
IRE1α Inhibitors as a Promising Therapeutic Strategy in Blood Malignancies

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
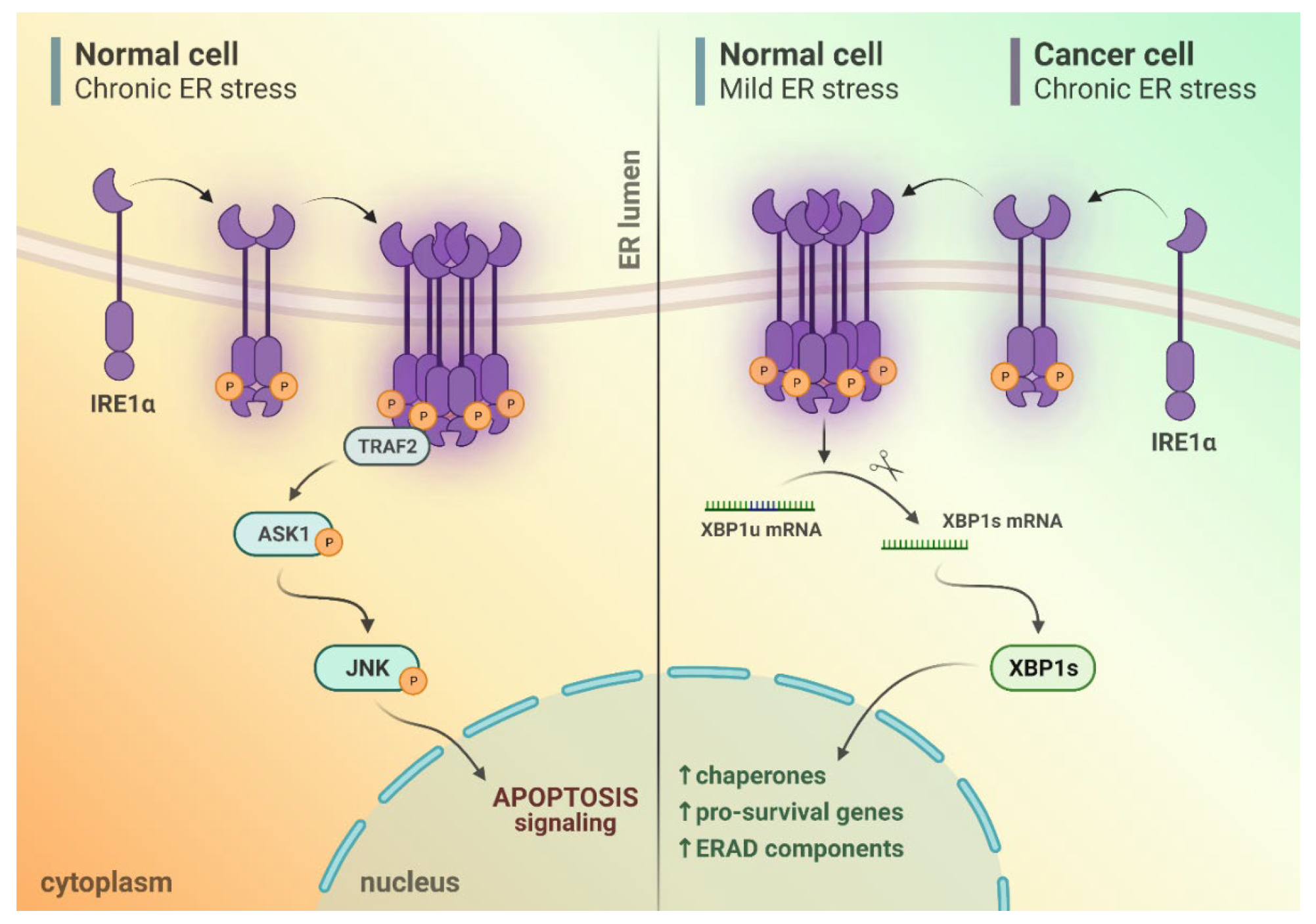
2. IRE1α Activation upon Endoplasmic Reticulum (ER) Stress Conditions
3. Different Outputs of IRE1α Activity upon ER Stress Conditions
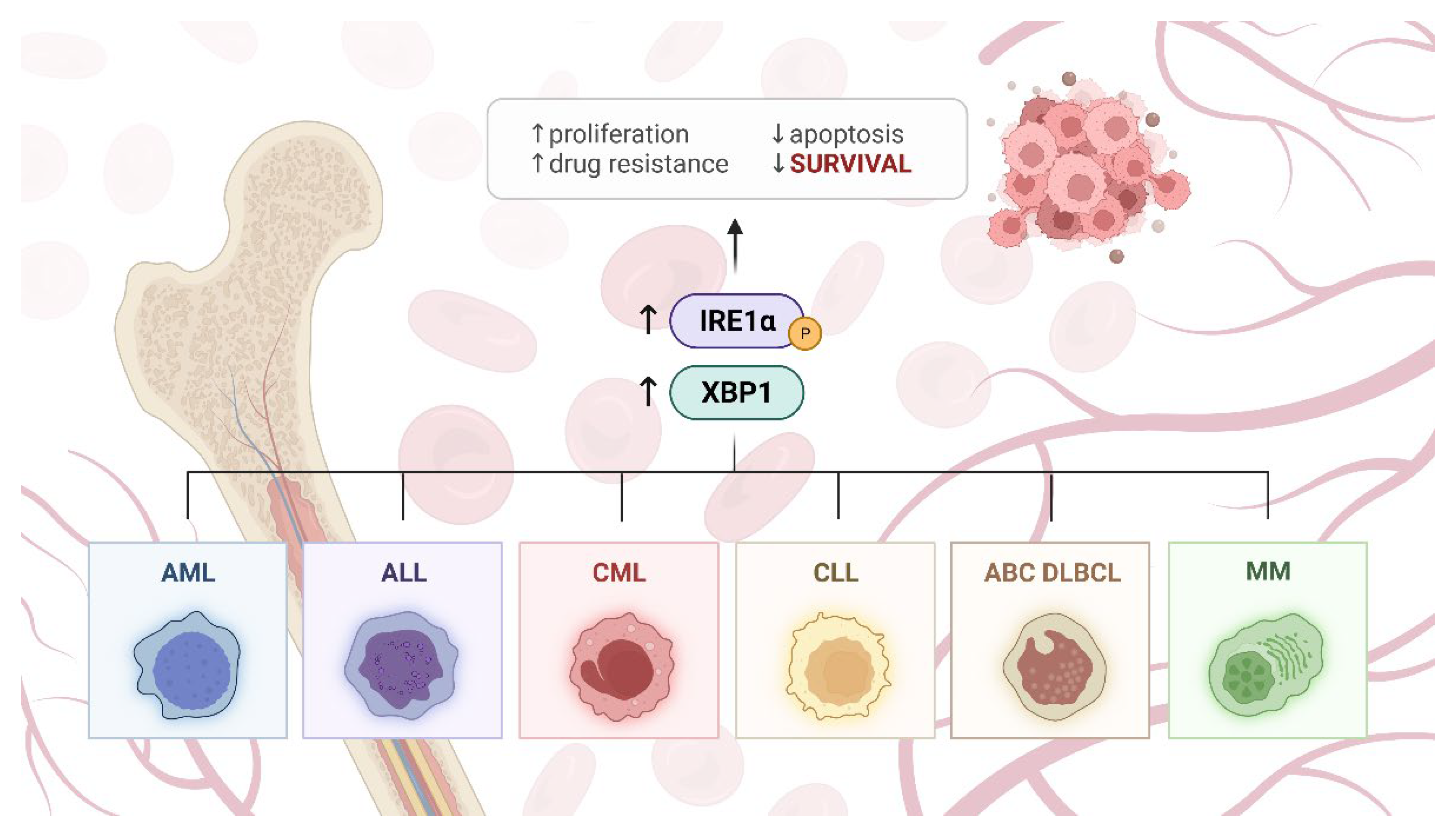
4. IRE1α in Blood Malignancies
4.1. Chronic Myelogenous Leukemia
4.2. Chronic Lymphocytic Leukemia
4.3. Acute Myeloid Leukemia
4.4. Acute Lymphoblastic Leukemia
4.5. Diffuse Large B-Cell Lymphoma
4.6. Other Lymphomas
4.7. Multiple Myeloma
5. Potential Application of IRE1α Inhibitors in Blood Malignancies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Naghavi, M.; Allen, C.; Barber, R.M.; Carter, A.; Casey, D.C.; Charlson, F.J.; Chen, A.Z.; Coates, M.M.; Coggeshall, M.; et al. Global, Regional, and National Life Expectancy, All-Cause Mortality, and Cause-Specific Mortality for 249 Causes of Death, 1980–2015: A Systematic Analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar] [CrossRef] [Green Version]
- Karakosta, M.; Delicha, E.M.; Kouraklis, G.; Manola, K.N. Association of Various Risk Factors with Chronic Lymphocytic Leukemia and Its Cytogenetic Characteristics. Arch. Environ. Occup. Health 2016, 71, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Bernatsky, S.; Ramsey-Goldman, R.; Labrecque, J.; Joseph, L.; Boivin, J.F.; Petri, M.; Zoma, A.; Manzi, S.; Urowitz, M.B.; Gladman, D.; et al. Cancer Risk in Systemic Lupus: An Updated International Multi-Centre Cohort Study. J. Autoimmun. 2013, 42, 130–135. [Google Scholar] [CrossRef] [Green Version]
- Mercer, L.K.; Davies, R.; Galloway, J.B.; Low, A.; Lunt, M.; Dixon, W.G.; Watson, K.D.; Symmons, D.P.M.; Hyrich, K.L. Risk of Cancer in Patients Receiving Non-Biologic Disease-Modifying Therapy for Rheumatoid Arthritis Compared with the UK General Population. Rheumatology 2013, 52, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Lebwohl, B.; Granath, F.; Ekbom, A.; Smedby, K.E.; Murray, J.A.; Neugut, A.I.; Green, P.H.R.; Ludvigsson, J.F. Mucosal Healing and Risk for Lymphoproliferative Malignancy in Celiac Disease: A Population-Based Cohort Study. Ann. Intern. Med. 2013, 159, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Ceribelli, M.; Pittaluga, S.; Wright, G.; Staudt, L.M. Oncogenic Mechanisms in Burkitt Lymphoma. Cold Spring Harb. Perspect. Med. 2014, 4, a014282. [Google Scholar] [CrossRef] [Green Version]
- Kaneda, A.; Matsusaka, K.; Aburatani, H.; Fukayama, M. Epstein–Barr Virus Infection as an Epigenetic Driver of Tumorigenesis: Figure 1. Cancer Res. 2012, 72, 3445–3450. [Google Scholar] [CrossRef] [Green Version]
- Van Nimwegen, F.A.; Ntentas, G.; Darby, S.C.; Schaapveld, M.; Hauptmann, M.; Lugtenburg, P.J.; Janus, C.P.M.; Daniels, L.; Van Leeuwen, F.E.; Cutter, D.J.; et al. Risk of Heart Failure in Survivors of Hodgkin Lymphoma: Effects of Cardiac Exposure to Radiation and Anthracyclines. Blood 2017, 129, 2257–2265. [Google Scholar] [CrossRef]
- Velez, M.P.; Richardson, H.; Baxter, N.N.; McClintock, C.; Greenblatt, E.; Barr, R.; Green, M. Risk of Infertility in Female Adolescents and Young Adults with Cancer: A Population-Based Cohort Study. Hum. Reprod. 2021, 36, 1981–1988. [Google Scholar] [CrossRef]
- Bhuller, K.S.; Zhang, Y.; Li, D.; Sehn, L.H.; Goddard, K.; McBride, M.L.; Rogers, P.C. Late Mortality, Secondary Malignancy and Hospitalisation in Teenage and Young Adult Survivors of Hodgkin Lymphoma: Report of the Childhood/Adolescent/Young Adult Cancer Survivors Research Program and the BC Cancer Agency Centre for Lymphoid Cancer. Br. J. Haematol. 2016, 172, 757–768. [Google Scholar] [CrossRef] [PubMed]
- Feldman, H.C.; Vidadala, V.N.; Potter, Z.E.; Papa, F.R.; Backes, B.J.; Maly, D.J. Development of a Chemical Toolset for Studying the Paralog-Specific Function of IRE1. ACS Chem. Biol. 2019, 14, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Lin, D.C.; Guo, X.; Masouleh, B.K.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1α-Driven pro-Survival Pathways Is a Promising Therapeutic Application in Acute Myeloid Leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef] [PubMed]
