1. Introduction
Despite the utilization of more aggressive systemic chemotherapy regimens, pancreas ductal adenocarcinoma (PDAC) remains a devastating disease and is the third leading cause of cancer related mortality [
1]. In 2022, pancreas cancer is expected to affect approximately 62,210 patients in the US and the incidence will continue to rise. Surgery remains the only option for cure but, unfortunately, only 15–20% of patients are candidates for resection and five-year overall survival remains less than 20% with surgery alone. Apart from surgery and chemotherapy, few effective treatment options exist. Antimetabolites, such as Gemcitabine (GEM) and 5-fluorouracil (5-FU) as well as microtubule depolymerizing agents, such as Nab-paclitaxel (Abraxane), which is a nanoparticle form of albumin-bound paclitaxel are among the most commonly used chemotherapeutic agents [
2]. Abraxane depletes tumor stroma, through interaction between albumin and secreted proteins that are acidic and rich in cysteine [
3]. However, the survival of patients with PDAC barely reaches one year. Immunotherapy, which has shown dramatic results in many gastrointestinal and cutaneous malignancies, thus far has shown minimal benefit in PDAC. Given these hurdles, novel treatment strategies are desperately needed.
Ion channels are emerging oncological targets, as altered expression and/or function of these druggable proteins [
1] is strictly linked to classical cancer hallmarks [
4]. Voltage-dependent K
+ channels (Kv) are a superfamily of ubiquitously expressed membrane proteins that are involved in maintaining membrane resting/action potentials, cell proliferation, immune activation, and cell death [
5]. Kv1.3 is a specific voltage-dependent K
+ channel located mainly in the plasma and inner mitochondrial membranes (mitoKv1.3). First discovered in the plasma membrane of human T lymphocytes, Kv1.3 is also found in tumor and immune cells where it regulates proliferation as well as apoptosis and is aberrantly expressed in malignancies [
6,
7,
8]. Our group has recently developed two specific mitoKv1.3 inhibitors that prevalently and specifically target the mitochondrial channel by virtue of a positively charged triphenylphosphonium group. These inhibitors (
PCARBTP and
PAPTP) were shown to selectively kill cancer cells but not normal healthy cells through a reactive oxygen species (ROS)-mediated cell death involving the respiratory chain complex I [
9,
10,
11].
Data in examining Kv1.3 in pancreas cancer are limited and the expression of Kv1.3 in PDAC specimens from cancer patients is unknown. Therefore, in this study we evaluated tissue expression of Kv1.3 in resected human PDAC and found that Kv1.3 is highly expressed. We assessed tumor growth inhibition using the novel inhibitors of mitoKv1.3 alone or in conjunction with cytotoxic chemotherapy in an immune-competent preclinical orthotopic mouse model and provide evidence that the combination therapy drastically reduces tumor size without significant side effects.
2. Materials and Methods
2.1. Cell Viability Assays
For the cell viability (MTT) assay, Pan02 cells (National Cancer Institute- Frederick Cancer Research and Development Center, Frederick, MD, USA) were seeded in 96-well plates at 0.005–0.01 × 106 cells/well and grown in DMEM + 10% FBS (100 µL/well) for 24 h. In the dark, the growth medium was replaced with a medium containing the desired compound (from a mother solution in DMSO) to the final concentration as shown in the figures. The final DMSO concentration was 0.1% or lower in all cases (including controls). After incubation for 48 h or 24 h for ALDH3A1 knockdown, a CyQUANT™ MTT Cell Viability Assay kit (Fisher, Hampton, NH, USA) was used to detect formazan formation. Formazan formation is a redox-dependent process and can be a confounding factor with ROS build up. MTT reagent was diluted at 1:10 with a culture medium and a 100 µL-well was added into the 96-well plate and cultured for 4 h. 100 µL/well DMSO was added to solve the formazan. Absorbance was measured at 570 nm to detect formazan formation using a Cytation5 plate reader (BioTek, Winooski, VT, USA).
2.2. Measurement of Mitochondrial Membrane Potential and ROS Release
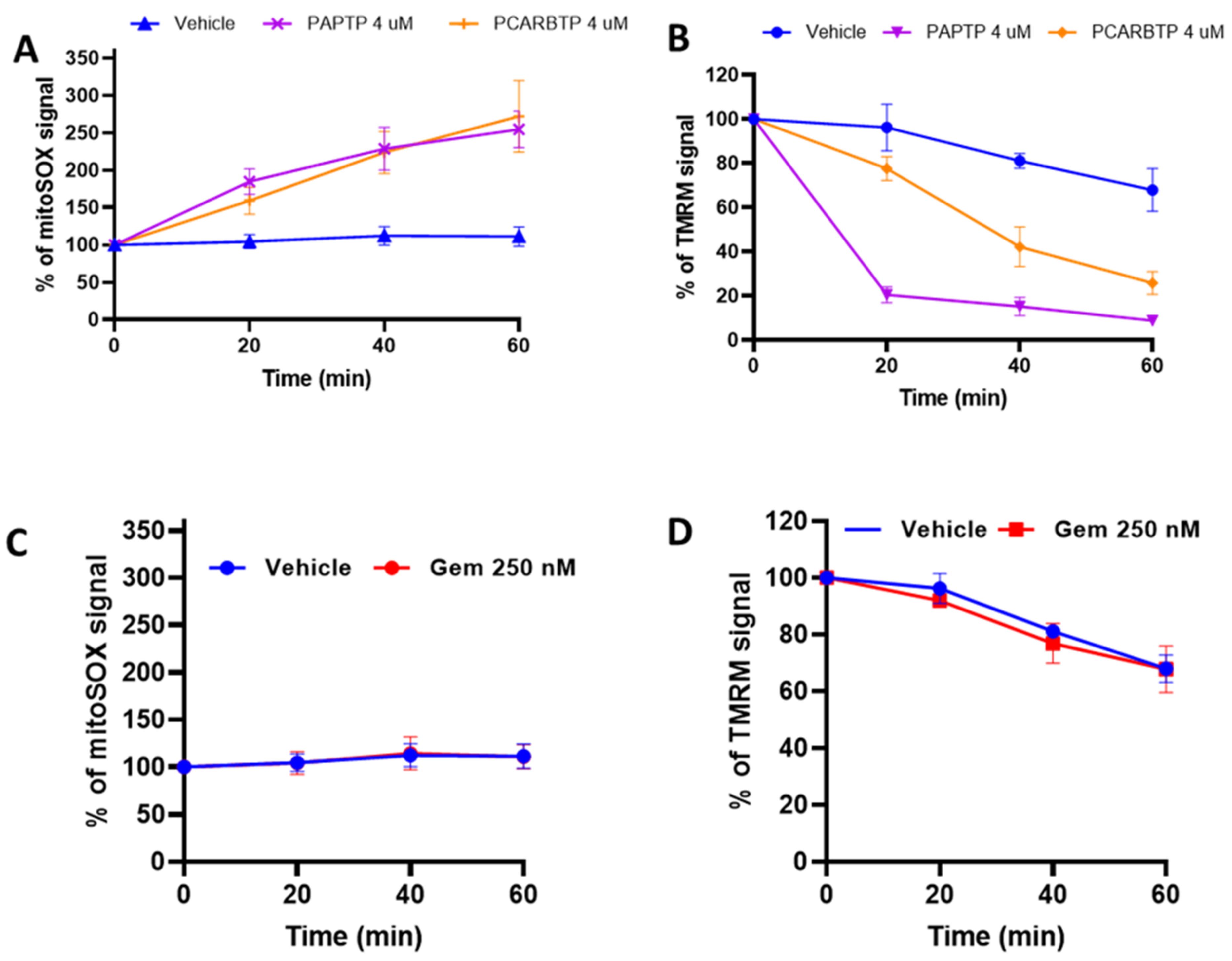
To measure mitochondrial membrane potential and ROS levels, Pan02 cells were incubated with 1 µM MitoSOX or 20 nM TMRM in HBSS (Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C for 20 min. After incubation, the compounds were added and the increase in MitoSOX fluorescence or the decrease in TMRM fluorescence was measured by flow cytometry. Median values of the fluorescence intensity distributions (5000 cells were counted) are presented in the data.
2.3. In Vitro Development of PCARBTP Resistant Clones
Pan02 cells were detached from tissue culture flasks before reaching confluence by removing the culture medium, adding trypsin-EDTA and incubating for 3 min at 37 °C and 5% CO2. After this incubation period, fresh medium was added, and the cells were spun at 200 g for 5 min. Supernatant was removed and fresh medium added. A cell count was carried out with the standard Trypan blue exclusion method. Pan02 cells were seeded at 1 cell per well in 200 µL of fresh medium in 96 flat-bottomed well plates. Clones were inspected regularly so those wells with more than 1 clone could be discarded. The addition of PCARBTP was carried out by replacing the medium containing PCARBTP at the different doses. If the cells survived at that dose for more than 3 days, we increased the dose, and obtained the 4 clones of Pan02 cells that survived under 10 µM PCARBTP in the medium. Resistant cells were amplified under 10 µM of PCARBTP, and then the proteins were collected for proteomic analysis.
