
Efficacy of Oncolytic Herpes Simplex Virus T-VEC Combined with BET Inhibitors as an Innovative Therapy Approach for NUT Carcinoma

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Treatment with BET Inhibitors (iBETs)
2.3. Virus Infection
2.4. Sulforhodamine B Cell Viability Assay
2.5. Real-Time Cell Proliferation Assay
2.6. Virus Growth Curve
2.7. GM-CSF ELISA
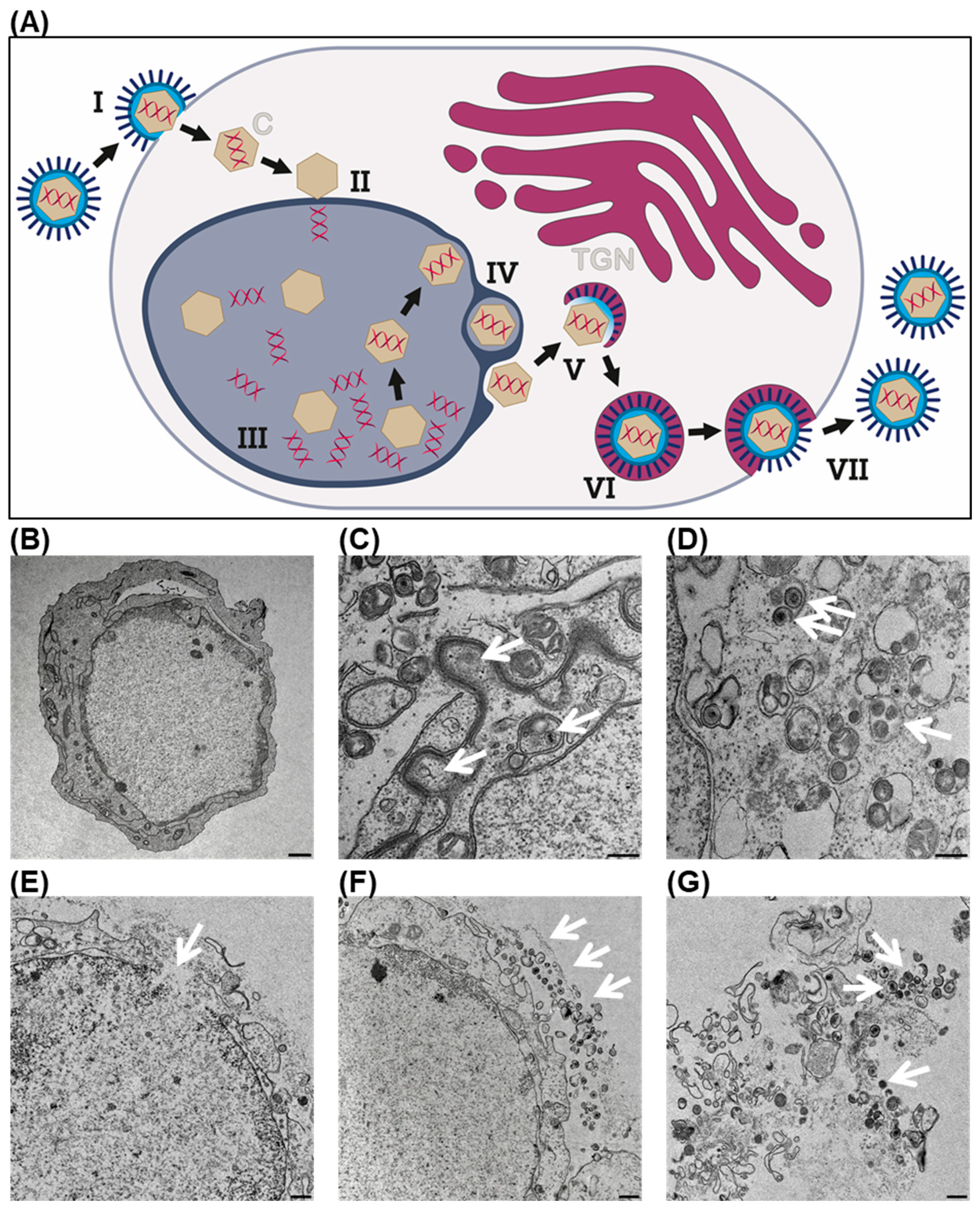
2.8. Transmission Electron Microscopy
2.9. Immunofluorescence Staining and Confocal Microscopy
2.10. Statistical Analysis
3. Results
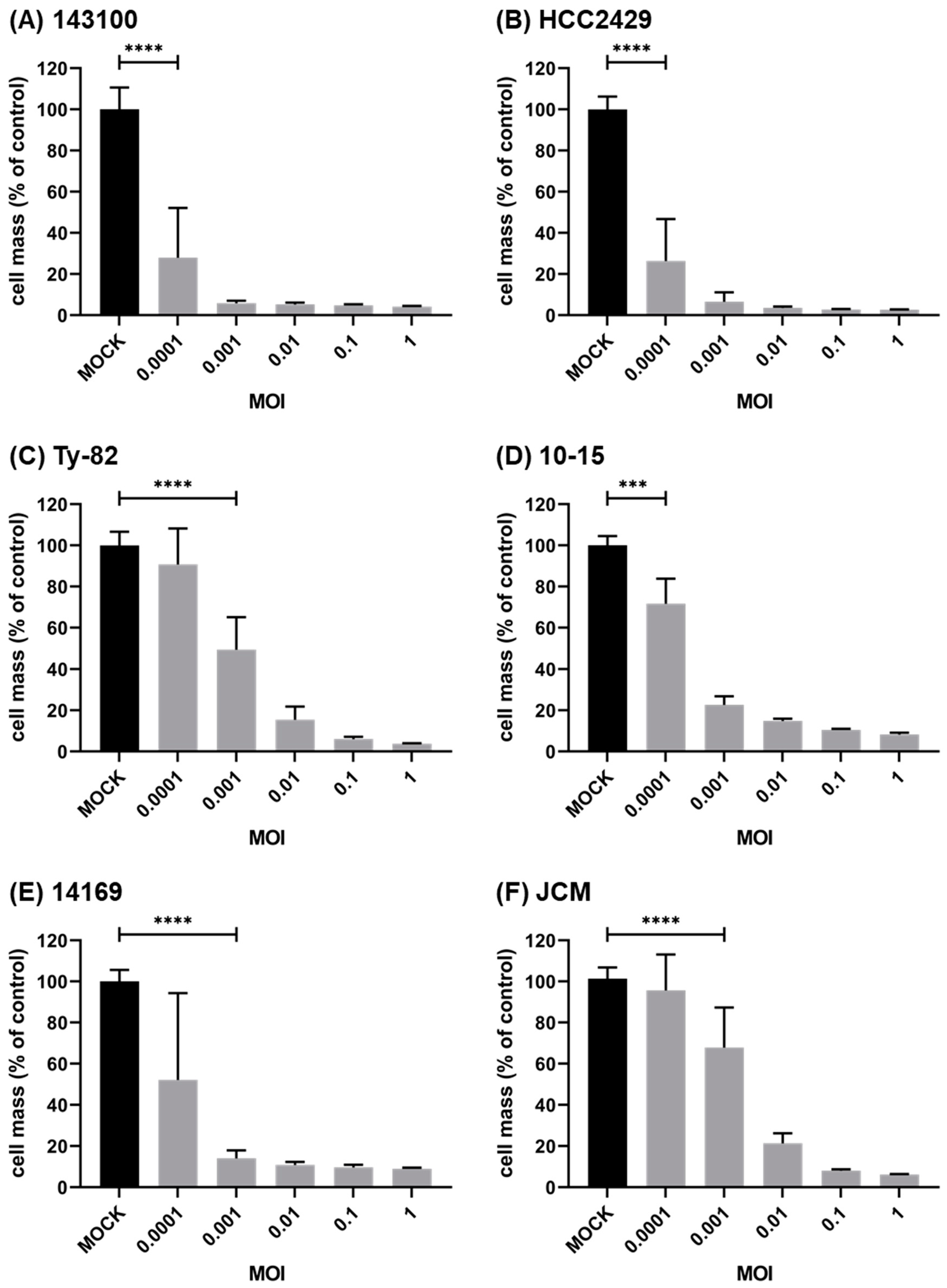
3.1. Oncolytic Virus T-VEC Efficiently Infects and Lyses NC Cells
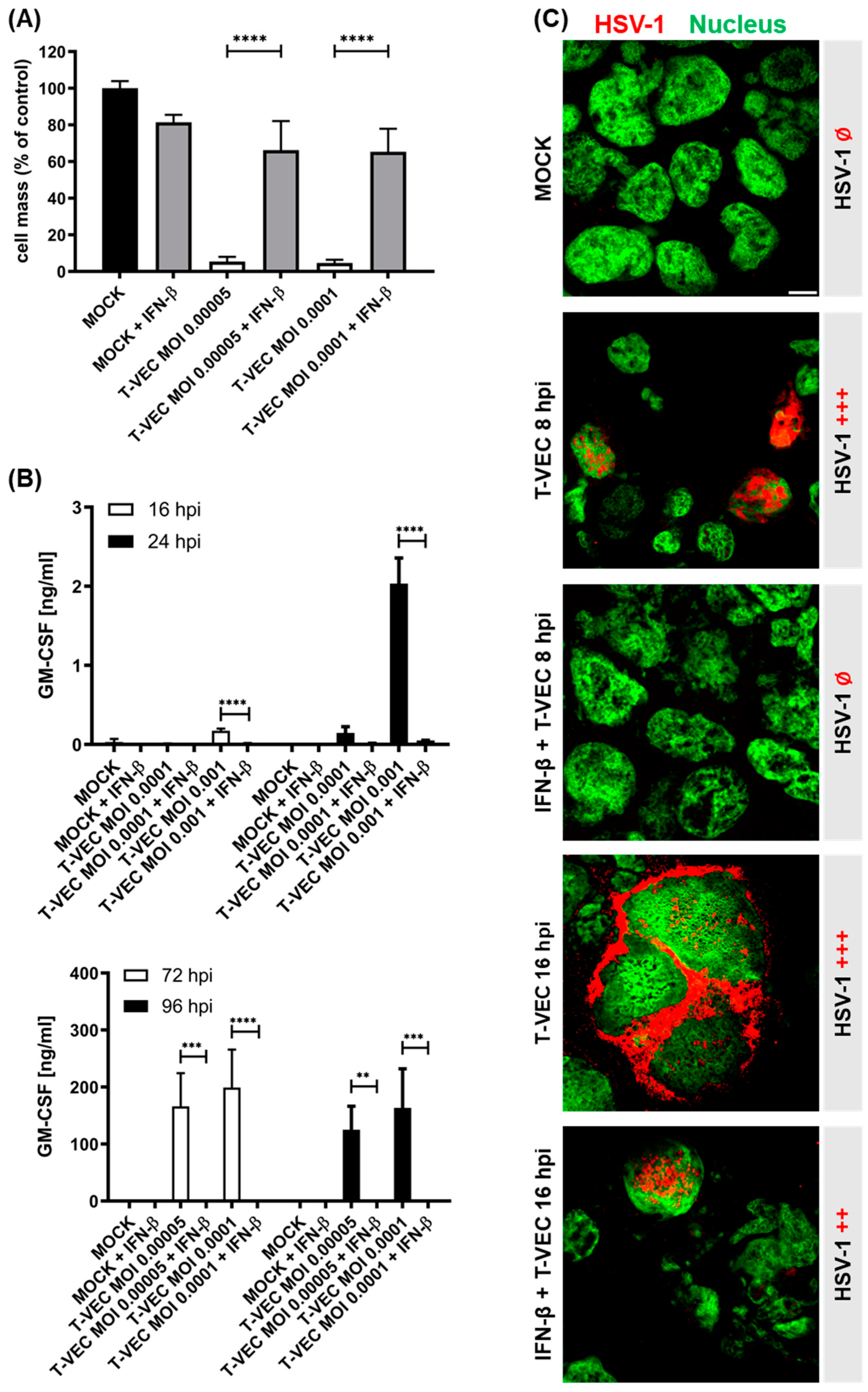
3.2. Pretreatment with IFN-β Heavily Impairs the Oncolytic Capacity of T-VEC in NC Cells
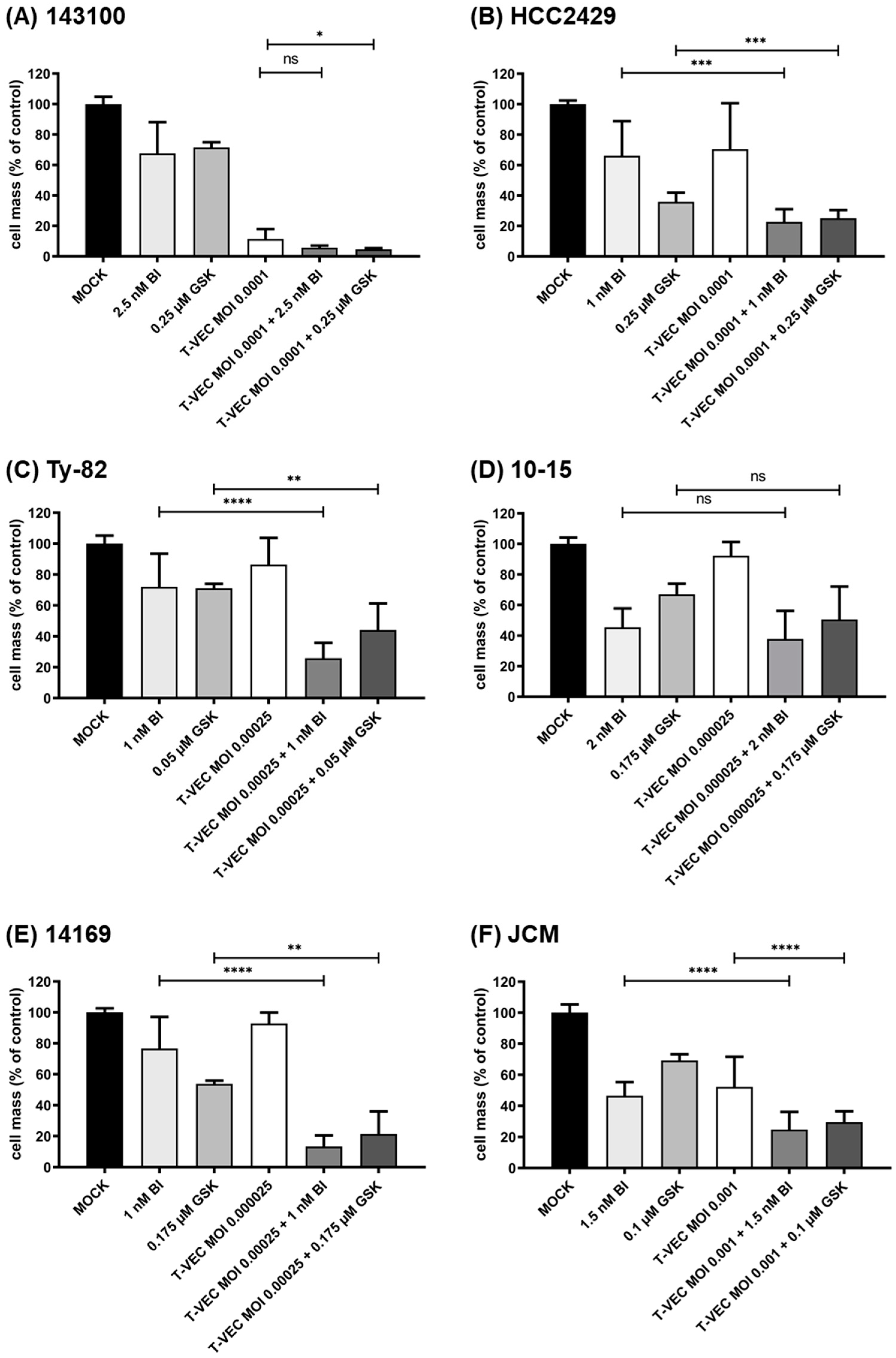
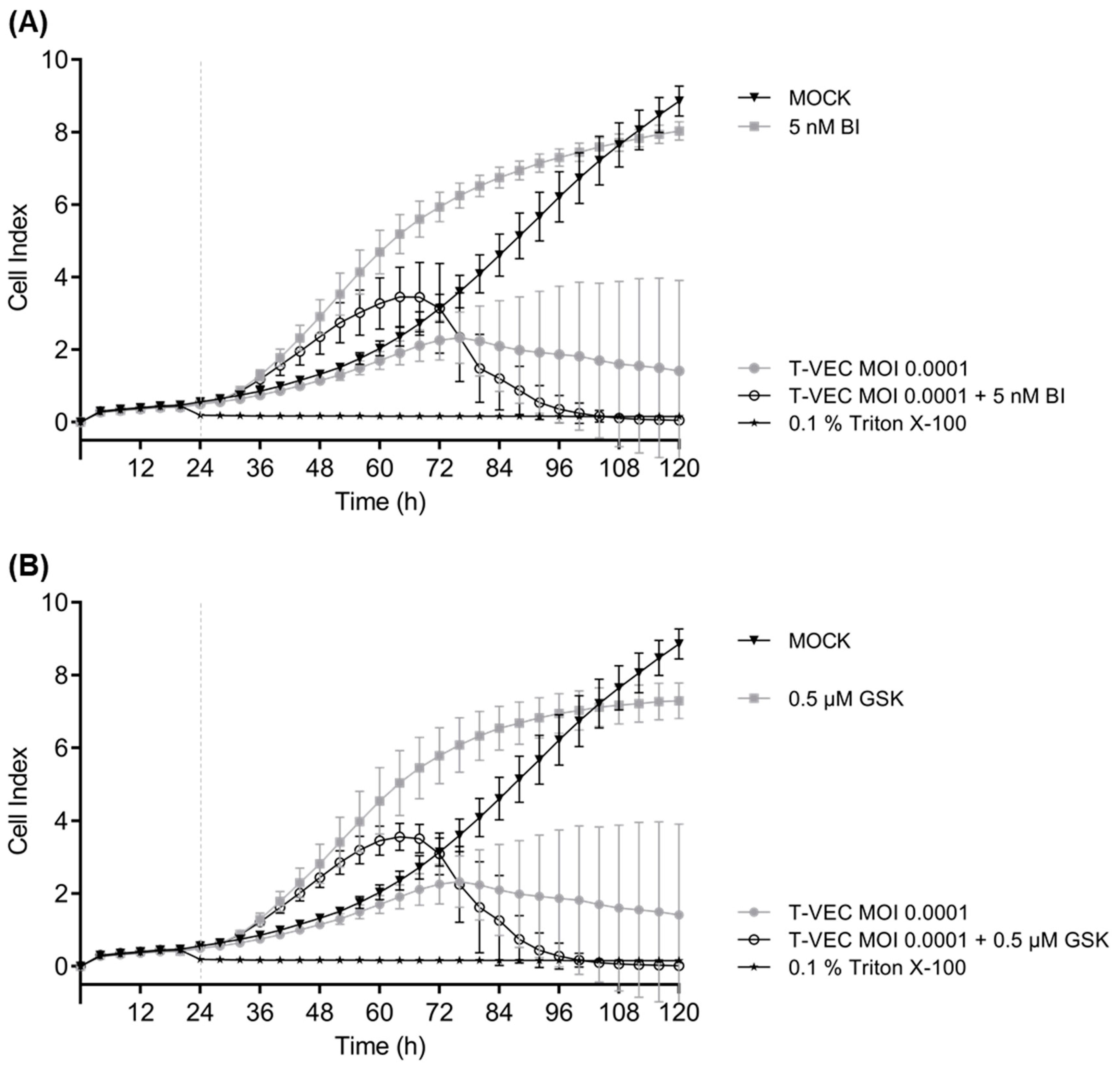
3.3. iBET Therapy in Addition to T-VEC Therapy Leads to an Enhanced Anti-Tumoral Effect
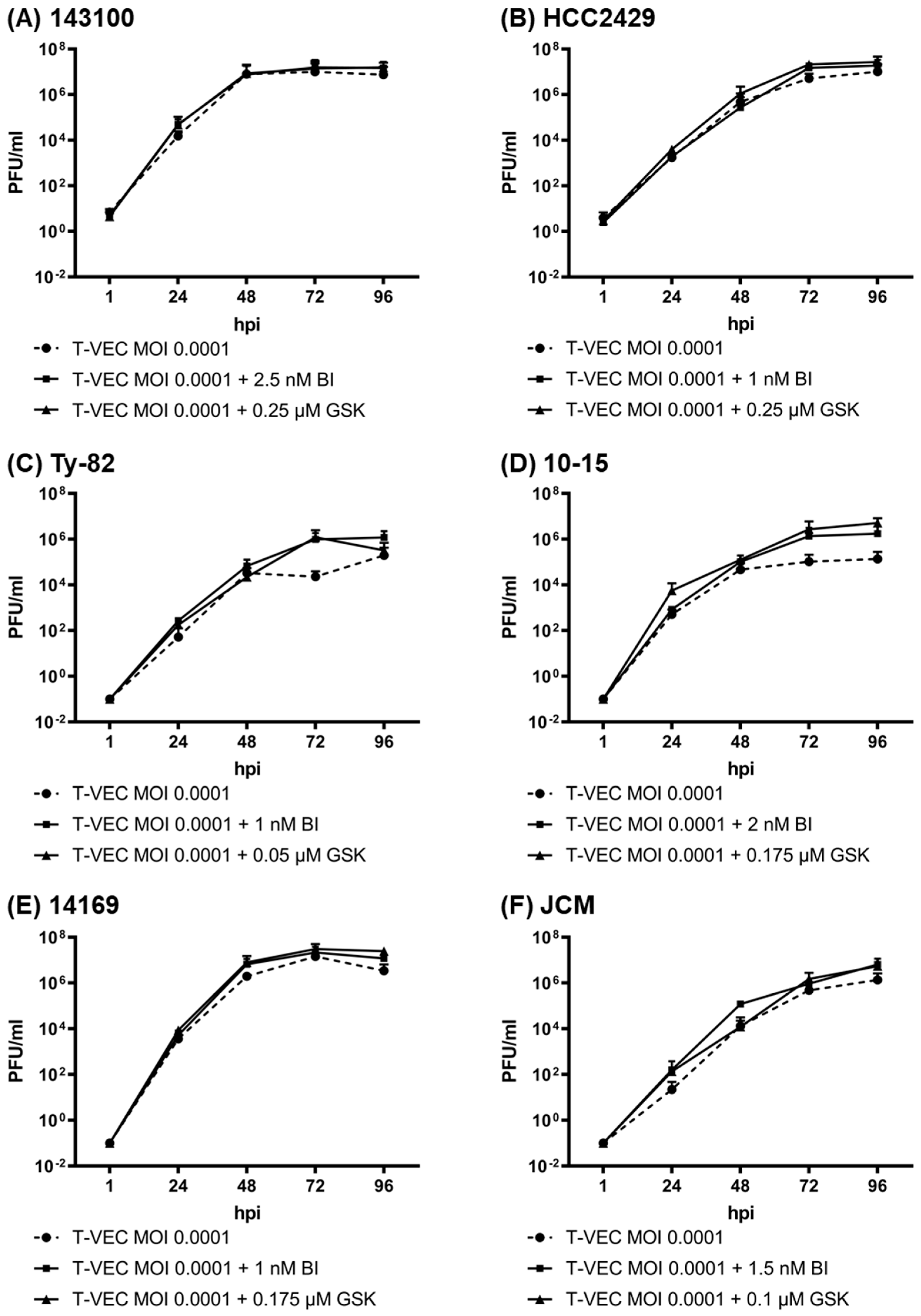
3.4. iBET Therapy Does Not Impair Viral Replication of T-VEC in NC Tumor Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- French, C.A. NUT Carcinoma: Clinicopathologic features, pathogenesis, and treatment. Pathol. Int. 2018, 68, 583–595. [Google Scholar] [CrossRef]
- French, C.A.; Ramirez, C.L.; Kolmakova, J.; Hickman, T.T.; Cameron, M.J.; Thyne, M.E.; Kutok, J.L.; Toretsky, J.A.; Tadavarthy, A.K.; Kees, U.R.; et al. BRD-NUT oncoproteins: A family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 2008, 27, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- French, C.A.; Rahman, S.; Walsh, E.M.; Kuhnle, S.; Grayson, A.R.; Lemieux, M.E.; Grunfeld, N.; Rubin, B.P.; Antonescu, C.R.; Zhang, S.; et al. NSD3-NUT fusion oncoprotein in NUT midline carcinoma: Implications for a novel oncogenic mechanism. Cancer Discov. 2014, 4, 928–941. [Google Scholar] [CrossRef] [Green Version]
- Alekseyenko, A.A.; Walsh, E.M.; Zee, B.M.; Pakozdi, T.; Hsi, P.; Lemieux, M.E.; Dal Cin, P.; Ince, T.A.; Kharchenko, P.V.; Kuroda, M.I.; et al. Ectopic protein interactions within BRD4-chromatin complexes drive oncogenic megadomain formation in NUT midline carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, E4184–E4192. [Google Scholar] [CrossRef] [Green Version]
- Reynoird, N.; Schwartz, B.E.; Delvecchio, M.; Sadoul, K.; Meyers, D.; Mukherjee, C.; Caron, C.; Kimura, H.; Rousseaux, S.; Cole, P.A.; et al. Oncogenesis by sequestration of CBP/p300 in transcriptionally inactive hyperacetylated chromatin domains. EMBO J. 2010, 29, 2943–2952. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.W.; He, L.J.; Zheng, S.; Liu, T.; Peng, B.N. An Overview of Molecular Mechanism, Clinicopathological Factors, and Treatment in NUT Carcinoma. BioMed Res. Int. 2019, 2019, 1018439. [Google Scholar] [CrossRef] [Green Version]
- Stevens, T.M.; Morlote, D.; Xiu, J.; Swensen, J.; Brandwein-Weber, M.; Miettinen, M.M.; Gatalica, Z.; Bridge, J.A. NUTM1-rearranged neoplasia: A multi-institution experience yields novel fusion partners and expands the histologic spectrum. Mod. Pathol. 2019, 32, 764–773. [Google Scholar] [CrossRef]