- Masouleh, B.K.; Geng, H.; Hurtz, C.; Chan, L.N.; Logan, A.C.; Chang, M.S.; Huang, C.; Swaminathan, S.; Sun, H.; Paietta, E.; et al. Mechanistic Rationale for Targeting the Unfolded Protein Response in Pre-B Acute Lymphoblastic Leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E2219–E2228. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wang, X.; Li, N.; Liu, J.; Zhang, L.; Hui, L.; Feng, A.; Wang, Z.; Wang, Y. Prolonged Unfolded Protein Reaction Is Involved in the Induction of Chronic Myeloid Leukemia Cell Death upon Oprozomib Treatment. Cancer Sci. 2021, 112, 133–143. [Google Scholar] [CrossRef]
- Zhang, D.; De Veirman, K.; Fan, R.; Jian, Q.; Zhang, Y.; Lei, L.; Evans, H.; Wang, Y.; Lei, L.; Wang, B.; et al. ER Stress Arm XBP1s Plays a Pivotal Role in Proteasome Inhibition-Induced Bone Formation. Stem Cell Res. Ther. 2020, 11, 516. [Google Scholar] [CrossRef]
- Anelli, T.; Sitia, R. Protein Quality Control in the Early Secretory Pathway. EMBO J. 2008, 27, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Byrd, A.; Brewer, J. Intricately Regulated: A Cellular Toolbox for Fine-Tuning XBP1 Expression and Activity. Cells 2012, 1, 738–753. [Google Scholar] [CrossRef] [Green Version]
- Blazanin, N.; Son, J.; Craig-Lucas, A.B.; John, C.L.; Breech, K.J.; Podolsky, M.A.; Glick, A.B. ER Stress and Distinct Outputs of the IRE1α RNase Control Proliferation and Senescence in Response to Oncogenic Ras. Proc. Natl. Acad. Sci. USA 2017, 114, 9900–9905. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the Cis-Acting Endoplasmic Reticulum Stress Response Element Responsible for Transcriptional Induction of Mammalian Glucose- Regulated Proteins: Involvement of Basic Leucine Zipper Transcription Factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [Green Version]
- Morl, K.; Ma, W.; Gething, M.J.; Sambrook, J. A Transmembrane Protein with a Cdc2+ CDC28-Related Kinase Activity Is Required for Signaling from the ER to the Nucleus. Cell 1993, 74, 743–756. [Google Scholar] [CrossRef]
- Huang, S.; Xing, Y.; Liu, Y. Emerging Roles for the ER Stress Sensor IRE1 in Metabolic Regulation and Disease. J. Biol. Chem. 2019, 294, 18726–18741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urano, F.; Wang, X.Z.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of Stress in the ER to Activation of JNK Protein Kinases by Transmembrane Protein Kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagöz, G.E.; Acosta-Alvear, D.; Nguyen, H.T.; Lee, C.P.; Chu, F.; Walter, P. An Unfolded Protein-Induced Conformational Switch Activates Mammalian IRE1. eLife 2017, 6, e30700. [Google Scholar] [CrossRef] [PubMed]
- Carlesso, A.; Hörberg, J.; Reymer, A.; Eriksson, L.A. New Insights on Human IRE1 Tetramer Structures Based on Molecular Modeling. Sci. Rep. 2020, 10, 17490. [Google Scholar] [CrossRef]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 Has Distinct Catalytic Mechanisms for XBP1/HAC1 Splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef] [Green Version]
- Belyy, V.; Tran, N.H.; Walter, P. Quantitative Microscopy Reveals Dynamics and Fate of Clustered IRE1α. Proc. Natl. Acad. Sci. USA 2020, 117, 1533–1542. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, Y.; Yi, P.; Dong, W.; Nalin, A.P.; Zhang, J.; Zhu, Z.; Chen, L.; Benson, D.M.; Mundy-Bosse, B.L.; et al. The IL-15–AKT–XBP1s Signaling Pathway Contributes to Effector Functions and Survival in Human NK Cells. Nat. Immunol. 2019, 20, 10–17. [Google Scholar] [CrossRef]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1α-XBP1s Pathway Promotes Prostate Cancer by Activating c-MYC Signaling. Nat. Commun. 2019, 10, 323. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Adams, N.M.; Xu, Y.; Cao, J.; Allan, D.S.J.; Carlyle, J.R.; Chen, X.; Sun, J.C.; Glimcher, L.H. The IRE1 Endoplasmic Reticulum Stress Sensor Activates Natural Killer Cell Immunity in Part by Regulating C-Myc. Nat. Immunol. 2019, 20, 865–878. [Google Scholar] [CrossRef]