2.4. Proteomic Analysis of Resistant Clones
The Pierce 660 nm Protein assay was performed on a 1:10 dilution of the samples to determine the protein concentration using BSA as a standard. Sufficient protein was present such that 50 µg was taken out to run on a short 1D gel for digestion. A total of 50 µg of each sample (non-resistant and resistant clones) in 40 µL of Laemmli buffer were run 1.5 cm into a 1D 1.5 mm 4–12% BT gel using MOPS running buffer. Pre-stained protein markers were used in surrounding lanes. The regions between the markers and the dye front were excised for trypsin digestion following the standard in gel protocol. The resulting peptides were extracted, dried, and prepared for mass spectrometry. A total of 2.5 µg of each sample was run on the nanoLC-MS/MS in DDA mode and the combined DDA runs were searched using Protein Pilot (SCIEX, AB Sciex Pte. Ltd., Framingham, MA, USA) to create the protein spectral library. A total of 720 proteins were identified with 99% confidence with an FDR of less than 1% at the peptide and protein levels. A matched SWATH-MS method in DIA mode of the samples was used to collect quantitative data for each of the samples for the comparative profiling. Three clones of PCARBTP resistant cells and three replicates of normal Pan02 were processed. SWATH-D data analysis workflow was used to validate the data set and detected significant quantitative changes.
2.5. Stable Downregulation of ALDH by Lentiviral Transduction
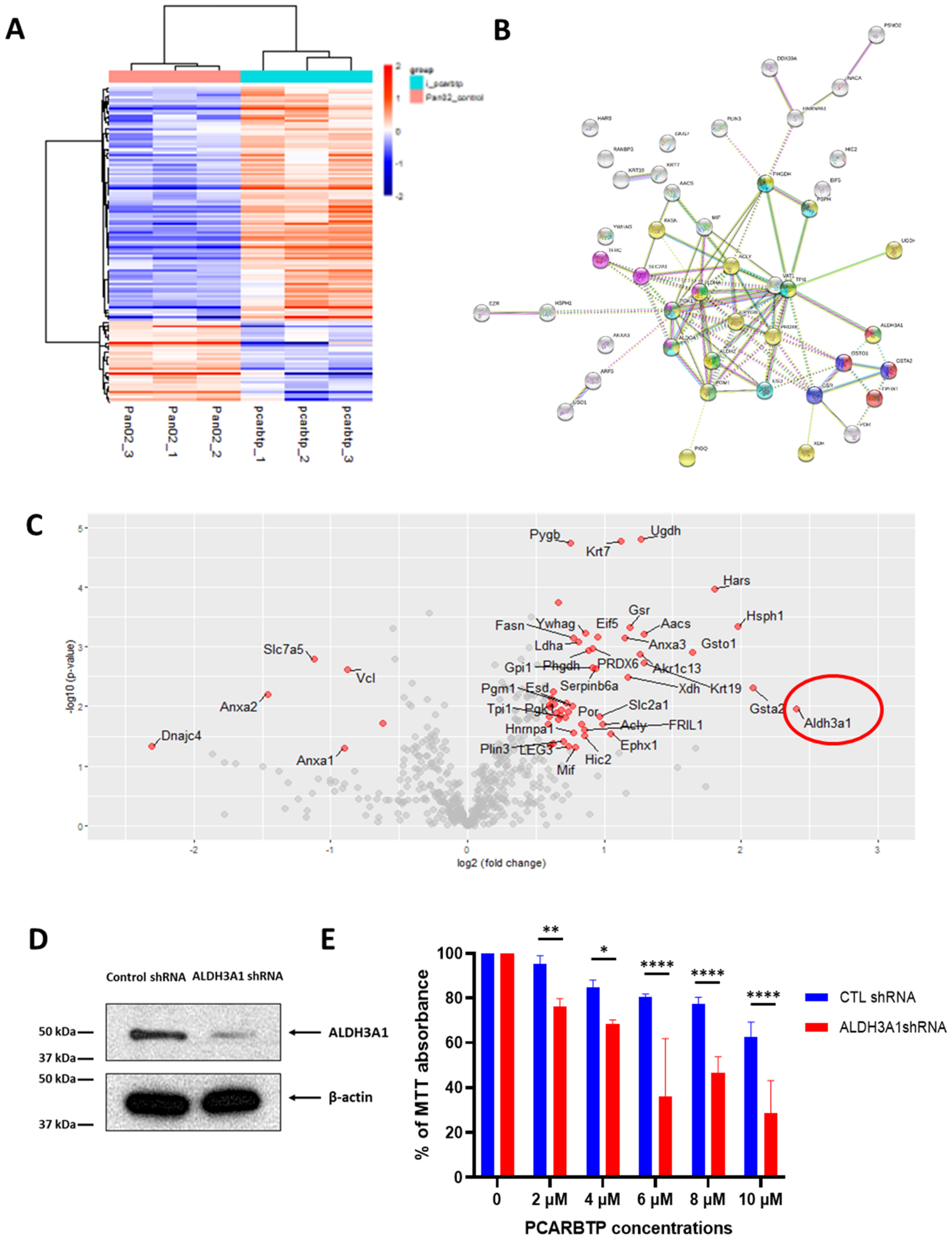
Pan02 cells were grown in a 12-well plate 24 h prior to viral infection. Cells were infected at approximately 50% confluency. To this end, the medium was replaced with medium containing 5 µg/mL of Polybrene® (sc-134220, Santa Cruz, CA, USA) and the cells were infected with 10 µL of ALDH3A1 shRNA (m) lentiviral particles (sc-72033-V, Santa Cruz, CA, USA) in the culture. The plate was swirled gently to mix and incubated overnight. The medium was replaced by a complete medium and cultured for an additional 48 h. Selection of the cells was started at day 5 with 10 µg/mL of puromycin dihydrochloride. The culture medium was replaced by fresh puromycin-containing medium every 3–4 days, until resistant colonies can be identified. Clones were identified, expanded and assayed. Three colonies were chosen, and Western blot analysis was performed to evaluate for ALDH3A1 knockdown (sc-376089, Santa Cruz, CA, USA).
2.6. Western Blot
Cells were lysed in whole-cell lysis buffer consisting of 50 mM of HEPES, 150 mM of NaCl, 1 mM of EGTA, 10 mM of sodium pyrophosphatate, 1.5 mM of MgCl
2, 100 mM of NaF, 10% glycerol and 1% Triton X-100, and pH 7.2, containing an inhibitor cocktail (1 mM of phenylmethylsulfonyl fluoride, 10 mg/mL of aprotinin and 1 mM of sodium orthovanadate) to extract total protein. Protein concentrations were determined using a standard bicinchoninic acid (BCA) assay (23225, Thermo Fisher Scientific, Waltham, MA, USA), and 50 µg of the total protein was subjected to 10% SDS-PAGE followed by electrotransfer onto nitrocellulose membranes. The membranes were blocked in 5% skimmed milk, and then incubated overnight at 4 °C with primary antibodies against human Kv1.3 (1:1000, P4497, Sigma, St. Louis, MO, USA), P-p38 MAPK (1:1000; CST, Framingham, MA, USA), ALDH3A1 (1:1000, sc-376089, Santa Cruz, CA, USA), and β-actin (1:2000; Abcam, Waltham, MA, USA). Membranes were then washed with TBST for 3 × 10 min. This was followed by incubation with horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature in 5% skimmed milk and washed with TBST for 3 × 10 min. Immunoreactive signals were detected using enhanced chemiluminescence (Pierce, Rockford, IL, USA). Three independent experiments were performed. Original western blots provided in
Supplementary Section.
2.7. Orthotopic Mouse Pancreatic Tumor Injection Model
All animal experiments were approved by the University of Cincinnati Ethic Committee and Institutional Animal Care and Use Committee. Eight-week-old, wild-type male, C57BL/6J mice were purchased from Jackson Labs (000664, Jackson Labs, Bar Harbor, ME, USA). Mice were anesthetized using 120 mg/kg of ketamine plus 20 mg/kg of xylazine. Orthotopic injection was performed as described by Tepal et al. [
12]. In detail, a left subcostal incision was made just below the rib cage and the pancreas was identified. The tumor cell suspension was created by mixing 25 µL of Matrigel with 25 µL of Pan02 cells containing 1 × 10
6 cells. Pan02 cells were cultured in DMEM + 10% FBS medium, under 37 °C and 5% CO
2, with no antibiotic added. The tumor suspension was slowly injected into the pancreas and the needle was left in place for 60 s to allow the Matrigel to set. After ensuring hemostasis, the abdomen was closed in 2 layers using 3-0 silk sutures.