- Chau, N.G.; Ma, C.; Danga, K.; Al-Sayegh, H.; Nardi, V.; Barrette, R.; Lathan, C.S.; DuBois, S.G.; Haddad, R.I.; Shapiro, G.I.; et al. An Anatomical Site and Genetic-Based Prognostic Model for Patients With Nuclear Protein in Testis (NUT) Midline Carcinoma: Analysis of 124 Patients. JNCI Cancer Spectr. 2020, 4, pkz094. [Google Scholar] [CrossRef]
- Bauer, D.E.; Mitchell, C.M.; Strait, K.M.; Lathan, C.S.; Stelow, E.B.; Luer, S.C.; Muhammed, S.; Evans, A.G.; Sholl, L.M.; Rosai, J.; et al. Clinicopathologic features and long-term outcomes of NUT midline carcinoma. Clin. Cancer Res. 2012, 18, 5773–5779. [Google Scholar] [CrossRef] [Green Version]
- Giridhar, P.; Mallick, S.; Kashyap, L.; Rath, G.K. Patterns of care and impact of prognostic factors in the outcome of NUT midline carcinoma: A systematic review and individual patient data analysis of 119 cases. Eur. Arch. Otorhinolaryngol. 2018, 275, 815–821. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef]
- Jang, J.E.; Eom, J.I.; Jeung, H.K.; Cheong, J.W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. AMPK-ULK1-Mediated Autophagy Confers Resistance to BET Inhibitor JQ1 in Acute Myeloid Leukemia Stem Cells. Clin. Cancer Res. 2017, 23, 2781–2794. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.C.; Galanis, E.; Kirn, D. Clinical trial results with oncolytic virotherapy: A century of promise, a decade of progress. Nat. Clin. Pr. Oncol. 2007, 4, 101–117. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef]