- Iwakoshi, N.N.; Lee, A.H.; Vallabhajosyula, P.; Otipoby, K.L.; Rajewsky, K.; Glimcher, L.H. Plasma Cell Differentiation and the Unfolded Protein Response Intersect at the Transcription Factor XBP-I. Nat. Immunol. 2003, 4, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Obiedat, A.; Seidel, E.; Mahameed, M.; Bernani, O.; Tsukerman, P.; Voutetakis, K.; Chatziioannou, A.; Mcmahon, M.; Avril, T.; Chevet, E.; et al. Transcription of the NKG2D Ligand MICA Is Suppressed by the IRE1/XBP1 Pathway of the Unfolded Protein Response through the Regulation of E2F1. FASEB J. 2019, 33, 3481–3495. [Google Scholar] [CrossRef] [PubMed]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma Cell Differentiation Requires the Transcription Factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; Dejeans, N.; Bouchecareilh, M.; Lhomond, S.; Pineau, R.; Higa, A.; Delugin, M.; Combe, C.; Loriot, S.; Cubel, G.; et al. Posttranscriptional Regulation of Per1 Underlies the Oncogenic Function of IREα. Cancer Res. 2013, 73, 4732–4743. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 Promotes Triple-Negative Breast Cancer by Controlling the HIF1α Pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Moore, K.; Hollien, J. Ire1-Mediated Decay in Mammalian Cells Relies on MRNA Sequence, Structure, and Translational Status. Mol. Biol. Cell 2015, 26, 2873–2884. [Google Scholar] [CrossRef]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel Mechanism of Enhancing IRE1α-XBP1 Signalling via the PERK-ATF4 Pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef] [Green Version]
- Hollien, J.; Weissman, J.S. Decay of Endoplasmic Reticulum-Localized MRNAs during the Unfolded Protein Response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.K.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1α Induces Thioredoxin-Interacting Protein to Activate the NLRP3 Inflammasome and Promote Programmed Cell Death under Irremediable ER Stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [Green Version]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1α Cleaves Select MicroRNAs during ER Stress to Derepress Translation of Proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1α Kinase Activation Modes Control Alternate Endoribonuclease Outputs to Determine Divergent Cell Fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [Green Version]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in Cell Fate Regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Aronson, L.I.; Davies, F.E. DangER: Protein OvERload. Targeting Protein Degradation to Treat Myeloma. Haematologica 2012, 97, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 Couples Endoplasmic Reticulum Load to Secretory Capacity by Processing the XBP-1 MRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.K.; Dey, M.; Neculai, D.; Cao, C.; Dever, T.E.; Sicheri, F. Structure of the Dual Enzyme Ire1 Reveals the Basis for Catalysis and Regulation in Nonconventional RNA Splicing. Cell 2008, 132, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian Endoplasmic Reticulum Stress Sensor IRE1 Signals by Dynamic Clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef] [Green Version]
- Bonventre, J.V.; Zuk, A. Ischemic Acute Renal Failure: An Inflammatory Disease? Kidney Int. 2004, 66, 480–485. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.; Strudwick, N.; Suwara, M.; Sutcliffe, L.K.; Mihai, A.D.; Ali, A.A.; Watson, J.N.; Schröder, M. An Initial Phase of JNK Activation Inhibits Cell Death Early in the Endoplasmic Reticulum Stress Response. J. Cell Sci. 2016, 129, 2317–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Reed, J.C. ER Stress-Induced Cell Death Mechanisms. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond—Mitochondrial Performance in Apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saveljeva, S.; Mc Laughlin, S.L.; Vandenabeele, P.; Samali, A.; Bertrand, M.J.M. Endoplasmic Reticulum Stress Induces Ligand-Independent TNfR1-Mediated Necroptosis in L929 Cells. Cell Death Dis. 2015, 6, e1587. [Google Scholar] [CrossRef] [PubMed]
