, B.A. Global prevalence of norovirus in cases of gastroenteritis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 725–730. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PROTEIN | Best Expression Cut off-p Score |
|---|---|
| ISG15 | 0.041 |
| KLC1 | 0.0012 |
| CAPN7 | 0.0022 |
| SEMA3A | 0.0029 |
| BMP6 | 0.0016 |
| C1QTNF12 (FAM132A) | 0.0027 |
| MDK | 0.0057 |
| DYRK4 | 0.020 |
| PLTP | 0.017 |
| HLA | Protein | Sequence | ViralProtein | AFF | Thalf | Binding |
|---|---|---|---|---|---|---|
| A*02:01 | ISG15 | MLAGNEFQV | 4.35 | 5.22 | SB | |
| MLAGNA FTA | capsid protein human calicivirus Seq ID: AAL18874.1 | 13.86 | 3.53 | WB | ||
| MDK | ALLALTSAV | 10.06 | 9.85 | SB | ||
| ALMAFTSAV | polyprotein HCV seq ID: AID60264.1 | 3.11 | 45.58 | SB | ||
| MDK | LLLTLLALL | 11.34 | 4.69 | WB | ||
| LLLTLLLLL | E3 14.5 kDa protein human adenovirus E4 Seq ID: AGT51280.1 | 17.75 | 4.41 | WB | ||
| C1QTNF12 | LLGPQLVLL | 37.45 | 2.61 | WB | ||
| LLGPLLVLL | S protein HBV Seq ID: AUF41974.1 | 20.91 | 3.38 | WB | ||
| A*24:02 | CAPN7 | VYSACSFTF | 3.83 | 61.9 | SB | |
| GSPACTFTF | protein UL29 CMV Seq ID: AFR55693.1 | 658.27 | 0.65 | WB | ||
| CAPN7 | IYTVSSFSI | 14.92 | 5.44 | SB | ||
| SYTVSSFQV | nonstructural protein 1 influenza A virus Seq ID: QEM33605.1 | 352.63 | 1.06 | WB | ||
| DYRK4 | FYFRNHFCI | 12.53 | 22.36 | SB | ||
| DYFRNQFKI | polymerase basic protein 2 influenza A virus Seq ID: AAV33795.1 | 77.75 | 1.39 | WB |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavalluzzo, B.; Mauriello, A.; Ragone, C.; Manolio, C.; Tornesello, M.L.; Buonaguro, F.M.; Tvingsholm, S.A.; Hadrup, S.R.; Tagliamonte, M.; Buonaguro, L. Novel Molecular Targets for Hepatocellular Carcinoma. Cancers 2022, 14, 140. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14010140
Cavalluzzo B, Mauriello A, Ragone C, Manolio C, Tornesello ML, Buonaguro FM, Tvingsholm SA, Hadrup SR, Tagliamonte M, Buonaguro L. Novel Molecular Targets for Hepatocellular Carcinoma. Cancers. 2022; 14(1):140. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14010140
Chicago/Turabian StyleCavalluzzo, Beatrice, Angela Mauriello, Concetta Ragone, Carmen Manolio, Maria Lina Tornesello, Franco M. Buonaguro, Siri Amanda Tvingsholm, Sine Reker Hadrup, Maria Tagliamonte, and Luigi Buonaguro. 2022. "Novel Molecular Targets for Hepatocellular Carcinoma" Cancers 14, no. 1: 140. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14010140