- Ning, J.; Rabkin, S.D. Translating Gene Therapy to the Clinic; Laurence, J., Franklin, M., Eds.; Science Direct: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Piha-Paul, S.A.; Hann, C.L.; French, C.A.; Cousin, S.; Brana, I.; Cassier, P.A.; Moreno, V.; de Bono, J.S.; Harward, S.D.; Ferron-Brady, G.; et al. Phase 1 Study of Molibresib (GSK525762), a Bromodomain and Extra-Terminal Domain Protein Inhibitor, in NUT Carcinoma and other Solid Tumors. JNCI Cancer Spectr. 2020, 4, pkz093. [Google Scholar] [CrossRef]
- Rehman, H.; Silk, A.W.; Kane, M.P.; Kaufman, H.L. Into the clinic: Talimogene laherparepvec (T-VEC), a first-in-class intratumoral oncolytic viral therapy. J. Immunother. Cancer 2016, 4, 53. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene 2003, 10, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Bechter, O.; Schoffski, P. Make your best BET: The emerging role of BET inhibitor treatment in malignant tumors. Pharmacol. Ther. 2020, 208, 107479. [Google Scholar] [CrossRef]
- Tontsch-Grunt, U.; Traexler, P.E.; Baum, A.; Musa, H.; Marzin, K.; Wang, S.; Trapani, F.; Engelhardt, H.; Solca, F. Therapeutic impact of BET inhibitor BI 894999 treatment: Backtranslation from the clinic. Br. J. Cancer 2022. [Google Scholar] [CrossRef] [PubMed]