- Shemorry, A.; Harnoss, J.M.; Guttman, O.; Marsters, S.A.; Kőműves, L.G.; Lawrence, D.A.; Ashkenazi, A. Caspase-Mediated Cleavage of IRE1 Controls Apoptotic Cell Commitment during Endoplasmic Reticulum Stress. eLife 2019, 8, e47084. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis Control by the Unfolded Protein Response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Akha, A.A.S.; Raden, D.; Kaufman, R.J. Adaptation to ER Stress Is Mediated by Differential Stabilities of Pro-Survival and pro-Apoptotic MRNAs and Proteins. PLoS Biol. 2006, 4, 2024–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; LaVail, M.M.; Walter, P. IRE1 Signaling Affects Cell Fate during the Unfolded Protein Response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwakoshi, N.N.; Pypaert, M.; Glimcher, L.H. The Transcription Factor XBP-1 Is Essential for the Development and Survival of Dendritic Cells. J. Exp. Med. 2007, 204, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Tavernier, S.J.; Osorio, F.; Vandersarren, L.; Vetters, J.; Vanlangenakker, N.; Van Isterdael, G.; Vergote, K.; De Rycke, R.; Parthoens, E.; Van De Laar, L.; et al. Regulated IRE1-Dependent MRNA Decay Sets the Threshold for Dendritic Cell Survival. Nat. Cell Biol. 2017, 19, 698–710. [Google Scholar] [CrossRef] [Green Version]
- Chitnis, N.S.; Pytel, D.; Bobrovnikova-Marjon, E.; Pant, D.; Zheng, H.; Maas, N.L.; Frederick, B.; Kushner, J.A.; Chodosh, L.A.; Koumenis, C.; et al. MiR-211 Is a Prosurvival MicroRNA That Regulates Chop Expression in a PERK-Dependent Manner. Mol. Cell 2012, 48, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol. Cell 2018, 71, 629–636.e5. [Google Scholar] [CrossRef] [Green Version]
- Sozen, E.; Yazgan, B.; Tok, O.E.; Demirel, T.; Ercan, F.; Proto, J.D.; Ozer, N.K. Cholesterol Induced Autophagy via IRE1/JNK Pathway Promotes Autophagic Cell Death in Heart Tissue. Metabolism 2020, 106, 154205. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek, W.; Pytel, D.; Wawrzynkiewicz, A.; Dziki, A.; Dziki, Ł.; Diehl, J.A.; Majsterek, I. Dual Role of Endoplasmic Reticulum Stress-Mediated Unfolded Protein Response Signaling Pathway in Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 4354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Wang, H.; Wei, S.; Wang, Z.; Ji, G. Inhibition of ER Stress-Related IRE1α/CREB/NLRP1 Pathway Promotes the Apoptosis of Human Chronic Myelogenous Leukemia Cell. Mol. Immunol. 2018, 101, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhao, M.; Jin, X.; Ney, G.; Yang, K.B.; Peng, F.; Cao, J.; Iwawaki, T.; Del Valle, J.; Chen, X.; et al. Adaptive Endoplasmic Reticulum Stress Signalling via IRE1α–XBP1 Preserves Self-Renewal of Haematopoietic and Pre-Leukaemic Stem Cells. Nat. Cell Biol. 2019, 21, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Tang, C.H.A.; Song, J.H.; Mancuso, A.; Del Valle, J.R.; Cao, J.; Xiang, Y.; Dang, C.V.; Lan, R.; Sanchez, D.J.; et al. IRE1α RNase-Dependent Lipid Homeostasis Promotes Survival in Myc-Transformed Cancers. J. Clin. Investig. 2018, 128, 1300–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.H.A.; Ranatunga, S.; Kriss, C.L.; Cubitt, C.L.; Tao, J.; Pinilla-Ibarz, J.A.; Del Valle, J.R.; Hu, C.C.A. Inhibition of ER Stress-Associated IRE-1/XBP-1 Pathway Reduces Leukemic Cell Survival. J. Clin. Investig. 2014, 124, 2585–2598. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, A.; Yujiri, T.; Tanaka, Y.; Tanaka, M.; Mitani, N.; Nakamura, Y.; Ariyoshi, K.; Tanizawa, Y. Activation of the Unfolded Protein Response in Primary Acute Myeloid Leukemia Cells. Int. J. Hematol. 2011, 94, 300–302. [Google Scholar] [CrossRef]
- Schardt, J.A.; Weber, D.; Eyholzer, M.; Mueller, B.U.; Pabst, T. Activation of the Unfolded Protein Response Is Associated with Favorable Prognosis in Acute Myeloid Leukemia. Clin. Cancer Res. 2009, 15, 3834–3841. [Google Scholar] [CrossRef] [Green Version]
- Doron, B.; Abdelhamed, S.; Butler, J.T.; Hashmi, S.K.; Horton, T.M.; Kurre, P. Transmissible ER Stress Reconfigures the AML Bone Marrow Compartment. Leukemia 2019, 33, 918–930. [Google Scholar] [CrossRef]
- Zhou, C.; Martinez, E.; Di Marcantonio, D.; Solanki-Patel, N.; Aghayev, T.; Peri, S.; Ferraro, F.; Skorski, T.; Scholl, C.; Fröhling, S.; et al. JUN Is a Key Transcriptional Regulator of the Unfolded Protein Response in Acute Myeloid Leukemia. Leukemia 2017, 31, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, T.; Bick, F.; Peters, K.; Mohta, V.; Tirosh, B.; Patterson, J.B.; Kharabi-Masouleh, B.; Huber, M. Infliction of Proteotoxic Stresses by Impairment of the Unfolded Protein Response or Proteasomal Inhibition as a Therapeutic Strategy for Mast Cell Leukemia. Oncotarget 2018, 9, 2984–3000. [Google Scholar] [CrossRef] [Green Version]