2.8. In Vivo Kv1.3 Inhibitor and Cytotoxic Chemotherapy Administration
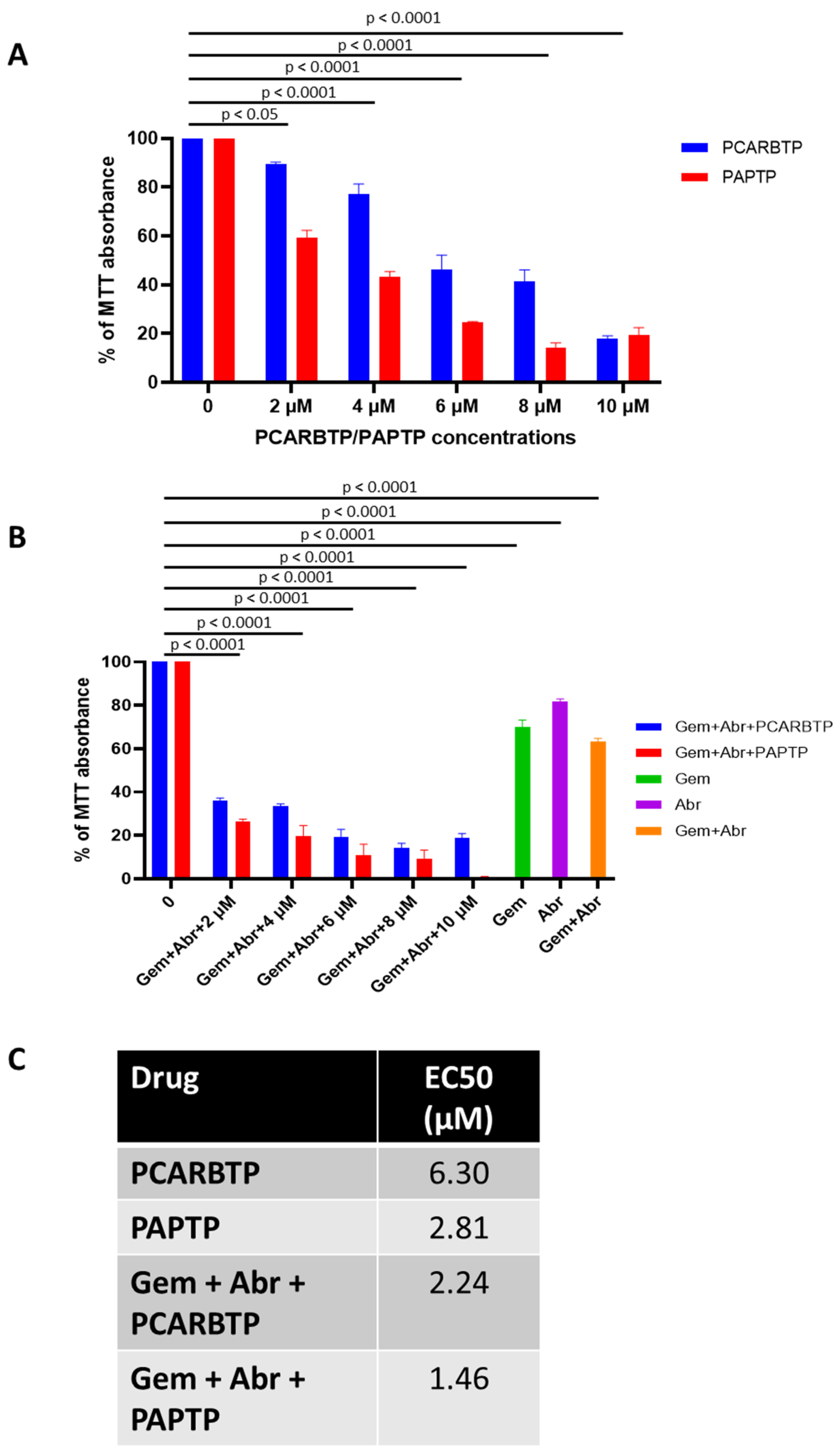
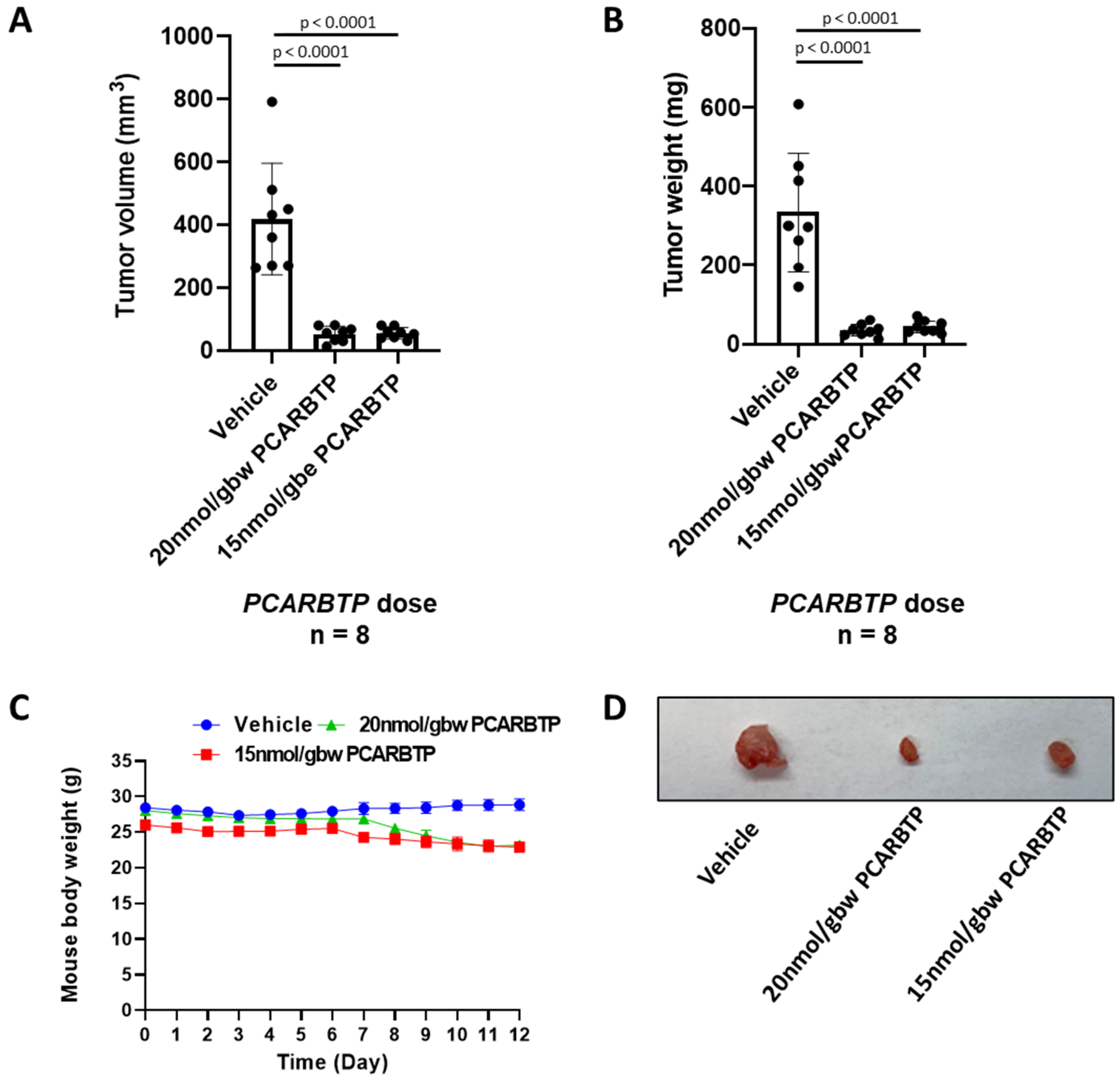
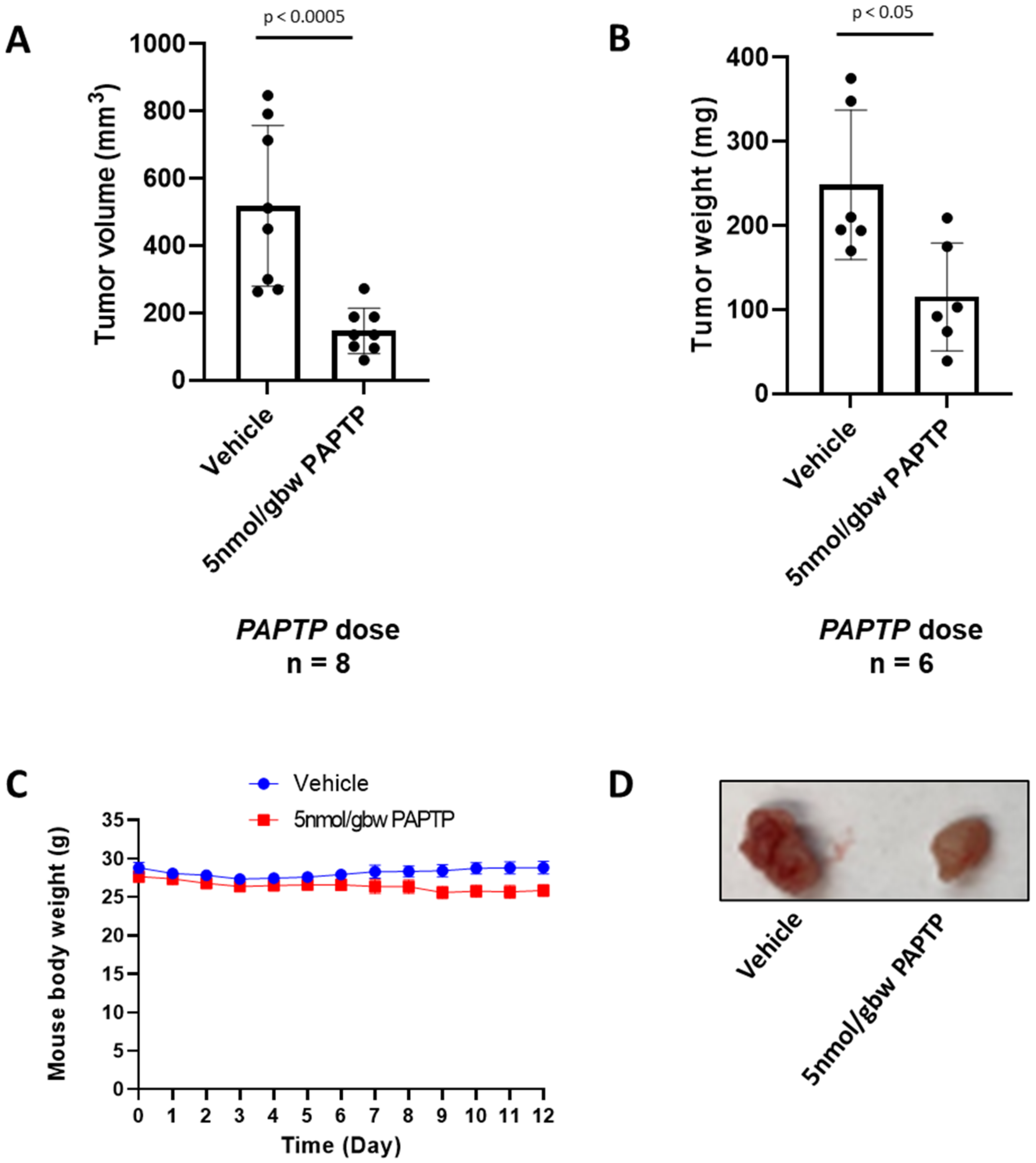
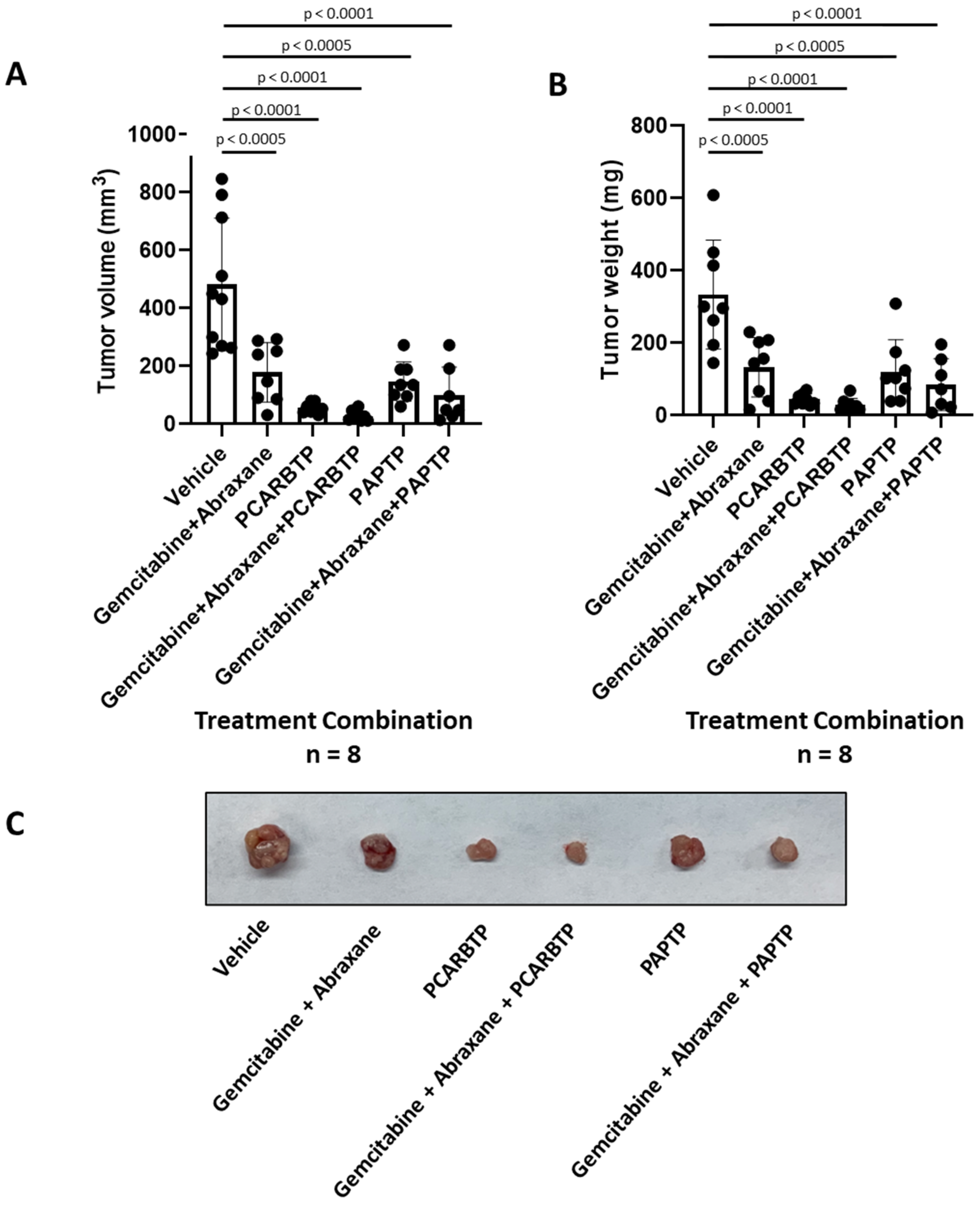
PCARBTP was suspended in 50% DMSO and injected into the peritoneal cavity at a dose of 15 nmol/gbw on day 6 after tumor injection. Similarly, PAPTP was administrated at a dose of 5 nmol/gbw. Gemcitabine was dissolved in ddH2O and injected into the intraperitoneal cavity at dose of 190 nmol/gbw 6 days after tumor injection. Abraxane was dissolved in DMSO and intraperitoneally injected at a dose of 23.4 nmol/gbw. Tumor volume was calculated using the formula: volume = length × width × depth.