- Kloker, L.D.; Berchtold, S.; Smirnow, I.; Schaller, M.; Fehrenbacher, B.; Krieg, A.; Sipos, B.; Lauer, U.M. The Oncolytic Herpes Simplex Virus Talimogene Laherparepvec Shows Promising Efficacy in Neuroendocrine Cancer Cell Lines. Neuroendocrinology 2019, 109, 346–361. [Google Scholar] [CrossRef]
- Markman, R.L.; Webber, L.P.; Nascimento Filho, C.H.V.; Reis, L.A.; Vargas, P.A.; Lopes, M.A.; Zanella, V.; Martins, M.D.; Squarize, C.H.; Castilho, R.M. Interfering with bromodomain epigenome readers as therapeutic option in mucoepidermoid carcinoma. Cell. Oncol. 2019, 42, 143–155. [Google Scholar] [CrossRef]
- French, C.A.; Cheng, M.L.; Hanna, G.J.; DuBois, S.G.; Chau, N.G.; Hann, C.L.; Storck, S.; Salgia, R.; Trucco, M.; Tseng, J.; et al. Report of the First International Symposium on NUT Carcinoma. Clin. Cancer Res. 2022, OF1–OF13. [Google Scholar] [CrossRef]
- Toda, M.; Rabkin, S.D.; Kojima, H.; Martuza, R.L. Herpes Simplex Virus as an in Situ Cancer Vaccine for the Induction of Specific Anti-Tumor Immunity. Hum. Gene Ther. 1999, 10, 385–393. [Google Scholar] [CrossRef]
- Noll, M.; Berchtold, S.; Lampe, J.; Malek, N.P.; Bitzer, M.; Lauer, U.M. Primary resistance phenomena to oncolytic measles vaccine viruses. Int. J. Oncol. 2013, 43, 103–112. [Google Scholar] [CrossRef]
- Berchtold, S.; Beil, J.; Raff, C.; Smirnow, I.; Schell, M.; D’Alvise, J.; Gross, S.; Lauer, U.M. Assessing and Overcoming Resistance Phenomena against a Genetically Modified Vaccinia Virus in Selected Cancer Cell Lines. Int. J. Mol. Sci. 2020, 21, 7618. [Google Scholar] [CrossRef]