- Feizi, F.; Allahbakhshian Farsani, M.; Mirzaeian, A.; Takhviji, V.; Hajifathali, A.; Hossein Mohammadi, M. Triangle Collaboration Assessment of Autophagy, ER Stress and Hypoxia in Leukemogenesis: A Bright Perspective on the Molecular Recognition of B-ALL. Arch. Physiol. Biochem. 2021, 127, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Balague, O.; Mozos, A.; Martinez, D.; Hernandez, L.; Colomo, L.; Mate, J.L.; Teruya-Feldstein, J.; Lin, O.; Campo, E.; Lopez-Guillermo, A.; et al. Activation of the Endoplasmic Reticulum Stress-Associated Transcription Factor X Box-Binding Protein-1 Occurs in a Subset of Normal Germinal-Center B Cells and in Aggressive B-Cell Lymphomas with Prognostic Implications. Am. J. Pathol. 2009, 174, 2337–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giuli, M.V.; Diluvio, G.; Giuliani, E.; Franciosa, G.; Di Magno, L.; Pignataro, M.G.; Tottone, L.; Nicoletti, C.; Besharat, Z.M.; Peruzzi, G.; et al. Notch3 Contributes to T-Cell Leukemia Growth via Regulation of the Unfolded Protein Response. Oncogenesis 2020, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Bujisic, B.; De Gassart, A.; Tallant, R.; Demaria, O.; Zaffalon, L.; Chelbi, S.; Gilliet, M.; Bertoni, F.; Martinon, F. Impairment of Both IRE1 Expression and XBP1 Activation Is a Hallmark of GCB DLBCL and Contributes to Tumor Growth. Blood 2017, 129, 2420–2428. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Zhang, W.; Wang, J.; Gao, L.; Jiao, L.; Wang, L.; Zheng, J.; Cai, Z.; Yang, J. Aggregative Perivascular Tumor Cell Growth Pattern of Primary Central Nervous System Lymphomas Is Associated with Hypoxia-Related Endoplasmic Reticulum Stress. J. Cancer 2021, 12, 3841–3852. [Google Scholar] [CrossRef]
- Zhang, X.-T.; Hu, X.-B.; Wang, H.-L.; Kan, W.-J.; Xu, L.; Wang, Z.-J.; Xiang, Y.-Q.; Wu, W.-B.; Feng, B.; Li, J.-N.; et al. Activation of Unfolded Protein Response Overcomes Ibrutinib Resistance in Diffuse Large B-Cell Lymphoma. Acta Pharmacol. Sin. 2021, 42, 814–823. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Kung, J.T. Rapid Death of Follicular B Cells and Burkitt Lymphoma Cells Effectuated by Xbp1s. J. Immunol. 2020, 204, 3236–3247. [Google Scholar] [CrossRef]
- Shigemi, Z.; Baba, Y.; Hara, N.; Matsuhiro, J.; Kagawa, H.; Watanabe, T.; Fujimuro, M. Effects of ER Stress on Unfolded Protein Responses, Cell Survival, and Viral Replication in Primary Effusion Lymphoma. Biochem. Biophys. Res. Commun. 2016, 469, 565–572. [Google Scholar] [CrossRef]
- Gonnella, R.; Gilardini Montani, M.S.; Guttieri, L.; Romeo, M.A.; Santarelli, R.; Cirone, M. IRE1 Alpha/XBP1 Axis Sustains Primary Effusion Lymphoma Cell Survival by Promoting Cytokine Release and STAT3 Activation. Biomedicines 2021, 9, 118. [Google Scholar] [CrossRef]
- Bednarska, K.; Gunawardana, J.; Vari, F.; Cui, Q.; Thillaiyampalam, G.; Stehbens, S.; Mujaj, S.; Nourse, J.; Cristino, A.; Gandhi, M.K. The IRE1-XBP1s Pathway Impairment Underpins NK Cell Dysfunction in Hodgkin Lymphoma, That Is Partly Restored by PD-1 Blockade. Blood 2019, 134, 2795. [Google Scholar] [CrossRef]
- Cross, B.C.S.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The Molecular Basis for Selective Inhibition of Unconventional MRNA Splicing by an IRE1-Binding Small Molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambella, M.; Rocci, A.; Passera, R.; Gay, F.; Omede, P.; Crippa, C.; Corradini, P.; Romano, A.; Rossi, D.; Ladetto, M.; et al. High XBP1 Expression Is a Marker of Better Outcome in Multiple Myeloma Patients Treated with Bortezomib. Haematologica 2014, 99, e14–e16. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, Y.; Morita, S.; Hosoi, H.; Kobata, H.; Kishimoto, S.; Ishibashi, T.; Mishima, H.; Kinoshita, A.; Backes, B.J.; Yoshiura, K.-I.; et al. Targeting Adaptive IRE1α Signaling and PLK2 in Multiple Myeloma: Possible Anti-Tumor Mechanisms of KIRA8 and Nilotinib. Int. J. Mol. Sci. 2020, 21, 6314. [Google Scholar] [CrossRef] [PubMed]