2.9. Immunohistochemistry
Immunohistochemistry (IHC) staining was performed using a biotin-streptavidin-peroxidase (SP) kit (AB64269, Abcam, Waltham, MA, USA) and a diaminobenzidine kit (DAB) as previously described [
13]. Institutional Review Board approval was obtained to obtain human resected pancreas ductal adenocarcinoma specimens and associated clinicopathologic data (IRB 2019-0324). Tumor specimens were obtained from the University of Cincinnati Department of Pathology. Five-micrometer sections were deparaffinized and rehydrated in xylene and gradients of ethanol. Slides were boiled in citrate buffer (10 mM of sodium citrate, 10 mM of citric acid, pH 6.0) at 92–98 °C for 10 min to retrieve the antigen. The sections were then incubated with 3% H
2O
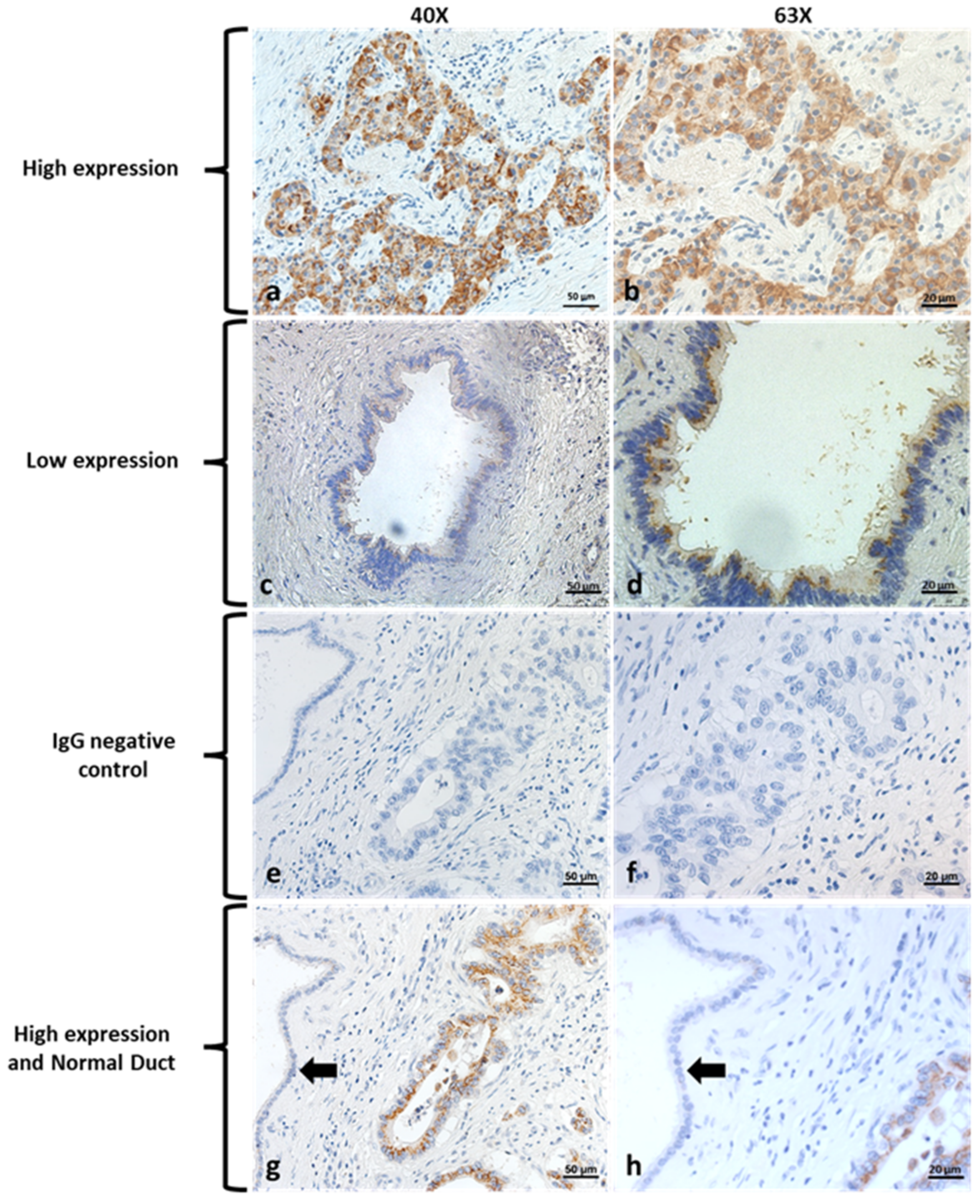
2 in methanol for 10 min to quench endogenous peroxidase and blocked with normal goat serum for 20 min. Sections were incubated with specific primary antibodies against Kv1.3 (1:200; P4497, Sigma, MO, USA) at 4 °C overnight. The sections were then incubated with biotinylated goat-anti-rabbit IgG secondary antibody and stained with DAB working reagent (per manufacturer’s instructions) for 30–60 s. They were then counterstained with hematoxylin. Finally, sections were mounted with Permount (SP15-500, Thermo Fisher Scientific, Waltham, MA, USA) onto slides. Negative control was performed using unconjugated rabbit IgG (011-000-003, Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Images were acquired with a ZEISS AXIO microscope (Carl ZEISS, Jena, Germany). Slides were scored by a gastrointestinal pathologist who specializes in evaluating pancreas cancer. He was blinded to all clinicopathologic data and determined the percent and intensity of staining (scored 0–3). A final score of high versus low expression was determined if patients had >70% of tumor cells staining with a score of 3 for intensity. The remaining group was classified as low.
2.10. Hematoxylin & Eosin Staining
Slides containing paraffin sections were passed using the following steps: 3 × 5 min in Xylene (blot excess xylene before going into ethanol), 2 × 5 min in 100% ethanol, 1 × 5 min in 95% ethanol, 1 × 5 min in 70% ethanol, 1 × 5 min deionized H2O, and 1 × 3 min Hematoxylin. They were then rinsed with deionized water 1 × 5 min, tap water and ethanol to destain. After subsequent rinse, they were treated for 1 × 30 sec with Eosin, for 3 × 5 min with 95% ethanol, for 3 × 5 min with 100% ethanol, for 3 × 5 min with Xylene, and a coverslip was placed using Permount mounting medium (SP15-500, Thermo Fisher Scientific, Waltham, MA, USA).
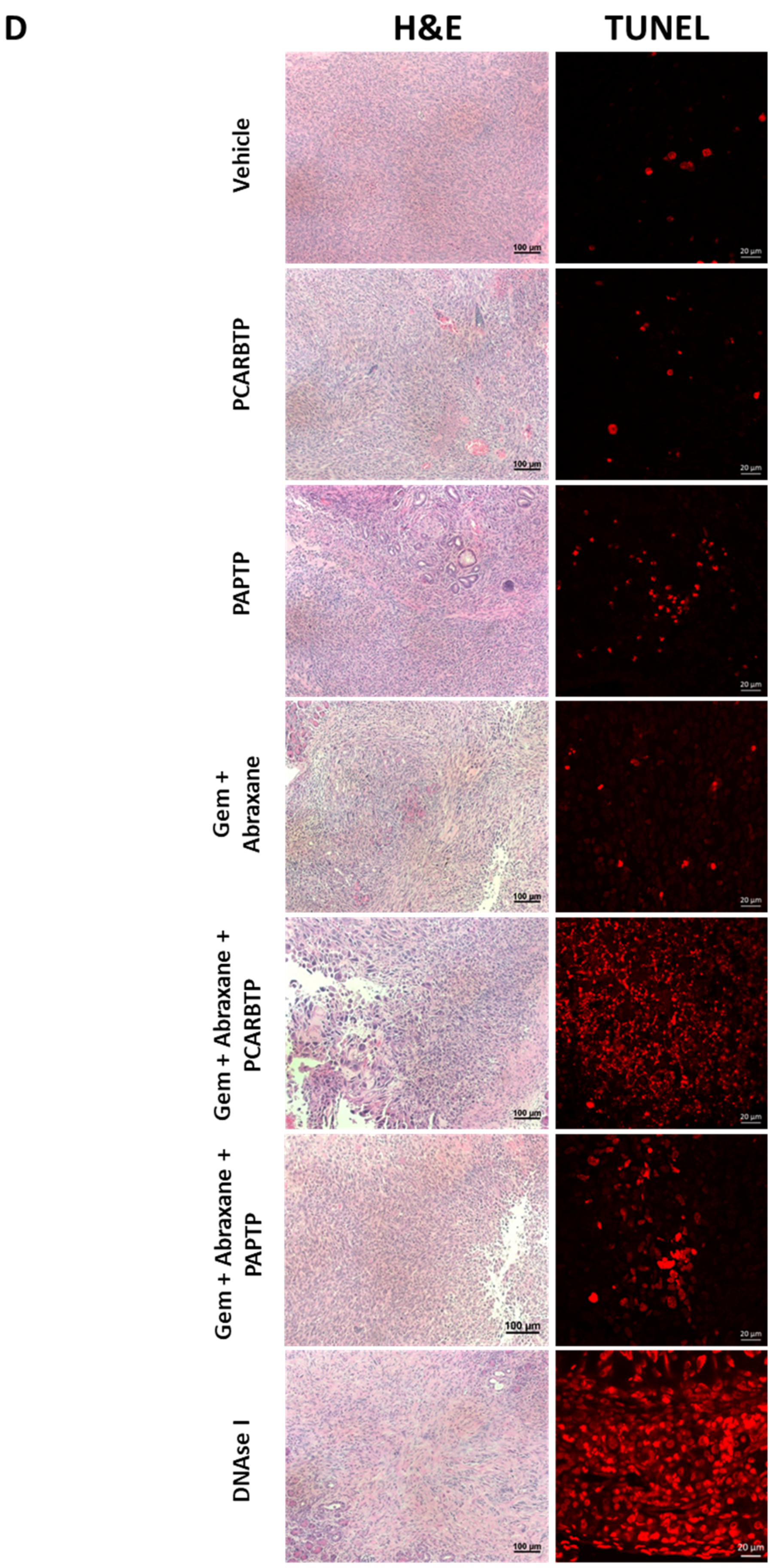
2.11. TUNEL Assay
Animals were sacrificed and immediately perfused via the right heart with 0.9% NaCl for 2 min followed by 10% formalin for 10 min. Organs, including the heart, lung, liver and kidney, were then removed and further fixed in 10% formalin for 48 h. TUNEL staining was performed with an In Situ Cell Death Detection Kit as instruction by the supplier. Briefly, the tissues were dehydrated, embedded in paraffin, and sectioned at a thickness of 5 µm. Sections were then dewaxed, rehydrated and incubated for 5 min in 0.1 M citrate buffer (pH 6.0) at 350 W in a microwave. After this, samples were immediately cooled in PBS and incubated with TMR coupled dUTP in the presence of terminal deoxynucleotidyl-transferase (Roche, Basel, Switzerland) at 37 °C for 60 min. Samples were embedded with mounting medium with DAPI prior to analysis. An excitation wavelength of 488 nm was used and evaluated using a ZEISS AXIO microscope (Carl ZEISS, Jena, Germany).