- Schoggins, J.W.; Rice, C.M. Interferon-stimulated genes and their antiviral effector functions. Curr. Opin. Virol. 2011, 1, 519–525. [Google Scholar] [CrossRef]
- Matveeva, O.V.; Chumakov, P.M. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev. Med. Virol. 2018, 28, e2008. [Google Scholar] [CrossRef]
- Shapiro, G.I.; Dowlati, A.; LoRusso, P.M.; Eder, J.P.; Anderson, A.; Do, K.T.; Kagey, M.H.; Sirard, C.; Bradner, J.E.; Landau, S.B. Abstract A49: Clinically efficacy of the BET bromodomain inhibitor TEN-010 in an open-label substudy with patients with documented NUT-midline carcinoma (NMC). Mol. Cancer Ther. 2015, 14, A49. [Google Scholar] [CrossRef]
- Lewin, J.; Soria, J.-C.; Stathis, A.; Delord, J.-P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial with Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients with Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef] [PubMed]
- Doroshow, D.B.; Eder, J.P.; LoRusso, P.M. BET inhibitors: A novel epigenetic approach. Ann. Oncol. 2017, 28, 1776–1787. [Google Scholar] [CrossRef]
- Adeegbe, D.O.; Liu, S.; Hattersley, M.M.; Bowden, M.; Zhou, C.W.; Li, S.; Vlahos, R.; Grondine, M.; Dolgalev, I.; Ivanova, E.V.; et al. BET Bromodomain Inhibition Cooperates with PD-1 Blockade to Facilitate Antitumor Response in Kras-Mutant Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2018, 6, 1234–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison-Smith, C.D.; Knox, T.M.; Filic, I.; Soroko, K.M.; Eschle, B.K.; Wilkens, M.K.; Gokhale, P.C.; Giles, F.; Griffin, A.; Brown, B.; et al. Combined targeting of the BRD4-NUT-p300 axis in NUT midline carcinoma by dual selective bromodomain inhibitor, NEO2734. Mol. Cancer Ther. 2020, 9, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Lee, A.Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [Google Scholar] [CrossRef] [Green Version]
- Stirnweiss, A.; Oommen, J.; Kotecha, R.S.; Kees, U.R.; Beesley, A.H. Molecular-genetic profiling and high-throughput in vitro drug screening in NUT midline carcinoma-an aggressive and fatal disease. Oncotarget 2017, 8, 112313–112329. [Google Scholar] [CrossRef] [Green Version]
- Sun, K.; Atoyan, R.; Borek, M.A.; Dellarocca, S.; Samson, M.E.S.; Ma, A.W.; Xu, G.-X.; Patterson, T.; Tuck, D.P.; Viner, J.L.; et al. Dual HDAC and PI3K Inhibitor CUDC-907 Downregulates MYC and Suppresses Growth of MYC-dependent Cancers. Mol. Cancer Ther. 2017, 16, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Maertens, O.; Cichowski, K.; Elledge, S.J. Genetic modifiers of the BRD4-NUT dependency of NUT midline carcinoma uncovers a synergism between BETis and CDK4/6is. Genes Dev. 2018, 32, 1188–1200. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogas, H.J.; Ribas, A.; Chesney, J.; Long, G.V.; Kirkwood, J.M.; Dummer, R.; Puzanov, I.; Hoeller, C.; Gajewski, T.F.; Gutzmer, R.; et al. 1037O-MASTERKEY-265: A phase III, randomized, placebo (Pbo)-controlled study of talimogene laherparepvec (T) plus pembrolizumab (P) for unresectable stage IIIB–IVM1c melanoma (MEL). In Proceedings of the ESMO Congress, Paris, France, 18 September 2021; pp. S867–S905. [Google Scholar]