- Harnoss, J.M.; Le Thomas, A.; Shemorry, A.; Marsters, S.A.; Lawrence, D.A.; Lu, M.; Chen, Y.C.A.; Qing, J.; Totpal, K.; Kan, D.; et al. Disruption of IRE1α through Its Kinase Domain Attenuates Multiple Myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 16420–16429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Zou, J.; Zhang, H.; Fu, W.; Zeng, T.; Huang, H.; Zhou, F.; Hou, J. Unfolded Protein Response Inducers Tunicamycin and Dithiothreitol Promote Myeloma Cell Differentiation Mediated by XBP-1. Clin. Exp. Med. 2015, 15, 85–96. [Google Scholar] [CrossRef]
- Raimondi, L.; De Luca, A.; Fontana, S.; Amodio, N.; Costa, V.; Carina, V.; Bellavia, D.; Raimondo, S.; Siragusa, S.; Monteleone, F.; et al. Multiple Myeloma-Derived Extracellular Vesicles Induce Osteoclastogenesis through the Activation of the XBP1/IRE1α Axis. Cancers 2020, 12, 2167. [Google Scholar] [CrossRef]
- Borjan, B.; Kern, J.; Steiner, N.; Gunsilius, E.; Wolf, D.; Untergasser, G. Spliced XBP1 Levels Determine Sensitivity of Multiple Myeloma Cells to Proteasome Inhibitor Bortezomib Independent of the Unfolded Protein Response Mediator GRP78. Front. Oncol. 2020, 9, 1530. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.W.; Chen, W.; Culpepper, J.; Jiang, H.; Ball, L.E.; Mehrotra, S.; Blumental-Perry, A.; Tew, K.D.; Townsend, D.M. Altered Redox Regulation and S-Glutathionylation of BiP Contribute to Bortezomib Resistance in Multiple Myeloma. Free Radic. Biol. Med. 2020, 160, 755–767. [Google Scholar] [CrossRef]
- Xu, X.; Liu, J.; Huang, B.; Chen, M.; Yuan, S.; Li, X.; Li, J. Reduced Response of IRE1α/Xbp-1 Signaling Pathway to Bortezomib Contributes to Drug Resistance in Multiple Myeloma Cells. Tumori J. 2017, 103, 261–267. [Google Scholar] [CrossRef]
- Bright, M.D.; Itzhak, D.N.; Wardell, C.P.; Morgan, G.J.; Davies, F.E. Cleavage of BLOC1S1 MRNA by IRE1 Is Sequence Specific, Temporally Separate from XBP1 Splicing, and Dispensable for Cell Viability under Acute Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2015, 35, 2186–2202. [Google Scholar] [CrossRef] [Green Version]
- Vincenz, L.; Jäger, R.; O’Dwyer, M.; Samali, A. Endoplasmic Reticulum Stress and the Unfolded Protein Response: Targeting the Achilles Heel of Multiple Myeloma. Mol. Cancer Ther. 2013, 12, 831–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Kaufman, R.J. The Impact of the Endoplasmic Reticulum Protein-Folding Environment on Cancer Development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Siwecka, N.; Rozpędek-Kamińska, W.; Wawrzynkiewicz, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. The Structure, Activation and Signaling of Ire1 and Its Role in Determining Cell Fate. Biomedicines 2021, 9, 156. [Google Scholar] [CrossRef]
- Ali, M.M.U.; Bagratuni, T.; Davenport, E.L.; Nowak, P.R.; Silva-Santisteban, M.C.; Hardcastle, A.; McAndrews, C.; Rowlands, M.G.; Morgan, G.J.; Aherne, W.; et al. Structure of the Ire1 Autophosphorylation Complex and Implications for the Unfolded Protein Response. EMBO J. 2011, 30, 894–905. [Google Scholar] [CrossRef] [Green Version]
- Harrington, P.E.; Biswas, K.; Malwitz, D.; Tasker, A.S.; Mohr, C.; Andrews, K.L.; Dellamaggiore, K.; Kendall, R.; Beckmann, H.; Jaeckel, P.; et al. Unfolded Protein Response in Cancer: IRE1α Inhibition by Selective Kinase Ligands Does Not Impair Tumor Cell Viability. ACS Med. Chem. Lett. 2015, 6, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Ri, M.; Tashiro, E.; Oikawa, D.; Shinjo, S.; Tokuda, M.; Yokouchi, Y.; Narita, T.; Masaki, A.; Ito, A.; Ding, J.; et al. Identification of Toyocamycin, an Agent Cytotoxic for Multiple Myeloma Cells, as a Potent Inhibitor of ER Stress-Induced XBP1 MRNA Splicing. Blood Cancer J. 2012, 2, e79. [Google Scholar] [CrossRef]