2.12. Statistical Analysis
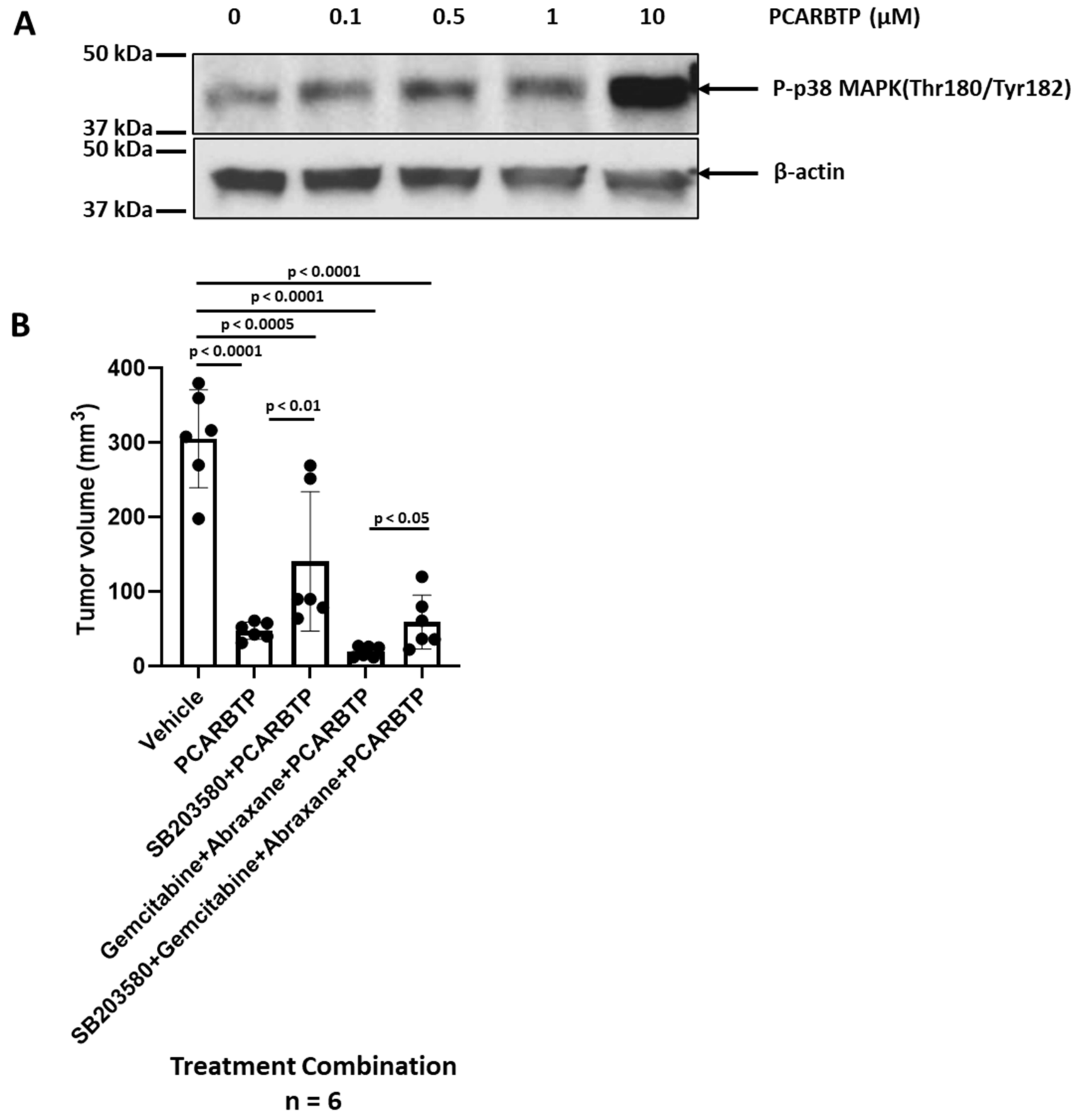
The tumor volume and mass and mouse body weight were analyzed by t-test and one-way ANOVA using GraphPad Prism 9.0, each experiment has more than 6 mice in 1 group, indicated in Results and Figure legends. p < 0.05 was considered as statistically significant. * p < 0.05; ** p < 0.01, *** p < 0.0005, **** p < 0.0001.
2.13. Immunofluoresence
Immunofluorescence staining was performed using Pan02 cells. These were seeded on a coverslip at 50% confluency 12 h before staining. Cells were washed with cold PBS × 3 then fixed by 4% paraformaldehyde for 30 min. Cells were again rinsed with cold PBS × 3, followed by 1% Triton-100 for 30 min. Cells were blocked with 5% donkey serum for 1 h at room temperature and washed with cold PBS for 10 min × 3. Primary antibodies Kv1.3 (1:100, APC-101, Alomone labs, Jerusalem, Israel) and TOM20 (1:100, MABT166, Sigma, Burlington, MA, USA) were incubated overnight at 4 °C. Cells were washed with cold PBS for 10 min × 3, secondary antibodies goat anti-mouse antibody Alexa 488 (1:2000, A11001, Thermo Fisher Scientific, Waltham, MA, USA) and goat anti-rabbit antibody Alexa Fluor™ 594 (1:2000, A11012, Thermo Fisher Scientific, Waltham, MA, USA) were incubated for 1 h at room temperature. Cells were washed with cold PBS for 10 min × 3 and mounting medium with DAPI was added. Carl Zeiss LSM 710 confocal laser-scanning microscope (Carl Zeiss, Jena, Germany) was used to capture the images.