- Riess, J.W.; Rahman, S.; Kian, W.; Edgerly, C.; Heilmann, A.M.; Madison, R.; Ramkissoon, S.H.; Klaitman, S.S.; Chung, J.H.; Trabucco, S.E.; et al. Genomic profiling of solid tumors harboring BRD4-NUT and response to immune checkpoint inhibitors. Transl. Oncol. 2021, 14, 101184. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohnesorge, P.V.; Berchtold, S.; Beil, J.; Haas, S.A.; Smirnow, I.; Schenk, A.; French, C.A.; Luong, N.M.; Huang, Y.; Fehrenbacher, B.; et al. Efficacy of Oncolytic Herpes Simplex Virus T-VEC Combined with BET Inhibitors as an Innovative Therapy Approach for NUT Carcinoma. Cancers 2022, 14, 2761. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14112761
Ohnesorge PV, Berchtold S, Beil J, Haas SA, Smirnow I, Schenk A, French CA, Luong NM, Huang Y, Fehrenbacher B, et al. Efficacy of Oncolytic Herpes Simplex Virus T-VEC Combined with BET Inhibitors as an Innovative Therapy Approach for NUT Carcinoma. Cancers. 2022; 14(11):2761. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14112761
Chicago/Turabian StyleOhnesorge, Paul V., Susanne Berchtold, Julia Beil, Simone A. Haas, Irina Smirnow, Andrea Schenk, Christopher A. French, Nhi M. Luong, Yeying Huang, Birgit Fehrenbacher, and et al. 2022. "Efficacy of Oncolytic Herpes Simplex Virus T-VEC Combined with BET Inhibitors as an Innovative Therapy Approach for NUT Carcinoma" Cancers 14, no. 11: 2761. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14112761