- Vieri, M.; Preisinger, C.; Schemionek, M.; Salimi, A.; Patterson, J.B.; Samali, A.; Brümmendorf, T.H.; Appelmann, I.; Kharabi Masouleh, B. Targeting of BCR-ABL1 and IRE1α Induces Synthetic Lethality in Philadelphia-Positive Acute Lymphoblastic Leukemia. Carcinogenesis 2021, 42, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Vieri, M.; Preisinger, C.; Schemionek, M.; Salimi, A.; Patterson, J.B.; Samali, A.; Brümmendorf, T.H.; Appelmann, I.; Kharabi Masouleh, B. Synergistic Dual Inhibition of BCR-ABL1 and the Unfolded Protein Response Causes P38 MAPK-Mediated Cell Death and Sensitizes BCR-ABL1+ Acute Lymphoblastic Leukemia to Dexamethasone. Blood 2018, 132, 4674. [Google Scholar] [CrossRef]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 Splicing by Inhibition of IRE1α Is a Promising Therapeutic Option in Multiple Myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef]
- Jiang, D.; Tam, A.B.; Alagappan, M.; Hay, M.P.; Gupta, A.; Kozak, M.M.; Solow-Cordero, D.E.; Lum, P.Y.; Denko, N.C.; Giaccia, A.J.; et al. Acridine Derivatives as Inhibitors of the IRE1α-XBP1 Pathway Are Cytotoxic to Human Multiple Myeloma. Mol. Cancer Ther. 2016, 15, 2055–2065. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Denko, N.C.; Olson, M.; Van Melckebeke, H.; Lust, S.; Tam, A.; Solow-Cordero, D.E.; Bouley, D.M.; Offner, F.; Niwa, M.; et al. Identification of an Ire1alpha Endonuclease Specific Inhibitor with Cytotoxic Activity against Human Multiple Myeloma. Blood 2011, 117, 1311–1314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the Unfolded Protein Response in Disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Suh, D.H.; Kim, M.K.; Kim, H.S.; Chung, H.H.; Song, Y.S. Unfolded Protein Response to Autophagy as a Promising Druggable Target for Anticancer Therapy. Ann. N. Y. Acad. Sci. 2012, 1271, 20–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C.; et al. Potent and Selective Inhibitors of the Inositol-Requiring Enzyme 1 Endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [Google Scholar] [CrossRef] [Green Version]
- Korennykh, A.V.; Egea, P.F.; Korostelev, A.A.; Finer-Moore, J.; Zhang, C.; Shokat, K.M.; Stroud, R.M.; Walter, P. The Unfolded Protein Response Signals through High-Order Assembly of Ire1. Nature 2009, 457, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Sanches, M.; Duffy, N.M.; Talukdar, M.; Thevakumaran, N.; Chiovitti, D.; Canny, M.D.; Lee, K.; Kurinov, I.; Uehling, D.; Al-Awar, R.; et al. Structure and Mechanism of Action of the Hydroxy-Aryl-Aldehyde Class of IRE1 Endoribonuclease Inhibitors. Nat. Commun. 2014, 5, 4202. [Google Scholar] [CrossRef] [Green Version]
- Kriss, C.L.; Pinilla-Ibarz, J.A.; Mailloux, A.W.; Powers, J.J.; Tang, C.H.A.; Kang, C.W.; Zanesi, N.; Epling-Burnette, P.K.; Sotomayor, E.M.; Croce, C.M.; et al. Overexpression of TCL1 Activates the Endoplasmic Reticulum Stress Response: A Novel Mechanism of Leukemic Progression in Mice. Blood 2012, 120, 1027–1038. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome Inhibitors Disrupt the Unfolded Protein Response in Myeloma Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef] [Green Version]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Lee, K.P.; Boise, L.H. Proteasome Inhibitors Induce a Terminal Unfolded Protein Response in Multiple Myeloma Cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef] [Green Version]
- Gugliotta, G.; Sudo, M.; Cao, Q.; Lin, D.C.; Sun, H.; Takao, S.; Le Moigne, R.; Rolfe, M.; Gery, S.; Müschen, M.; et al. Valosin-Containing Protein/P97 as a Novel Therapeutic Target in Acute Lymphoblastic Leukemia. Neoplasia 2017, 19, 750–761. [Google Scholar] [CrossRef]
- Nguyen, T.K.; Grant, S. Dinaciclib (SCH727965) Inhibits the Unfolded Protein Response through a CDK1- And 5-Dependent Mechanism. Mol. Cancer Ther. 2014, 13, 662–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moia, R.; Diop, F.; Favini, C.; Kodipad, A.A.; Gaidano, G. Potential of BCL2 as a Target for Chronic Lymphocytic Leukemia Treatment. Expert Rev. Hematol. 2018, 11, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Patriarca, A.; Gaidano, G. Investigational Drugs for the Treatment of Diffuse Large B-Cell Lymphoma. Expert Opin. Investig. Drugs 2021, 30, 25–38. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Disease Name | The Role of XBP1 |
|---|---|
| Chronic myelogenous leukemia (CML) | XBP1 promotes the survival of hematopoietic stem cells (HSCs) under ER stress [63]. |
| Chronic lymphocytic leukemia (CLL) | Myc-overexpression-activated XBP1 sustains cell proliferation and viability [64]. |