4. Discussion
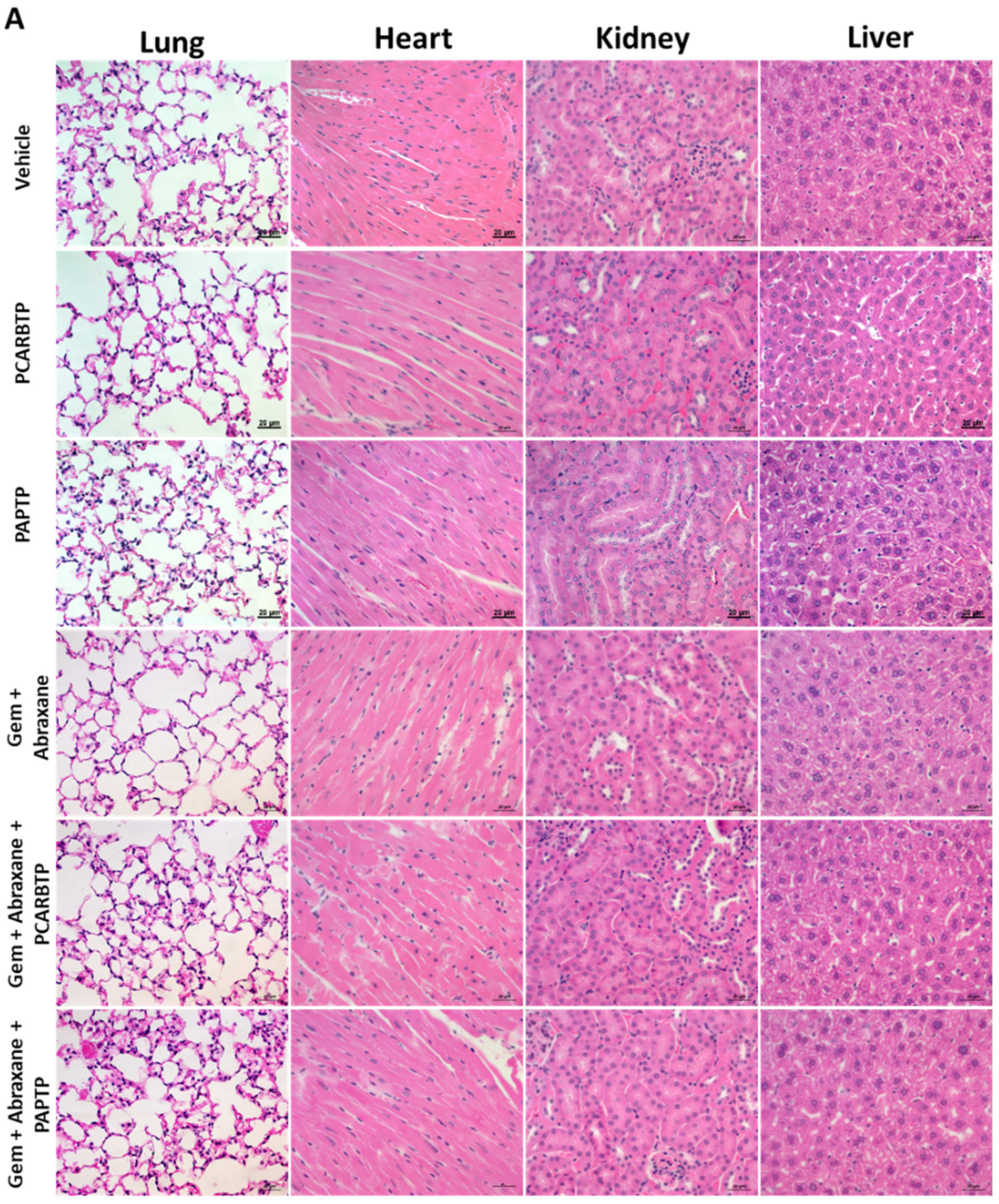
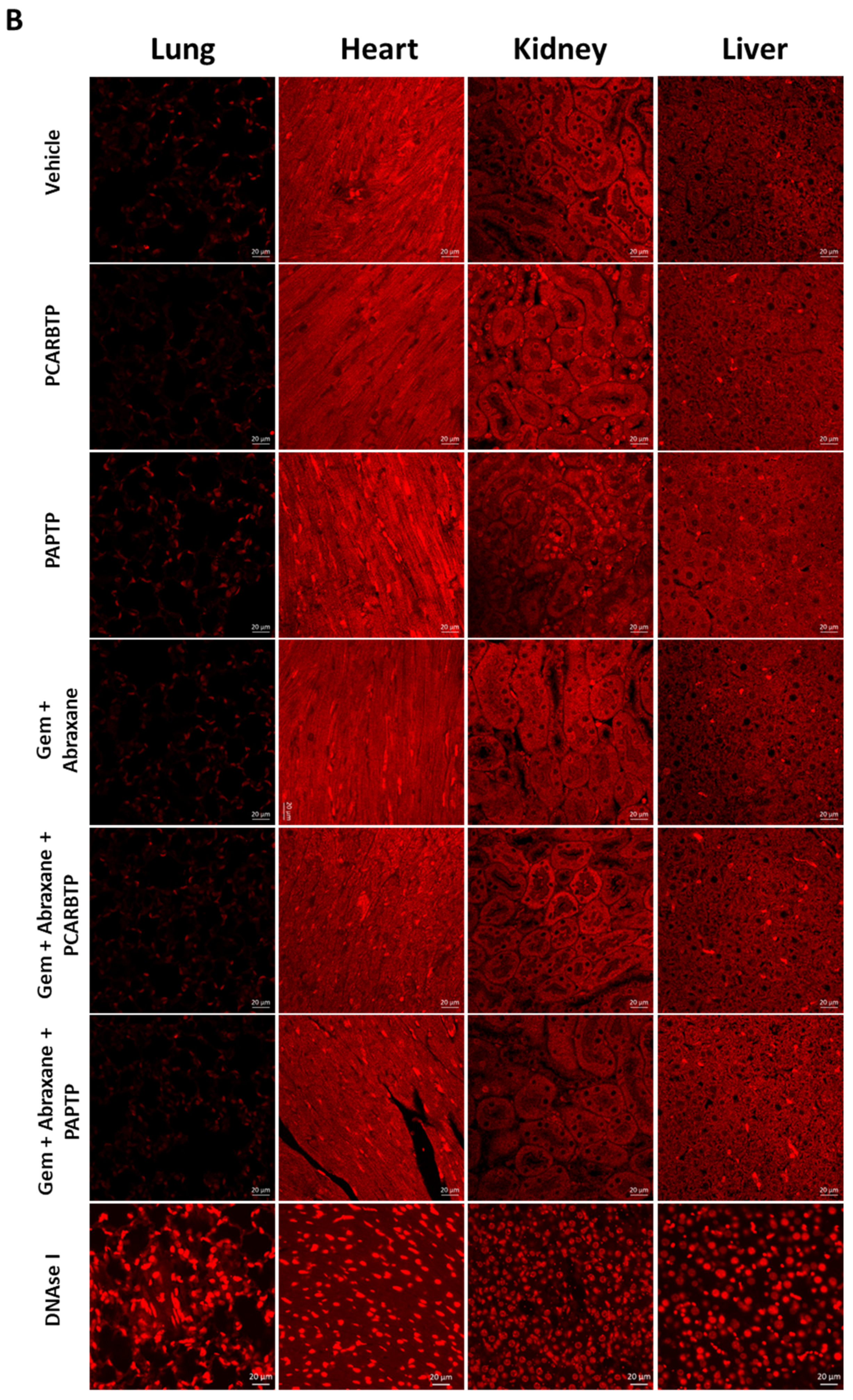
In the present study, we show that treatment with mitochondrial Kv1.3 inhibitors
PAPTP and
PCARBTP in a syngeneic orthotopic mouse model using mouse Pan02 cells resulted in a reduction in tumor size by almost 90% (please check results. The single treatment is not given there). When combined with cytotoxic chemotherapy Gemcitabine and Abraxane, tumor size reduction reached 95%, which is a reduction that has not been observed so far to our knowledge. Most importantly, this treatment strategy did not result in visible organ toxicity. We chose to use Gemcitabine and Abraxane as it is one of the common cytotoxic chemotherapy regimens used in the treatment of PDAC, has been used in the MPACT trial and is a two-drug regimen in comparison to FOLFIRINOX (5-fluorouracil, oxaliplatin, irinotecan), which involves three drugs and associated with different toxicity profile [
23,
24]. Although we employed Abraxane along with the other inhibitors, in our Pan02 model, the relevance of Abraxane may be lower than in human PDAC, given that in the present animal model stroma is less dense than in the human pathology [
15].
We found that Kv1.3 was highly expressed in a large number of resectable human pancreatic cancer specimens (55). This is consistent with previous data from us showing by Affymetrix as well as by Western blots that Kv1.3 is highly expressed in various, largely chemoresistant human PDAC cell lines harboring p53 mutations (PANC-1, AsPC-1, BxPC-3, Capan-1, Colo-357, MiaPaCa2) [
9]. The channel is present also in the mitochondrial fraction of these cells [
25]. A previous investigation by Bielanska et al. showed that Kv1.3 was under-expressed in human PDAC specimens, but these immunohistochemical studies were performed in a very limited sample (
n = 2) of patients [
26]. Another study examined 18 patients and found a correlation between Kv1.3 expression decrease and metastasis [
27]. Importantly, both studies were performed on tumor samples from patients with metastatic PDAC, which is biologically different from those with resectable, non-metastatic disease. Given that our study found that Kv1.3 is highly expressed in resected PDAC, it is possible that during the transition to a metastatic phenotype, pancreas cancer cells are able to down regulate Kv1.3, possibly due to methylation of the promoter region of the
Kcna3 gene encoding for Kv1.3, or another unexplained mechanism [
27]. The findings reported here are consistent with previous studies showing that K
+ channels can promote proliferation [
1], while downregulation of Kv1.3 renders cells resistant to apoptotic stimuli [
28]. In addition, overexpression of the channel in our patient samples suggests Kv1.3 is a novel therapeutic target to treat pancreas cancer, although the specific role of mitochondrial Kv1.3 in patients with PDAC has not been addressed here. All tumor specimens showed Kv1.3 expression, at least at a low level. Even in patient samples with low Kv1.3 expression, there are some cells that express Kv1.3 and are likely to respond to Kv1.3 inhibitors, although whether these specific cells correspond to cancer cells with specific characteristics or eventually to cancer stem cells remains to be determined.
When examining if Kv1.3 expression holds prognostic value, we found no association with overall or recurrence free survival. It is plausible that in this patient population with resectable PDAC, compared to metastatic PDAC, alterations at the DNA and/or protein level have not occurred to Kv1.3 to render the channel a prognostic biomarker. We also examined if there was an association of Kv1.3 expression with presence of nodal metastases, LVI, and PNI and found there was none. This is likely due to the high rates of PNI (91%), LVI (56%), and positive lymph nodes (84%) in these patients even with localized disease. The predictive value of mitochondrial Kv1.3 expression to
PAPTP and
PCARBTP treatment in mice was not tested in this study but we have previously shown a positive correlation between Kv1.3 expression and PCARBTP/PAPTP-induced death in human PDAC lines and between the expression of the channel in mitochondria and the plasma membrane [
9].