| XBP1s supports cell growth and increases IgM production and BCR signaling [65]. | |
| Acute myeloid leukemia (AML) | XBP1s regulates AML cell survival [13,69] and expansion [69]. |
| Activation of XBP1 is associated with a more favorable course of the disease [67]. | |
| XBP1 induction in the AML niche contributes to adaptive changes in stromal cells of the bone marrow [68]. | |
| Mast cell leukemia (MCL) | Splicing of XBP1 is crucial for cell proliferation and survival [70]. |
| Pre-B acute lymphoblastic leukemia (ALL) | XBP1 is highly expressed in patients, induces cancer survival and proliferation, and is associated with poor outcomes [14]. |
| Diffuse large B-cell lymphoma (DLBCL) | Activated XBP1s correlates with poorer clinical outcome and shorter overall survival [72,75] and is associated with more invasive phenotypes [75]. |
| Activated B-cell (ABC) DLBCL | Lower XBP1 levels induce resistance to ibrutinib [76]. |
| Germinal center B-cell–like (GCB) DLBCL | Downregulation of XBP1 is pro-survival and supports tumor growth/XBP1s activity and negatively impacts tumor growth [74]. |
| Burkitt’s lymphoma (BL) | XBP1 splicing is enhanced in Myc-overexpressing cells and has a protective role [64]. |
| Overexpression of XBP1s is lethal to BL cells [77]. | |
| Primary effusion lymphoma (PEL) | Basal activation of XBP1 is essential for PEL cell survival, the release of cytokines, and autophagy regulation [79]. |
| Reduced basal splicing of XBP1 makes cells susceptible to ER-stress-induced apoptosis [78]. | |
| Multiple myeloma (MM) | XBP1s is highly expressed and has pro-survival effects on MM cells [83]; it is essential for MM growth, chemoresistance [84], differentiation, and maturation [85]. |
| XBP1s is a key regulator of osteoblast differentiation induced by proteasome inhibitors [16]. | |
| Splicing of XBP1 is involved in MM-cell-derived small extracellular vesicle (EV)-induced osteoclast differentiation [86]. | |
| High levels of XBP1 correlate with a better response to bortezomib [82]. | |
| Low levels of XBP1s induce resistance to bortezomib [87]. | |
| Change in XBP1 expression determines the effectiveness of bortezomib treatment [89]. |
| Name of the Inhibitor | Mechanism of Action | Study Model | First Scientific Evidence |
|---|---|---|---|
| Sunitinib | Type I kinase inhibitor | MM (H929 and U266 cells) [94] | [105] |
| KIRA8 | Type II kinase inhibitor | MM and B-cell lymphoma cell lines [84] | [95] |
| 4μ8C | RNase inhibitor | MM (MM1.R cells) [81] | [81] |
| Toyocamycin | RNase inhibitor | MM (cell lines, patient samples, mouse xenografts) [96], AML (patient samples) [13] | [96] |
| MKC-8866 | RNase inhibitor | Ph+ ALL (SUP-B15 and TOM-1 cells, genetic mouse model) [97,98] | [106] |
| MKC-3946 | RNase inhibitor | AML (patient samples) [13], MM (MM.1S and MM.1R cells) [99] | [99] |
| 3,6-DMAD | Unknown | MM (RPMI 8226 and MM1.R cells and xenografts) [100] | [100] |
| STF-083010 | RNase inhibitor | AML (patient samples) [13], pre-B ALL and Ph+ ALL (genetic and patient-derived xenografts) [14], MM (cell lines, xenografts) [101] | [101] |
| A106/HNA | RNase inhibitor | AML (patient samples) [13], pre-B ALL and Ph+ ALL (genetic and patient-derived xenografts) [14] | [107] |
| B-I09 | RNase inhibitor | BL (human and mouse cells), CLL (human [64] and mouse cells [65] | [65] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiese, W.; Siwecka, N.; Wawrzynkiewicz, A.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. IRE1α Inhibitors as a Promising Therapeutic Strategy in Blood Malignancies. Cancers 2022, 14, 2526. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102526
Wiese W, Siwecka N, Wawrzynkiewicz A, Rozpędek-Kamińska W, Kucharska E, Majsterek I. IRE1α Inhibitors as a Promising Therapeutic Strategy in Blood Malignancies. Cancers. 2022; 14(10):2526. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102526
Chicago/Turabian StyleWiese, Wojciech, Natalia Siwecka, Adam Wawrzynkiewicz, Wioletta Rozpędek-Kamińska, Ewa Kucharska, and Ireneusz Majsterek. 2022. "IRE1α Inhibitors as a Promising Therapeutic Strategy in Blood Malignancies" Cancers 14, no. 10: 2526. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14102526