Mechanistically, the signaling pathway of
PCARBTP or
PAPTP-induced tumor cell death was associated with ROS production and the ability of these drugs to drive the cells above a critical ROS level, as indicated by the finding that N-acetyl-cysteine, a molecule able to boost the antioxidant system, prevented the in vivo tumor reducing effect of both drugs [
9]. Here, we show that additional mechanisms also come into play, namely an increase in the phosphorylation of p38/MAPK. When treated with the p38 inhibitor, SB203580, there was a reduction in
PCARBTP-mediated in vivo effect, suggesting that mitoKv1.3 inhibition triggers, at least in part, the p38-mediated death pathway. These findings are consistent with existing data that show activation of the p38/MAPK pathway to be a favorable prognostic marker and associated with improved overall survival in patients with PDAC [
29,
30,
31]. Interestingly, apoptosis induced by a ROS-producing agent (MC3 a Thioredoxin Reductase inhibitor), was also associated with mitochondrial dysfunction and activation of p38-MAPK in a PDAC line. ROS scavengers or inhibition of p38 signaling with SB203580 attenuated MC3-induced apoptosis [
32], indicating a relationship between redox balance, p38MAPK activation and cell death in PDAC cells. This connection is further indicated by the finding that p38-MAPK is activated also by GEM [
13]. In our case, ROS release is triggered at mitochondria by mitoKv1.3 inhibitors. Given that ROS production due to NOX activation upon GEM treatment [
21] has previously been reported, it is reasonable to propose that the synergistic effect of
PAPTP/PCARBTP and gemcitabine on cell death can be ascribed to an enhanced p38-MAPK activation and downstream signalling and high oxidative stress upon combined treatment.
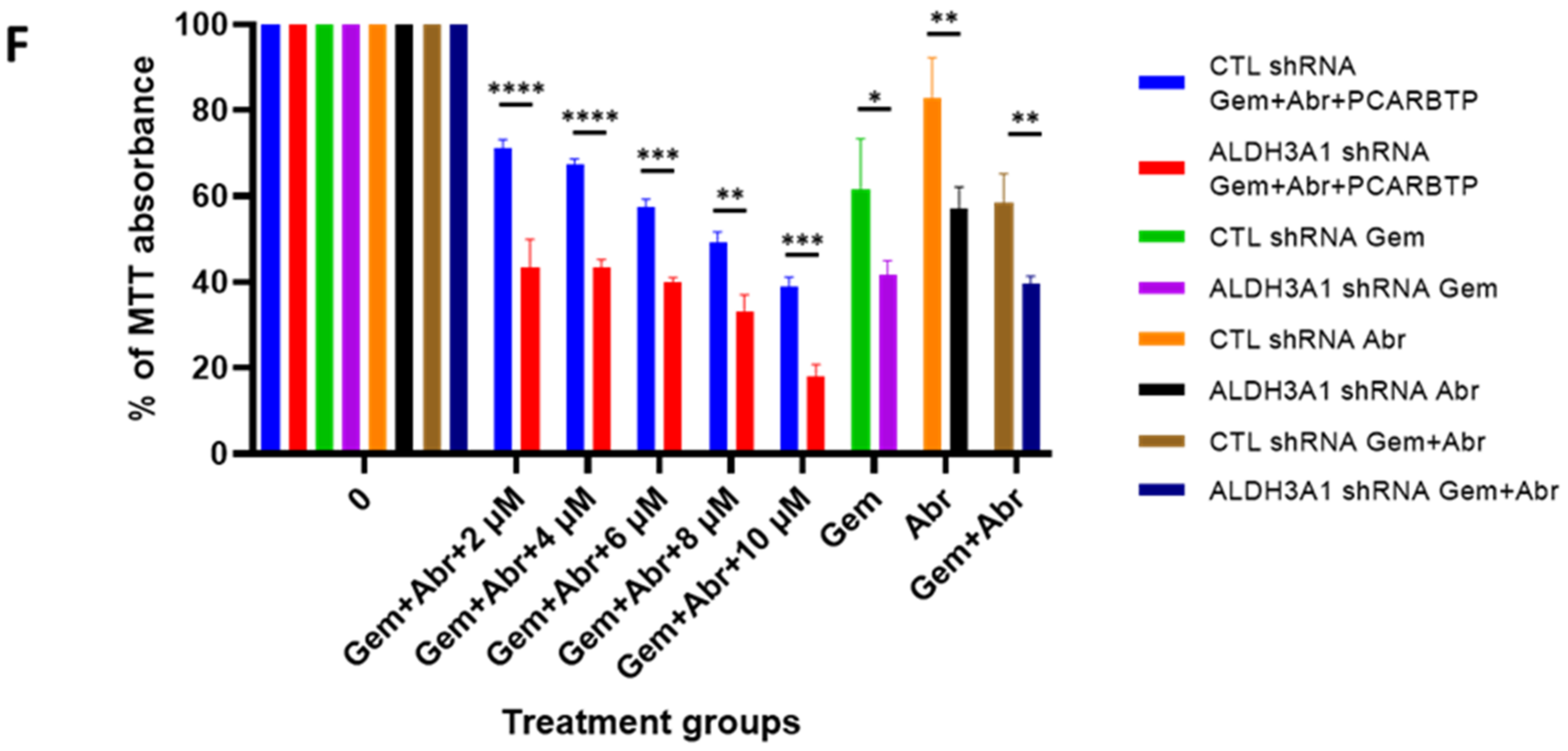
Despite an unprecedented 95% tumor growth inhibition using a combination of mitoKv1.3 inhibitors and cytotoxic chemotherapies, there were still viable tumor cells. After developing a
PCARBTP treatment resistant cell line, proteomic analysis revealed there was an almost a four-fold increase in the antioxidant system. Compared to normal human cells, cancer cells have increased ROS generation due to reduced ability to produce scavengers [
14,
33]. On the other hand, adaptive mechanisms enable cancer cells to escape from oxidative damage by means of overexpressing ROS scavengers. Inhibition of mitoKv1.3 can initiate a cascade of events that leads to transient hyperpolarization of the inner mitochondrial membrane, formation of ROS, stimulation of permeability transition pore, release of cytochrome c, and resulting apoptosis [
14,
28]. In addition, our recent findings indicate that PAP-1 derivatives bound to mitoKv1.3 are positioned in a way that their psoralenic moieties can directly accept electrons from complex I of the respiratory chain and donate these electrons to molecular oxygen, further boosting ROS [
11]. Given the importance of ROS generation to the mechanism of mitoKv1.3 inhibitor mediated cell death, it is not surprising to see that cancer cells resistant to
PCARBTP upregulated the antioxidant system (GSR, GSTO1, GSTA2, ALDH3A1). An intrinsic resistance to GEM has been linked to upregulation of antioxidant defense system also in AsPc-1 human pancreatic tumor cells and in related orthotopic xenograft mouse model [
21]. Indeed, GEM in combination with β-phenylethyl isothiocyanate (PEITC) is able to deplete the cells of glutathione and thus enhance oxidative stress, and it caused the tumor size to reduce to a higher extent than GEM alone. Nonetheless, a reduction of up to 60% occurred in vivo. Thus, even though the principle underlying the effect of GEM along with agents that induce oxidative stress is similar in the two studies (the one by Ju et al. and us), the present paper demonstrates that combination is more efficient when using a drug that is able to release ROS rapidly and to great extent by modulating mitochondrial function. Proteins linked to the cytochrome P450-mediated detoxification system are upregulated (EPHX1, GSTO1, GSTA2), as this system is mainly responsible for xenobiotic metabolism in the cells [
34]. In addition, an upregulation of proteins linked to metabolic processes such as glycolysis, carboxylic acid biosynthesis and amino acid synthesis can be observed. This result can be interpreted in light of the inhibition of the mitochondrial respiration and ATP production [
9].
In summary, our study indicates that a combination treatment using GEM and mitoKv1.3 inhibitors along with Abraxane represents a promising way to defeat PDAC and opens the possibility to identify further drug combinations that allow complete eradication of the tumor. Importantly, the fact that the pro-oxidant activity of mitoKv1.3 inhibitors is linked to the expression of Kv1.3, offers an important layer of selectivity towards killing of PDAC cells, given that only 8% of normal PDAC specimens showed expression of the channel to a detectable level. Future studies should focus on using mitoKv1.3 inhibitors to selectively kill cells with higher Kv1.3 expression, which may result in not only the direct killing of cancer cells but also enhance the activity of cytotoxic chemotherapies and potential immunotherapies.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











